

Abstract
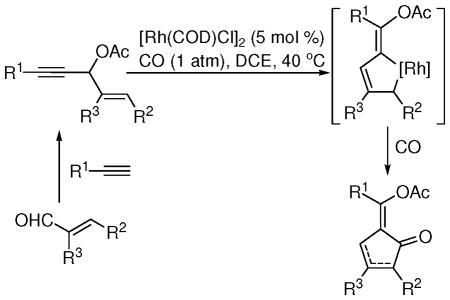
Functionalized cyclopentenones were synthesized by a Rh-catalyzed carbonylation of 3-acyloxy-1,4-enynes, derived from alkynes and α,β-unsaturated aldehydes. The reaction involved a Saucy-Marbet 1,3-acyloxy migration of propargyl esters and a [4+1] cycloaddition of the resulting acyloxy substituted vinylallene with CO.
The transition metal-catalyzed cycloaddition reaction is one of the most efficient ways to access ring systems.1 Formation of cyclopentenones via Pauson-Khand cycloaddition has proven to be extremely valuable for the synthesis of natural products and pharmaceutical agents.2 However, transition metal-catalyzed intermolecular Pauson-Khand reactions are still challenging for the synthesis of various monocyclic cyclopentenones. Even in the case of intramolecular reactions, the scope of alkenes is often limited. Efficient synthesis of cyclopentenones with diverse substitutions and functionalities are still highly desirable and continue to stimulate the development of novel cycloaddition reactions.2f
We herein report an efficient synthesis of highly functionalized cyclopentenones 4 and 5 from 3-acyloxy-1,4-enyne 3 via a Rh(I)-catalyzed carbonylation reaction (Scheme 1). Since substrate 3 could be conveniently prepared from the addition of terminal alkyne 1 to α,β-unsaturated aldehyde 2 followed by esterification, this carbonylation reaction offered an efficient protocol to access functionalized cyclopentenones, which are present in many bioactive natural products and pharmaceuticals. For examples, 5-alkylidene-cyclopent-2-enones such as clavulones2g and punaglandins2h display anti-inflammatory and anti-tumor activities respectively.
Scheme 1.

Carbonylation of 3-acyloxy-1,4-enyne for the synthesis of cyclopentenones
We and others have demonstrated that 3-acyloxy-1,4-enynes with a terminal alkyne (3, R1=H) can serve as a five-carbon building block for [5+1]3 and [5+2]4 cycloadditions with CO and alkynes respectively. Both cycloadditions involved a Rh-catalyzed 1,2-acyloxy migration of propargyl esters, a process first described by Rautenstrauch in 1984 using a Pd(II) catalyst.5
We found that propargyl esters with an internal alkyne underwent 1,3-acyloxy migration to form an acyloxy substituted allene intermediate in several Rh-catalyzed cascade reactions.6 The 1,3-acyloxy migration of propargyl esters was first discovered by Saucy and Marbet using a Ag(I) catalyst.7 Trapping the resulting allene intermediate has been realized in tandem reactions catalyzed by Ag(I), Cu(I), Pt(II), and Au(I) complexes.8 We envisioned that a new carbonylation method could be realized for the synthesis of cyclopentenones if a Rh(I) complex was able to catalyze the Saucy-Marbet rearrangement of 3-acyloxy-1,4-enynes with an internal alkyne (3, R1≠H) and a [4+1] cycloaddition of the resulting vinylallene with CO.9
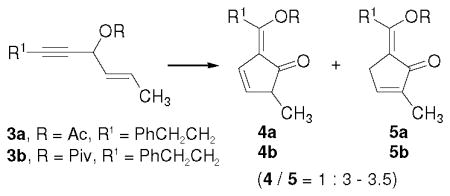
We were pleased to find that [Rh(CO)2Cl]2 was indeed able to facilitate both the 1,3-acyloxy migration of propargyl ester 3a and [4+1] cycloaddition of the resulting vinylallene with CO (entry 1, Table 1). A mixture of isomeric alkylidene cyclopentenones 4a and 5a was observed. We then examined various other Rh(I) catalysts (entries 2–5). A slightly better yield was obtained using [Rh(COD)Cl]2 catalyst (entry 2). We also tried substrate 3b bearing a pivalate group (entry 6). The combined yield and the ratio of 4b and 5b were similar to results from acetate shown in entry 2. Since acetate is generally easier to be removed, we decided to focus on acetate 3a for further optimization of the conditions. Dichloroethane (DCE) provided the best results among solvents we screened (entries 2 and 7–9). We then examined the effect of temperature (entries 10–12). The best result was obtained at 40 °C (entry 11). Product 5a could be separated from minor isomer 4a and isolated in 70% yield.
