

Abstract
Objective
Endothelial transcription factors KLF4 and KLF2 are implicated in protection against atherogenesis. Steady state microRNA (miR) regulation of KLFs in vivo is accessible by screening region-specific endothelial miRs and their targets.
Methods and Results
A subset of differentially expressed endothelial miRs were identified in athero-susceptible vs protected regions of normal swine aorta. In silico analyses predicted highly conserved binding sites in the 3′-untranslated region (3′UTR) of KLF4 for five miRs of the subset (-26a,b, -29a, -92a and -103), and a single binding site for a miR-92a complex in the 3′UTR of KLF2. Of these, only miR-92a knock-down and knock-in resulted in responses of KLF4 and KLF2 expression in human arterial endothelial cells (HAEC). Dual luciferase reporter assays demonstrated functional interactions of miR-92a with full length 3′UTR sequences of both KLFs and with the specific binding elements therein. Two evolutionarily-conserved miR-92a sites in KLF4 3′UTR and one site in KLF2 3′UTR were functionally validated. Knock-down of miR-92a in vitro resulted in partial rescue from cytokine-induced pro-inflammatory marker expression (MCP-1, VCAM-1, E-Selectin and eNOS) that was attributable to enhanced KLF4 expression. Leukocyte-HAEC adhesion experiments supported this conclusion. In swine aortic arch endothelium, a site of athero-susceptibility where miR-92a expression was elevated, both KLFs were expressed at low levels relative to protected thoracic aorta.
Conclusions
miR-92a co-regulates KLF4 and KLF2 expression in arterial endothelium and contributes to phenotype heterogeneity associated with regional athero-susceptibility and protection in vivo.
Keywords: KLF4, KLF2, miR-92a, athero-susceptibility, endothelium in vivo
Atherosclerotic lesions preferentially originate and develop at arterial sites of curvatures, branches, and bifurcations where complex hemodynamic conditions of disturbed flow are associated with chronically ER-stressed endothelial phenotypes expressing pro-inflammatory and pro-coagulant susceptibility1-5. The internal curvature of the aortic arch (AA) is athero-susceptible while the nearby descending thoracic aorta (DT) is protected. Endothelial transcriptome profiling at AA and DT sites in vivo, as well as exposure to various arterial flows in vitro have identified a repertoire of transcription factors that regulate downstream pathways important in endothelial function relevant to atherogenesis1, 6-8. These include transcription factors activator protein 1 (AP-1) and nuclear factor κB (NFκB) that mediate gene sets associated with proinflammatory and procoagulant endothelial phenotypes7, 9. Conversely, Krüppel-like factor 2 (KLF2), Krüppel-like factor 4 (KLF4), and nuclear factor erythroid 2-like 2 (Nrf2) regulate gene networks that confer athero-protective properties to the endothelium through an anti-inflammatory/antioxidant and anti-coagulant phenotype profile6, 10-14.
MicroRNAs (miRNAs), highly conserved noncoding small RNAs of 19–26 nucleotides which posttranscriptionally suppress their target genes, have recently emerged as important pathophysiological mediators of the vascular system15, 16,17, 18. We reported that a cohort of endothelial microRNAs (miRs) are differentially-expressed between athero-susceptible AA and nearby athero-protected DT in normal swine19. One of these, endothelial miR-10a, which is suppressed in AA, serves as a negative regulator of NF-κB signaling and therefore participates in modulating the pro- and anti-inflammatory endothelial phenotypes in vivo19. In addition, flow-sensitive miR-663 and miR-21 have been demonstrated to provoke endothelial inflammation 20, 21, the latter promoting AP-1 expression.
We postulated that other differentially expressed miRs may target molecules known to be important in endothelial regions predisposed to athero-susceptibility or protection; the Krüppel-like family of transcription factors, particularly KLF4 and KLF2, are attractive candidates. Common up-stream stimuli (e.g. laminar flow, statins, MAPK activators, and proteasome inhibitors) and mediators (e.g. MEK5/MEF2-dependent signaling pathway) of endothelial KLF4 and KLF2 have been reported10-12, 22, 23. Very recently, miR regulation of flow-induced endothelial KLF2 expression in vitro has been reported24 but little is known about KLF4 regulation by miRs. Here we characterize miR-92a recognition sites in the 3′UTR of KLF4 and KLF2 that regulate coupled post-transcriptional control of these two genes.
Methods
Expanded methods are provided in online supplemental materials.
Regional Isolation of Arterial Endothelium
Immediately following death of the animal in accordance with normal abattoir procedures, endothelium was isolated from normal swine aortic arch (AA) and descending thoracic aorta (DT) as previously described3, 19 and in the Online Data Supplement.
Endothelial Protein Extraction and Western Blots
Briefly, HAECs in a 60-mm Petri dish and fresh endothelial scrapes from individual swine were collected in ice cold radioimmunoprecipitation assay (RIPA) buffer (Millipore) containing protease inhibitors, phosphatase inhibitors, and proteasome inhibitors. Total protein was isolated by sonication and centrifugation of the cell lysates and measured by bicinchoninic acid protein assay (Pierce). Supplement Table I lists the primary antibodies used.
Transfection of miR Mimetics and Inhibitors in Human Aortic Endothelial Cells
Endogenous expression of individual miRs in human aortic endothelial cells (HAECs, Lonza) was inhibited by transfection with 50 nM miR hairpin inhibitors (miRIDIAN, Dharmacon) targeting hsa-miR-26a, miR-26b, miR-29a, miR-92a, and miR-103 with reference to miR inhibitor negative controls for 48h. To over-express miR-92a in HAEC, cells were transfected with 10 nM hsa-miR-92a mimetics or miRNA mimetic negative controls (Dharmacon) using Lipofectamine RNAiMAX transfection reagent.
MicroRNA and cDNA Quantitative Real-Time PCR
Expression of selected miRs was quantified by two-step quantitative real-time PCR (Applied Biosystems) and normalized to endogenous small nuclear U6 RNA. cDNA was quantified using LightCycler® 480 SYBR Green I Master (Roche). PCR primers for genes of interest are listed in Supplement Table I.
Luciferase Reporter Assay-Full-length 3′ UTRs
Full-length wild-type and mutated 3′ UTRs of human KLF4 and KLF2 were inserted downstream of the firefly luciferase reporter gene in the pEZX-MT01 vectors (GeneCopoeia). Mutant clones were generated employing Polymerase cycling assembly (Assembly PCR) and confirmed by DNA sequencing (GeneCopoeia). The Renilla luciferase encoded by the same vector was used as internal controls for the dual-luciferase assays (GeneCopoeia). To express miR-92a mimetics, HEK 293 cells transfected with a given pEZX-MT01 vector were knocked-in 100 nM hsa-miR-92a mimetics or miR mimetic negative controls (Dharmacon). Alternatively, HEK cells were cotransfected with pEZX-MT01 vectors and pEZM-MR01 vectors which express human miR-92a precursors or scrambled controls (GeneCopoeia).
Luciferase Reporter Assay-miR-92a Binding Elements
MiR-92a binding elements and the corresponding mutants were synthesized and cloned into a pRL-TK vector (Promega) as described in our previous study19. HEK 293 cells were cotransfected with pRL-TK vectors, control pGL3 vectors, and 100 nM hsa-miR-92a mimetics or miR mimetic negative controls, followed by dual-luciferase assays (Promega).
TNFα Stimulation
Six hour incubations of 5 ng/ml recombinant human TNFα (BD and Company) were applied to stimulate endothelial inflammation in control HAECs, cells with knockdown of miR-92a, and cells with knockdown of miR-92a and KLF4. Inflammatory responses were measured by comparing endothelial expression of MCP-1, VCAM-1, E-SEL, and eNOS to corresponding non-treated cells.
Leukocyte Adhesion Assay
HAECs were transfected with miR-92 inhibitors or miR-92a inhibitors/ KLF4-targeting siRNAs for 48h and then replated for overnight growth in 48 well plates. 10ng/ml recombinant human TNFα was added to the confluent endothelial monolayer for 6 h and the leukocyte-endothelial interaction was measured employing the CytoSelect™ Leukocyte-Endothelium Adhesion Assay (Cell Biolabs). THP-1 cells were a gift from Dr. Ellen Pure (Wistar Institute). Briefly, Fluorescence-labeled THP-1 cells were incubated with the TNFα-induced endothelial cells for 1 h before the washes of the non-adherent cells. Adherent leukocytes were visualized under an inverted fluorescence microscope and the fluorescence was measured with a fluorescence plate reader at 480nm/520nm.
Results
In silico prediction of miRNAs putatively regulating endothelial KLF4 and KLF2
The bioinformatics tool TargetScan (Human 5.1) was employed to search the 3′ UTR of KLF4 for the presence of conserved 8mer and 7mer sites that match the seed region of each known miR. The in silico analyses identified twelve evolutionarily conserved putative binding sites for twenty-seven distinct mammalian miRs (Supplement Figure IA). The miRs predicted to interact with KLF4 were cross-referenced with a list of thirty-four endothelial miRs that show differential (up or down) regional expression in swine AA and DT in vivo19. Five miRs (miR-26a, miR-26b, miR-29a, miR-92a, and miR-103; highlighted in bold) were common to both groups. Three (miR-29a, 92a, and 103; solid black boxes) showed higher expression in aortic arch endothelium where KLF4 expression is low while two (miR-26a, 26b; white box) demonstrated low expression in aortic arch. Given the phylogenetic proximity as well as the common upstream stimuli and downstream targets of KLF4 and KLF225, the same approach was employed to probe candidate miRs that may regulate endothelial KLF2. TargetScan identified one evolutionarily-conserved binding site for six distinct mammalian miRs in the 3′UTR of KLF2 (Supplement Figure IB) of which only one, miR-92a, was identified as differentially-expressed between AA and DT endothelial cells. Thus, miR-92a was a predicted common regulator for both KLFs. A less conserved second binding site for miR-92a in the 3′UTR of KLF4 was also identified and investigated by experiment (cross-hatch box).