Table 1.
Screening of catalysts and conditions for carbonylation of 3-acyloxy-1,4-enynesa
 | ||
|---|---|---|
| entry | conditions | yield (4 + 5) |
| 1 | [Rh(CO)2Cl]2 (5 mol %), DCE, 60 °C | 53% |
| 2 | [Rh(COD)Cl]2 (5 mol %), DCE, 60 °C | 55% |
| 3 | [Rh(CO)2(acac)] (5 mol %), DCE, 60 °C | 10% |
| 4 | [Rh(PPh3)3Cl] (5 mol %), DCE, 60 °C | 30% |
| 5 | [Rh(COD)2]BF4 (5 mol %), DCE, 60 °C | trace |
| 6b | [Rh(COD)Cl]2 (5 mol %), DCE, 60 °C | 56% |
| 7 | [Rh(COD)Cl]2 (5 mol %), toluene, 60 °C | 43% |
| 8 | [Rh(COD)Cl]2 (5 mol %), dioxane, 60 °C | 46% |
| 9 | [Rh(COD)Cl]2 (5 mol %), CH3CN, 60 °C | 33% |
| 10 | [Rh(COD)Cl]2 (5 mol %), DCE, 80 °C | 38% |
| 11 | [Rh(COD)Cl]2 (5 mol %), DCE, 40 °C | 81% (70%, Z/E=7:1)c |
| 12 | [Rh(COD)Cl]2 (5 mol %), DCE, rt | 40% |
Unless noted otherwise, substrate 3a was employed. The combined yield and the ratio of 4a and 5a was determined by 1H NMR. The ratio of 4a/5a ranged from 1:3 to 1:3.5. All reactions were carried out under a CO balloon for 8 h.
Substrate 3b was employed. The ratio of 4b/5b was 1:3.5.
Isolated yield of 5a. The Z/E ratio was determined by 1H NMR.
For all entries in Table 1, the ratios of 4a/5a did not change significantly. They ranged from 1:3 to 1:3.5. We observed no obvious change to the ratio of 4a/5a when the mixture of two products was treated with bases (e.g. DBU, DBN, DMAP, DABCO, DIPEA, or Et3N). When dppf was employed as the ligand, the ratio of 4a/5a became 1:1 after the carbonylation. Most other ligands (e.g. dppe, dppp, and dppb) had no effect on the ratio.
Similar results (yield of 5c and ratios of 4c/5c) were observed for substrate 3c (Scheme 2). The conversion was low for substrate 3d with a phenyl substituted alkene. After 40 h, product 5d was isolated in 20% yield and 60% starting material was recovered. The yield of product 5d could not be improved by varying the temperature or CO pressure.
Scheme 2.

Carbonylation of 3-acyloxy-1,4-enyne with di- and trisubstituted alkenes
For substrate 3e with a tri-substituted olefin, isomer 4e became the major product (condition a, Scheme 2). The ratio of isomers 4e/5e was about 7:1 based on the 1H NMR of the crude product. After purification by silica gel column chromatography, however, we found that the ratio of 4e/5e varied each time. We suspected that isomer 5e with a tetrasubstituted olefin might become more stable and the isomerization from 4e to 5e occurred during purification. Indeed, when the crude product from the Rh-catalyzed carbonylation was treated with 2.0 equiv of triethylamine, isomer 4e was not detected and product 5e was isolated in 86% yield. This greatly simplified the purification and characterization of the cyclopentenone product.