Modulation of endothelial KLF4 and KLF2 expression by miR-92a
Inhibitors individually introduced into human aortic endothelial cells (HAECs) to target miR-26a, 26b, 29a, 92a, and 103 suppressed their corresponding endogenous miRs by 60-90% (Figure 1A). Only the inhibition of endogenous miR-92a significantly up-regulated endothelial KLF4 mRNA expression (by 4-fold; p<0.001; Figure 1B). In contrast, inhibition of the other four miRs failed to modulate KLF4. Inhibition of miR-92a also showed a statistically significant (1.6-fold; p<0.002) up-regulation of KLF2 (Figure 1C). Knock-down of miR-92a stimulated expression of KLF4 protein but expression of KLF2 protein remained unchanged (Figure 1D). However, knock-in of miR-92a suppressed transcript expression (Figure 1E) and protein expression (Figure 1F) of both KLF4 and KLF2.
Figure 1.
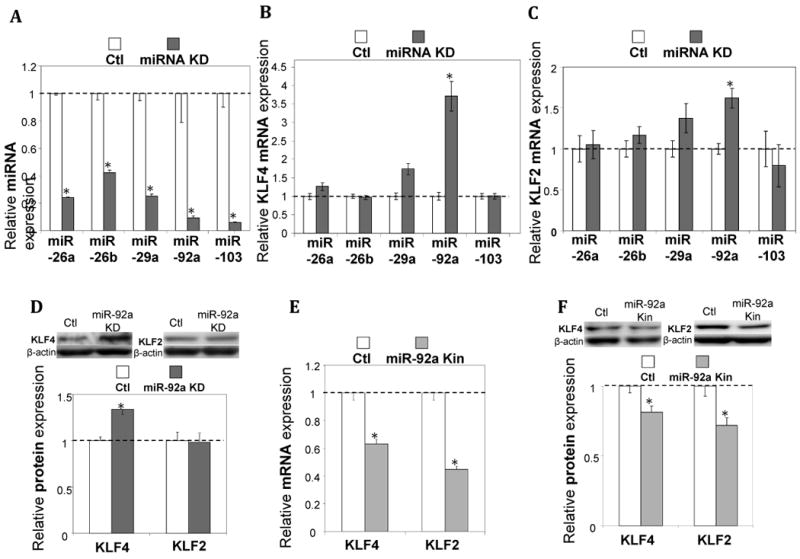
miR-92a regulation of endothelial KLF4 and KLF2. A-D: Knock-down of miRs (A) selectively influenced expression of KLF4/KLF2 mRNA (B, C) and protein (D) in HAECs (n=4-6). E, F: miR-92a knock-in suppressed expression of KLF4 and KLF2 in HAECs (n=4). Data represent mean ± SEM. *p < 0.05. KD: knock-down; Kin: knock-in.
miR-92a inhibits KLF expression through 3′ UTR binding
The functional interaction of miR-92a with the 3′UTR sequences of KLF4 and KLF2 was investigated by dual-luciferase reporter assay. The full-length 3′UTRs containing the predicted miR-92a recognition elements present in KLF4 (2 sites) and KLF2 (single site) were inserted downstream of the firefly luciferase in the pEZX-MT01 vectors (Figure 2A). The vectors also express Renilla luciferase which was utilized for normalization in the dual-luciferase assay. Intracellular delivery of miR-92a mimetics repressed the firefly luciferase activity in HEK293 cells expressing KLF4 3′UTR- or KLF2 3′UTR-containing luciferase transcripts (Figure 2B). In contrast, firefly luciferase without the KLF4- or KLF2-3′UTR insertion was not responsive to the miR-92a knock-in. To further validate the functional interaction between miR-92a and KLF4 and KLF2, the 3′ UTRs containing luciferase vectors were co-transfected with vectors expressing the miR-92a precursors or scrambled controls. Overexpression of miR-92a precursors, but not the scrambled sequence, significantly inhibited the activity of KLF4 3′UTR- or KLF2 3′UTR-containing luciferase in HEK293 cells (Figure 2B) without effect upon the control luciferase. Collectively, the data demonstrate the presence of functional miR-92a binding site(s) in the 3′ UTRs of both human KLF4 and KLF2.
Figure 2.

miR-92a targeting of the 3′ UTRs of KLF4 and KLF2. A: Evolutionarily-conserved putative miR-92a binding sites in 3′ UTRs of KLF4 and KLF2. B: Reduced luciferase activity in HEK293 cells overexpressing miR-92a mimetics or miR-92a precursors following insertion of the full-length 3′UTRs cloned from human KLF4 and KLF2 (n=4-5). Ctl plasmids express wild-type firefly luciferase without 3′ UTR insertions. Data represent mean ± SEM. *p < 0.05.
The function of miR-92a seed-pairing sequences in the full-length 3′UTRs of human KLF4 and KLF2 was tested in mutated clones (Figure 3A). Two evolutionarily-conserved miR-92a seed-pairing sites in human KLF4 3′UTR were mutated as was the single miR-92a seed-pairing site in human KLF2 3′UTR. Overexpression of miR-92a precursors significantly reduced the activity of luciferase that contained the single mutant site in the KLF4 3′UTR (one wild-type miR-92a seed-pairing site remained), suggesting that both of the evolutionarily-conserved in silico-predicted miR-92a pairing-sequences contribute to KLF4 regulation. Mutation of both miR-92a seed-pairing sites in the human KLF4 full-length 3′UTR fully abolished the sensitivity of the luciferase to miR-92a knock-in (Figure 3B). Similarly, mutations that disrupt the base-paired complement between the human KLF2 3′UTR and miR-92a seed region eliminated the miR-92a-mediated suppression of the luciferase (Figure 3B). The sensitivity to miR-92a of the less evolutionarily-conserved binding site identified in the 3′UTR of KLF4 suggests an additive contribution to KLF4 regulation.
Figure 3.

miR-92a negatively regulates KLF4 and KLF2 through the seed-pairing sequence(s) in the 3′UTR binding site(s). A: Schematic representation of the luciferase vectors containing the full-length 3′ UTR(s) of KLF4 and KLF2 and mutation(s) in the miR-92a seed-pairing site(s). B: Reduced luciferase activity in HEK293 cells overexpressing miR-92a precursors following insertion of the mutant clones (n=3-5). Data represent mean ± SEM. *p < 0.05.
Inhibition through miR-92a recognition elements in the 3′ UTR
To further test the function of predicted miR-92a recognition elements present in KLF4 or KLF2 3′UTRs, one evolutionarily-conserved and one less-conserved site in KLF4 and a single evolutionarily-conserved site of KLF2 were inserted into the 3′ UTR of Renilla luciferase (Figure 4A). Intracellular delivery of miR-92a mimetics significantly repressed the activity of Renilla luciferase containing highly evolutionarily-conserved mR-92a recognition elements cloned from the 3′ UTR of KLF4 and KLF2 (Figure 4B). Renilla luciferase with putative let-7b binding sites (control) cloned from LIN-24 3′ UTR, was unresponsive to miR-92a knock-in (Figure 4B). Moreover, the functional assay demonstrated the responsiveness of the less evolutionarily-conserved miR-92a putative binding element, consistent with measurements in constructs expressing human KLF4 full-length 3′ UTR.
Figure 4.


miR-92a negatively regulates KLF4 and KLF2 through the miR-92a recognition elements in the 3′UTR binding site(s). A: Schematic representation of the luciferase vectors containing the corresponding miR-92a binding sites. B: Reduced luciferase activity in HEK293 cells overexpressing miR-92a mimetics following insertion of miR-92a putative binding elements cloned from KLF4 and KLF2 3′ UTRs. Renilla luciferase inserted with putative let-7b binding sites, cloned from LIN-28 3′ UTR, served as negative control (n=3-5). *p < 0.05. C-E: Seed sequence-mediated regulation of KLF4 by miR-92a. C: Point mutations in the miR-92a binding elements that disrupt the interaction between miR-92a seed region and the 3′ UTR of KLF4. D: Schematic representation of the luciferase vectors containing the combinations of wild-type/mutated conserved and less-conserved miR-92a binding sites cloned from 3′ UTR of KLF4. E: Reduced luciferase activity in HEK293 cells overexpressing miR-92a following insertion of miR-92a binding elements cloned from wild-type conserved and less-conserved miR-92a binding sites (n=3-5). Data represent mean ± SEM. *p < 0.05.
To investigate synergy between KLF4 binding sites, a series of luciferase constructs was created containing combinations of the conserved and less-conserved miR-92a binding sites (wild type; wt, and mutant; mut) as outlined in Figure 4C,D. Maximum repression of luciferase activity was associated with the double wt, with incrementally less effects when either site was mutated (Figure 4E); double mut eliminated luciferase repression.