The scope of the carbonylation was examined more extensively for substrates with a tri-substituted olefin (Table 2). Substrates 3e—3k could be easily prepared from the addition of various terminal alkynes to commercially available tiglic aldehyde followed by acetylation. For substrate 3f with a phenyl substituted alkyne, a complex mixture was observed. Enynes 3g—3k with various alkyl groups on the alkyne termini all participated in the tandem reaction. The yield became lower for substrate 3k with a siloxy group on the propargylic position.
Table 2.
Carbonylation of 3-acyloxy-1,4-enynesa
| substrates | products | yield,b (Z/E) |
|---|---|---|
 3f |
a complex mixture | - |
 3g |
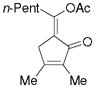 5g |
72%,c (8:1) |
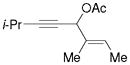 3h |
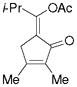 5h |
55%, (3:1) 12%d |
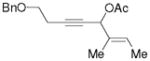 3i |
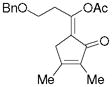 5i |
68%, (8:1) |
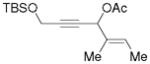
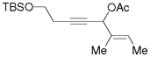 3j |
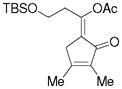 5j |
66%, (17:1) |
 3k |
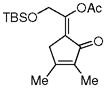 5k |
48%, (13:1) |
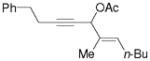 3l |
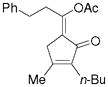 5l |
56%, (7:1) |
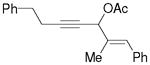 3m |
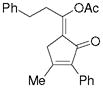 5m |
51%,c (6:1) 65%,c,e (6:1) |
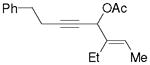 3n |
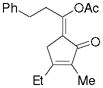 5n |
75%,c (11:1) |
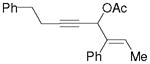 3o |
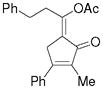 5o |
67%, (12:1) |
|
|
|
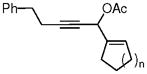
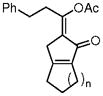
| 3p, n = 1 | 5p, n = 1 | 84%,c (7:1) |
| 3q, n = 2 | 5q, n = 2 | 82%,c (7:1) |
| 3r, n = 3 | 5r, n = 3 | 70%, c (5:1) |
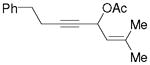 3s |
- | 0% |
|
3t |
- | 0% |
Conditions: [Rh(COD)Cl]2 (5 mol %), CO (1 atm), 40 °C, DCE, 8–20 h, then Et3N (2.0 equiv). The ratios of Z/E isomer were determined by 1H NMR of the crude product.
Isolated yield of the Z-isomer unless noted otherwise.
Isolated yield of the mixture of Z- and E-isomers.
Isolated yield of E-isomer.
The reaction was run at 5 atm of CO.
Substrate 3l with a n-Bu group on the alkene could also be tolerated. Based on results for substrate 3d in Scheme 2, it appeared that the introduction of a phenyl substituent to the terminal position of the alkene decreased the reactivity of the substrate significantly. We were pleased to find that product 5m could be prepared in 51% yield under similar conditions. The yield of this product could be further improved to 65% with higher CO pressure. The carbonylation worked smoothly for substrates 3n and 3o, where the internal position of the alkene had an ethyl or aryl substituent.
Bicyclic products 5p, 5q, and 5r were also prepared in good yields from the corresponding 3-acyloxy-1,4-enynes. This provided an easy access to 5-5, 5-6, and 5-7 fused compounds with a highly functionalized cyclopentenone.
No desired cyclopentenone product was observed for substrates 3s and 3t. When we tried to prepare tertiary propargyl esters with a trisubstituted alkene, they underwent isomerization to form a mixture of conjugated enynes during the preparation.
For many cyclopentenone products in Table 2, the major Z-isomer was separated and isolated for clear characterization. The combined yield of both isomers would be slightly higher. The Z/E ratio of product 5 is actually inconsequential after hydrolysis of the enol ester. The mixture of carbonylation products derived from substrate 3q was then directly treated with K2CO3 in methanol (Scheme 3). Product 1,3-diketone 6q could be isolated in 68% yield after two steps. The NMR spectra of diketone 6q, however, was complex due to the presence of different enols. The 1,3-diketone was then further alkylated with methyl acrylate.10 Bicyclic compound 7 could be isolated in high yield and clearly characterized. Monocyclic cyclopentenone 8 was obtained in 50% overall yield from substrate 3e.
Scheme 3.

Functionalization of cyclpentenones derived carbonylation of 3-acyloxy-1,4-enynes
The mechanism for the carbonylation of 3-acyloxy-1,4-enynes was proposed in Scheme 4. The alkenyl substituted propargyl ester 3 could undergo a Rh-catalyzed 1,3-acyloxy migration to form allene intermediate 10.6 The coordination of the acyloxy group to Rh may favor the formation of metallocyclopentene intermediate 11, which will produce the Z-isomer of cyclopentenones 4 or 5 after CO insertion and reductive elimination.
Scheme 4.

Proposed mechamism for the carbonylation of 3-acyloxy-1,4-enynes
In summary, we have developed an efficient method for the synthesis of various highly functionalized monocyclic and bicyclic cyclopentenones from readily available 3-acyloxy-1,4-enynes. The combination of the novel reactivity of Rh(I) catalyst for promoting 1,3-acyloxy migration of propargyl esters and its ability to facilitate a carbonylation reaction made this tandem transformation possible. The acyloxy group in the propargyl ester starting material not only eliminates the need for the preformation of allenes, but also provides a useful handle for further selective functionalizations of the cyclopentenone products.
Supplementary Material
Acknowledgments
We thank the NIH (R01 GM088285), the Chinese Scholarship Council (to S.H.), and the University of Wisconsin for financial support and a Young Investigator Award (to W.T.) from Amgen.
Footnotes
Supporting Information Available 1H NMR, 13C NMR, IR, HRMS for starting materials and products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected reviews on transition metal catalyzed cycloadditions, see: Lautens M, Klute W, Tam W. Chem Rev. 1996;96:49. doi: 10.1021/cr950016l.Fruhauf HW. Chem Rev. 1997;97:523. doi: 10.1021/cr941164z.Aubert C, Buisine O, Malacria M. Chem Rev. 2002;102:813. doi: 10.1021/cr980054f.Evans PA. Modern Rhodium-Catalyzed Organic Reactions. Wiley-VCH; Weinheim: 2005. Michelet V, Toullec PY, Genet JP. Angew Chem Int Ed. 2008;47:4268. doi: 10.1002/anie.200701589.Yu Z, Wang Y, Wang Y. Chem Asian J. 2010;5:1072. doi: 10.1002/asia.200900712.Inglesby PA, Evans PA. Chem Soc Rev. 2010;39:2791. doi: 10.1039/b913110h.Aubert C, Fensterbank L, Garcia P, Malacria M, Simonneau A. Chem Rev. 2011;111:1954. doi: 10.1021/cr100376w.