Partial rescue from TNFα-induced endothelial inflammation and leukocyte adhesion by miR-92a-regulated KLF4
Given miR-92a suppression of KLF4 and KLF2 and the well-documented anti-inflammatory role of endothelial KLFs, the effects of miR-92a in endothelial inflammation were determined in miR-92a knockdown and in control HAECs stimulated with TNFα. Six hour incubations of 5ng/ml TNFα significantly induced endothelial inflammatory markers in HAECs compared to non-treated cells. Monocyte chemotactic protein 1 (MCP-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin were upregulated by 10.9, 13.9, 2.7 fold respectively, while athero-protective nitric oxide synthase 3 (eNOS) was inhibited by 47% (Figure 5A-D). Knock-down of endothelial miR-92a significantly dampened TNFα-induced inflammatory responses in HAECs. Specifically, miR-92a knock-down reduced the TNFα-stimulated up-regulation of MCP-1, VCAM-1, and E-SEL by 30% (10.9- vs 7.7- fold), 74% (13.9- vs 5.6- fold), and 24% (2.7- vs 2.05- fold), respectively (Figure 5A-C). In TNFα–stimulated HAECs, eNOS inhibition was attenuated from -47% to -28% (vs control cells) following miR-92a knock-down. Simultaneous siRNA inhibition of KLF4 reversed the miR-92a knock-down results and restored the stronger pro-inflammatory marker expression profile (column 4 of Figure 5A-D) strongly suggesting a dominant role of KLF4 over KLF2. This conclusion is supported by the contrasting responses of KLF4 and KLF2 to TNFα, which has been shown to upregulate KLF4 but downregulate KLF2 expression in human umbilical vein endothelial cells (HUVEC)12. We confirmed that TNFα decreased KLF2 and increased KLF4 expression in HAEC as shown in Supplement Figure IIA. Furthermore, in miR-92a knockdown HAEC, TNFα stimulation completely eliminated KLF2 upregulation but not that of KLF4 (Supplement Figure IIB) indicating that the rescue was primarily attributable to miR-92a-KLF4 interactions. KLF4-targeted siRNAs had no significant effect on KLF2 expression in the miR-92a knockdown cells (Supplement Figure IIIA,B).
Figure 5.
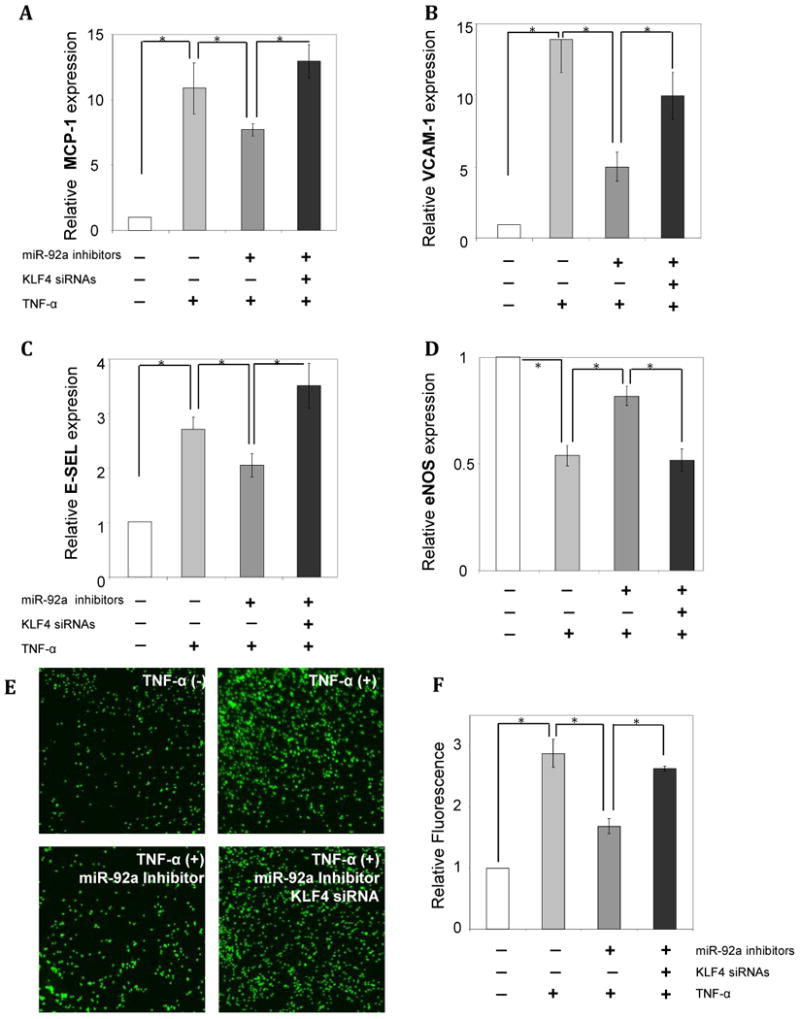
miR-92a primes endothelial inflammation by inhibiting athero-protective KLF4. A, B, C: Knockdown of endogenous miR-92a desensitizes the TNFα-induced inflammatory markers MCP-1 (A), VCAM-1 (B) and E-SEL (C). The rescue effect was reversed in cells in which both miR-92a and KLF4 (siRNA) were knocked-down (n=4). D: Rescue of TNFα-induced inhibition of eNOS in endothelial cells knockdown of endogenous miR-92a and reversal following knockdown of both miR-92a and KLF4 (n=4). E, F: Knockdown of endogenous miR-92a in HAECs decreases THP-1 cell adhesion to HAEC while simultaneous siRNA inhibition of KLF4 restores the level of leukocyte-endothelial interaction. Representative images of fluorescence-labeled THP-1 cells (E), and quantitative fluorescence measurements (F). n=4. Data represent mean ± SEM. *p < 0.05.
To assess the functional consequences of miR-92a knockdown in HAEC, leukocyte-endothelial interactions were determined by THP1-HAEC adhesion assays. As shown in Figure 5 E and F 10ng/ml TNFα markedly increased the numbers of THP-1 cells adherent to the endothelial monolayer. Knockdown of endogenous miR-92a in HAEC partially reduced the TNFα-induced adhesion of THP-1 cells. Moreover, siRNA inhibition of the KLF4 in HAEC simultaneous with miR-92a knockdown restored the strong pro-adhesive endothelial phenotype induced by TNFα (Figure 5 E, F). The data demonstrate that knockdown of endogenous miR-92a desensitizes TNFα-induced endothelial inflammation and adhesion principally through induction of anti-inflammatory KLF4.
In vivo expression of endothelial miR-92a and KLF4/KLF2 in swine aorta
The elevated expression of endothelial miR-92a in athero-susceptible endothelium in vivo is consistent with its athero-promoting role. To test for in vivo equivalence of the in vitro knockdown and knock-in experiments relating miR-92a to KLF4 and KLF2 expression, endothelial KLF4 and KLF2 expression was measured in athero-protected descending thoracic aorta (DT), a site of suppressed expression of miR-92a. Transcript and protein expression of both KLFs was up-regulated in DT compared with aortic arch endothelium (Figure 6) consistent with a steady-state reciprocal relationship between miR-92a and KLF4/KLF2 expression in vivo.
Figure 6.

Endothelial expression of miR-92a, KLF4, and KLF2 in swine athero-susceptible aortic arch (AA) and athero-resistant descending thoracic aorta (DT). A: Low expression of endothelial miR-92a in DT referenced to AA (n=7-9). B, C: Increased expression of endothelial KLF4 and KLF2 mRNA (B) and protein (C) in DT aorta referenced to AA (n=7). Data represent mean ± SEM. *p < 0.05
Discussion
Endothelial KLFs, particularly KLF4 and KLF2, are critical transcriptional regulators of endothelial homeostasis by establishing an anti-inflammatory, vasodilatory, and anti-thrombotic vascular phenotype10-12, 22, 26-28. In vivo regional expression, in silico predictions and experimental validation demonstrated that endothelial miR-92a is an upstream regulator of KLF4 biogenesis and also plays a role in KLF2 expression. Phylogenetic studies demonstrate that the closest evolutionary relationship of the seventeen human KLFs is between KLF4 and KLF225. The structural homology of KLF4 and KLF2 is correlated with functional similarity of regulation by miR-92a. To our knowledge, regulation of endothelial KLF4 by miRs has not previously been reported. Common up-stream stimuli and regulators of endothelial KLF4 and KLF2 have been reported10-12, 22, 23 but have not included co-regulation of these genes by miRNA(s). Approx 90% homology in the zinc finger domains has been identified between KLF4 and KLF2 although they share low homology in the 3′ UTR. The occurrence of the miR-92a seed-pairing sequences in both 3′UTRs of KLF4 and KLF2 regardless of low homology in the regions suggests the functional significance of a previously-unrecognized, coupled post-transcriptional control of these two important transcription factors.
The reduced TNFα-induced inflammation in miR-92a knockdown cells reported herein is consistent with the athero-protective role of endothelial KLF4 and its suppression by miR-92a. Endothelial KLF4 overexpression significantly reduces TNFα-induced VCAM-1 and E-SEL expression12. In contrast, KLF4-targeting siRNAs abolish the up-regulation of eNOS and down-regulation of VCAM-1 conferred by anti-inflammatory kallistatin in endothelial cells treated with TNFα29.