- 2.Khand IU, Knox GR, Pauson PL, Watts WE, Foreman MI. J Chem Soc Perkin Trans 1. 1973:977.For selected reviews on Pauson-Khand reactions, see: Gibson SE, Mainolfi N. Angew Chem Int Ed. 2005;44:3022. doi: 10.1002/anie.200462235.Park JH, Chang KM, Chung YK. Coord Chem Rev. 2009;253:2461.Habermann J. Curr Org Chem. 2010;14:1139.Lee HW, Kwong FY. Eur J Org Chem. 2010:789.For a recent review on synthesis of cyclopentenones, see: Gibson SE, Lewis SE, Mainolfi N. J Organomet Chem. 2004;689:3873.For selected bioactive cyclopentenones, see: Kikuchi H, Tsukitani Y, Iguchi K, Yamada Y. Tetrahedron Lett. 1982;23:5171.Baker BJ, Okuda RK, Yu PTK, Scheuer PJ. J Am Chem Soc. 1985;107:2976.
- 3.Brancour C, Fukuyama T, Ohta Y, Ryu I, Dhimane AL, Fensterbank L, Malacria M. Chem Commun. 2010;46:5470. doi: 10.1039/c0cc00747a. [DOI] [PubMed] [Google Scholar]
- 4.Shu XZ, Huang S, Shu D, Guzei IA, Tang W. Angew Chem Int Ed. 2011;50:8153. doi: 10.1002/anie.201103136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Rautenstrauch V. J Org Chem. 1984;49:950. [Google Scholar]; b) Rautenstrauch V. Tetrahedron Lett. 1984;25:3845. [Google Scholar]
- 6.a) Shu D, Li X, Zhang M, Robichaux PJ, Tang W. Angew Chem Int Ed. 2011;50:1346. doi: 10.1002/anie.201006881. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li X, Zhang M, Shu D, Robichaux PJ, Huang S, Tang W. Angew Chem Int Ed. 2011;50:10421. doi: 10.1002/anie.201104861. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang S, Li X, Lin CL, Guzei IA, Tang W. Chem Commun. 2012;48:2204. doi: 10.1039/c2cc17406e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saucy Von G, Marbet R, Lindlar H, Isler O. Helv Chim Acta. 1959;42:1945. [Google Scholar]
- 8.For a leading review on π-acidic metal catalyzed 1,3-acyloxy migration of propargyl esters, see: Wang S, Zhang G, Zhang L. Synlett. 2010:692.For selected related reviews on π-acidic metal catalyzed reactions, see: Marion N, Nolan SP. Angew Chem Int Ed. 2007;46:2750. doi: 10.1002/anie.200604773.Fürstner A, Davies PW. Angew Chem Int Ed. 2007;46:3410. doi: 10.1002/anie.200604335.Hashmi ASK. Chem Rev. 2007;107:3180. doi: 10.1021/cr000436x.Hashmi ASK. Angew Chem Int Ed. 2008;47:6754. doi: 10.1002/anie.200802517.Jimenez-Nunez E, Echavarren AM. Chem Rev. 2008;108:3326. doi: 10.1021/cr0684319.Gorin DJ, Sherry BD, Toste FD. Chem Rev. 2008;108:3351. doi: 10.1021/cr068430g.
- 9.For rhodium-catalyzed [4+1] cycloaddition of vinylallene with CO, see: Murakami M, Itami K, Ito Y. Angew Chem Int Ed. 1995;34:2691.Murakami M, Itami K, Ito Y. J Am Chem Soc. 1997;119:2950.Murakami M, Itami K, Ito Y. J Am Chem Soc. 1999;121:4130.Murakami M, Itami K, Ito Y. Organometallics. 1999;18:1326.For iron-mediated [4+1] cycloadditions, see: Sigman MS, Kerr CE, Eaton BE. J Am Chem Soc. 1993;115:7545.Sigman MS, Eaton BE. J Org Chem. 1994;59:7488.Sigman MS. Eaton BE, Heise JD, Kubiak CP. Organometallics. 1996;15:2829.Sigman MS, Eaton BE. J Am Chem Soc. 1996;118:11783.
- 10.Ye W, Xu J, Tan CT, Tan CH. Tetrahedron Lett. 2005;46:6875. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.