Dysregulated miR-92a is associated with various types of tumors, and plasma miR-92a level is correlated with coronary artery disease in patients30; however, the molecular and cellular functions of miR-92a remain largely unknown. Manni et al31 demonstrated miR-92 increased myeloid cell proliferation by negative regulation of an isoform of the cell-cycle regulator p63. The proapoptotic protein Bim has been shown to be directly suppressed by miR-92a in lymphocytes and monocytes32. A recent study has identified miR-92a as an endogenous repressor of the angiogenic program in endothelial cells by synergistically inhibiting the pro-angiogenic factors integrin subunits α5 and αv, sphingosine-1-phosphate receptor 1, and mitogen-activated kinase kinase 4 33. The same study reported down-regulation of endothelial nitric oxide synthase (eNOS) in human umbilical vein endothelial cells that over-expressed miR-92a; this relationship may also promote an atherogenic role for miR-92a in arterial endothelium where miR-92a is enhanced and eNOS is suppressed in athero-susceptible regions. Given the widespread expression of KLF4/2 in various tissues and their diverse cellular functions including proliferation, differentiation, somatic cell reprogramming, and responses to external stress25, miR-92a may participate in a variety of biological processes in addition to vascular health; miR-92a has been detected in various cell types including cardiomyocytes, fibroblasts, lymphocytes and hematopoietic stem cells.
Spatially-defined non-coding RNAs have emerged as novel posttranscriptional regulators controlling athero-relevant endothelial phenotypes. We reported endothelial down-regulated miR-10a in athero-susceptible aortic arch that promoted pro-inflammatory transcription factor NFκB 19. As shown here, miR-92a is upregulated at the same site and influences the pro-inflammatory cascade via KLFs. Other examples have been reported; Zhou et al21 recently reported that miR-21, which is up-regulated in the athero-susceptible endothelium in vivo19 and induced under athero-susceptible flow waveform in vitro21, suppressed the expression of PPARα expression and therefore up-regulated anti-inflammatory AP-1. Collectively these are likely to be components of a miR regulatory complex responsible for the differential regional susceptibility of the endothelium to overt pathological change. The relative contributions of individual parts to susceptibility are not known, although KLFs have been reported to be disproportionately important transcription factors for the promotion of athero-protection11, 13, 27, 34, and therefore miR-92a regulation is of some importance.
The reciprocal expression of endothelial miR-92a and KLF4/2 in arterial regions exposed to locally disturbed blood flow in vivo is consistent with underlying hemodynamic mechanisms. These include the well-established flow-sensitive nature of KLF4 and KLF26, 11, 22, 35. Wu et al 24 recently reported that an athero-prone flow waveform not only increased the level of endothelial miR-92a but also the association of miR-92a and KLF2 mRNA with both Ago1 and Ago2 proteins that are associated with the RNA-induced silencing complex (RISC), providing molecular clues to the regulation of endothelial KLFs by miR-92a. In addition to hemodynamic forces, pharmacological treatment of statin has been shown to robustly increase both endothelial KLF4 and KLF2 expression. We have tested the hypothesis that miR-92a participates in the statin-induced expression of endothelial KLF4 and KLF222, 36, 37. As shown in Supplement Figure IV, although simvastatin significantly up-regulated endothelial KLF4 and KLF2, it had no effect on the miR-92a expression in cells.
Given that both endothelial KLF4 and KLF2 are associated with athero-protection in vitro and in vivo, miR-92a may be as important in arterial homeostasis as in microcirculatory angiogenesis.
Supplementary Material
Acknowledgments
(a) We thank Drs. Zissimos Mourelatos, Mark Kahn and Richard Assoian of the University of Pennsylvania for critical discussions. Drs. Ellen Pure and Shu-Lin Liu of the Wistar Institute kindly provided THP-1 cells and advised in the adhesion assay. We thank Ms. Kathya Tappeta and Mr. Yuxiang Zhang for assisting in the simvastatin study.
(b) Supported by NIH grants P01 HL62250 and K99 HL103789 and AHA Beginning Grant-in-Aid 11BGIA7080012.
Footnotes
(c) The authors have no conflicts to disclose.
References
- 1.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Jr, Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci U S A. 2004;101:2482–2487. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr, Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ Res. 2009;105:453–461. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–2083. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr, Davies PF. Coronary artery endothelial transcriptome in vivo: identification of endoplasmic reticulum stress and enhanced reactive oxygen species by gene connectivity network analysis. Circ Cardiovasc Genet. 2011;4:243–252. doi: 10.1161/CIRCGENETICS.110.958926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJ. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2) Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 7.Lan Q, Mercurius KO, Davies PF. Stimulation of transcription factors NF kappa B and AP1 in endothelial cells subjected to shear stress. Biochem Biophys Res Commun. 1994;201:950–956. doi: 10.1006/bbrc.1994.1794. [DOI] [PubMed] [Google Scholar]
- 8.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, Garcia-Cardena G, Gimbrone MA., Jr Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci U S A. 2004;101:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, Chaudhury H, Luong le A, Mason JC, Udalova I, Gsell W, Jones H, Haskard DO, Krams R, Evans PC. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: a novel mode of NF-kappaB regulation that promotes arterial inflammation. Circ Res. 2011;108:950–959. doi: 10.1161/CIRCRESAHA.110.233841. [DOI] [PubMed] [Google Scholar]
- 10.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, Garcia-Cardena G, Jain MK. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, Garcia-Cardena G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;282:13769–13779. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- 13.Boon RA, Horrevoets AJ. Key transcriptional regulators of the vasoprotective effects of shear stress. Hamostaseologie. 2009;29:39–40. 41–33. [PubMed] [Google Scholar]
- 14.Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA., Jr Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101:723–733. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 15.Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–1173. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 16.Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59–68. doi: 10.1161/CIRCRESAHA.107.153916. [DOI] [PubMed] [Google Scholar]
- 17.Suarez Y, Sessa WC. MicroRNAs as novel regulators of angiogenesis. Circ Res. 2009;104:442–454. doi: 10.1161/CIRCRESAHA.108.191270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79:581–588. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 19.Fang Y, Shi C, Manduchi E, Civelek M, Davies PF. MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad Sci U S A. 2010;107:13450–13455. doi: 10.1073/pnas.1002120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni CW, Qiu H, Jo H. MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. Am J Physiol Heart Circ Physiol. 2011;300:H1762–1769. doi: 10.1152/ajpheart.00829.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou J, Wang KC, Wu W, Subramaniam S, Shyy JY, Chiu JJ, Li JY, Chien S. MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci U S A. 2011;108:10355–10360. doi: 10.1073/pnas.1107052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villarreal G, Jr, Zhang Y, Larman HB, Gracia-Sancho J, Koo A, Garcia-Cardena G. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem Biophys Res Commun. 2010;391:984–989. doi: 10.1016/j.bbrc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamo L, Zhang Y, Garcia-Cardena G. AICAR activates the pluripotency transcriptional network in embryonic stem cells and induces KLF4 and KLF2 expression in fibroblasts. BMC Pharmacol. 2009;9:2. doi: 10.1186/1471-2210-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu W, Xiao H, Laguna-Fernandez A, Villarreal G, Jr, Wang KC, Geary GG, Zhang Y, Wang WC, Huang HD, Zhou J, Li YS, Chien S, Garcia-Cardena G, Shyy JY. Flow-Dependent Regulation of Kruppel-Like Factor 2 Is Mediated by MicroRNA-92a. Circulation. 2011;124:633–641. doi: 10.1161/CIRCULATIONAHA.110.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–4363. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- 27.Atkins GB, Jain MK. Role of Kruppel-like transcription factors in endothelial biology. Circ Res. 2007;100:1686–1695. doi: 10.1161/01.RES.0000267856.00713.0a. [DOI] [PubMed] [Google Scholar]
- 28.Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr, Balasubramanian V, Garcia-Cardena G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48–57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- 29.Shen B, Smith RS, Jr, Hsu YT, Chao L, Chao J. Kruppel-like factor 4 is a novel mediator of Kallistatin in inhibiting endothelial inflammation via increased endothelial nitric-oxide synthase expression. J Biol Chem. 2009;284:35471–35478. doi: 10.1074/jbc.M109.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 31.Manni I, Artuso S, Careccia S, Rizzo MG, Baserga R, Piaggio G, Sacchi A. The microRNA miR-92 increases proliferation of myeloid cells and by targeting p63 modulates the abundance of its isoforms. Faseb J. 2009;23:3957–3966. doi: 10.1096/fj.09-131847. [DOI] [PubMed] [Google Scholar]
- 32.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 34.Atkins GB, Wang Y, Mahabeleshwar GH, Shi H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI, Jain MK. Hemizygous deficiency of Kruppel-like factor 2 augments experimental atherosclerosis. Circ Res. 2008;103:690–693. doi: 10.1161/CIRCRESAHA.108.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Methe H, Balcells M, Alegret Mdel C, Santacana M, Molins B, Hamik A, Jain MK, Edelman ER. Vascular bed origin dictates flow pattern regulation of endothelial adhesion molecule expression. Am J Physiol Heart Circ Physiol. 2007;292:H2167–2175. doi: 10.1152/ajpheart.00403.2006. [DOI] [PubMed] [Google Scholar]
- 36.Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–726. doi: 10.1161/CIRCULATIONAHA.104.525774. [DOI] [PubMed] [Google Scholar]
- 37.Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr, Garcia-Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–26719. doi: 10.1074/jbc.C500144200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.