

Abstract
Infection with Helicobacter pylori is associated with significantly reduced risks of oesophageal adenocarcinoma, however few studies have examined the association between H pylori and Barrett's oesophagus (BO), the precursor lesion. We explored the relationship between H pylori infection and BO and sought to identify potential modifiers. We compared the prevalence of positive H pylori serology among 217 adults with simple BO (without dysplasia), 95 with dysplastic BO and 398 population controls sourced from the metropolitan Brisbane area. We determined H pylori serostatus using enzyme-linked immunosorbent assay. To estimate relative risks, we calculated odds ratios (OR) and 95% confidence intervals (CI) using multivariable logistic regression in the entire sample and stratified by factors known to cause BO. The prevalence of H pylori seropositivity was 12%, 3% and 18% respectively, among patients with simple BO, dysplastic BO and population controls. BO patients were significantly less likely to have antibodies for H pylori (Simple BO: OR=0.51, 95% CI: 0.30-0.86; Dysplastic BO: OR=0.10, 95% CI: 0.03-0.33) than population controls. For simple BO, the association was diminished after adjustment for frequency of gastro-oesophageal reflux (GOR) symptoms. Adjustment for frequency of GOR symptoms did not substantially alter the observed effect for dysplastic BO. While there was some variation in the magnitude of risk estimates across strata of age, sex, GOR symptoms, and use of PPIs or H2-receptor antagonists, the differences were uniformly nonsignificant. H pylori infection is inversely associated with BO, and our findings suggest that decreased acid load is not the only mechanism underlying the H pylori protective effect.
Keywords: Barrett's oesophagus, environmental modifiers, epidemiology, Helicobacter pylori, gastro-oesophageal reflux
Introduction
Barrett's oesophagus (BO) is an acquired premalignant condition in which the oesophageal squamous epithelium is replaced by specialised intestinal metaplasia.1 BO is a recognised precursor lesion of oesophageal adenocarcinoma (OAC), the incidence of which is rising more rapidly than that of any other malignancy in many Western populations.2-6 People with BO have a 30- to 40-fold increased risk of OAC but there is currently no way of predicting which BO patients will progress to OAC.7, 8 Evaluation of potential risk factors for BO may provide information on early events in oesophageal carcinogenesis that are amenable to intervention, with the long term aim of reducing the morbidity and mortality associated with OAC.
Helicobacter pylori is a bacterium that colonises the human stomach.9 Epidemiological studies have shown that while infection with H pylori is causally associated with the development of gastric cancer,10 infection with this organism is associated with reduced risks of OAC.11-16 It is hypothesised that the reduction in risk is due to less frequent gastro-oesophageal reflux (GOR) resulting from diminished gastric acid secretion and the induction of atrophic gastritis in those infected with H pylori.9, 17 However, there is some evidence that not all the protective effect may be explained by reduced gastric acid production. H pylori colonisation is found to increase gastric acid secretion in some subgroups of the population, thus H pylori may in fact contribute to GOR in certain patients.18-21 It is postulated that the protective effect may act early in the oesophageal inflammation-metaplasia-dysplasia-adenocarcinoma sequence before BO. In comparison with OAC, few studies have examined the association between H pylori and BO, and the majority were conducted among referral populations of endoscopy patients and lacked a true population-based comparison group.22, 23 These studies may be biased due to differences in health-care seeking behaviour of people who come to medical attention and those who do not. Only a small number of studies to date have compared patients with BO to population controls.24, 25 Additionally, the magnitude of the association may differ across subgroups, however few studies have considered potential effect modifying by risk factors for BO.
Here, we report the findings of a population-based case-control study evaluating whether H pylori antibody status was associated with BO and, separately, BO with dysplasia. We also sought to identify potential modifiers of the associations.
Material and Methods
We compared the prevalence of circulating immunoglobulin G antibodies against H pylori using serum samples from participants in a population-based case-control study of BO conducted in Brisbane, Australia. Approval to undertake the study was obtained from the human research ethics committees of the Queensland Institute of Medical Research and from all participating hospitals. Case and control participants provided written informed consent to take part in the parent study and subsequent analyses. Patients who had died or who were mentally incompetent, too ill to participate, or unable to complete an English language questionnaire were excluded.
Study participants
The study population and methods have been described in detail previously.26 Briefly, eligible case patients were residents of metropolitan Brisbane aged 18–79 years with a new (incident) histologically confirmed diagnosis of BO between 1 February 2003 and 30 June 2006. BO was defined as the presence of specialised intestinal metaplasia (i.e. columnar epithelium with goblet cells) in an oesophageal biopsy taken from the tubular oesophagus by upper gastrointestinal endoscopy, irrespective of the length of involvement. This analysis was restricted to patients with newly diagnosed BO (for simple cases), or newly diagnosed dysplasia (for dysplastic cases). A total of 1714 patients with presumptive BO were approached through pathology laboratories servicing metropolitan Brisbane (population 1.5 million), of whom 1096 gave permission (64% response rate) to the pathology laboratories to release their contact details to the study investigators. Of these, 487 patients were found to have a previous diagnosis of BO and a further 86 patients had only intestinal metaplasia of the gastro-oesophageal junction; both groups were deemed ineligible for this analysis. A further 130 potentially eligible patients were excluded for various reasons (too sick, unable to complete a questionnaire, etc). Thus, a total of 393 patients returned a completed questionnaire, with data available for 285 simple BO and 108 dysplastic BO patients.
Population control participants from the same geographic region were randomly selected from the Australian Electoral Roll (enrolment is compulsory by law in Australia), frequency matched to the case series of simple BO by sex and 5-year age group. Of 1554 potentially eligible controls that were contacted and invited to participate, 2 were ineligible due to a diagnosis of BO, 30 were excluded (2 deceased, 4 too ill, 17 were unable to read or write in English, 7 other exclusion) and 404 failed to respond to the invitation. Of 1118 remaining controls, 746 accepted the invitation, and 644 returned the completed questionnaires (the response rate among controls was 72% of those contacted). Population controls were not required to undergo endoscopy as part of their participation in the study.
Data collection
Data were collected through structured, self-completed health and lifestyle questionnaires, followed by standardised telephone interviews with trained research nurses. We elicited a history of GOR symptoms by asking about experience of heartburn (“a burning pain behind the breastbone after eating”) or acid reflux (“a sour taste from acid or bile rising up into the mouth or throat”). For analysis, we used the highest reported frequency for either symptom occurring during the 10 years before diagnosis or reference date. Height and weight one year ago were self-reported by participants, and body mass index (BMI) was calculated (kg/m2). Participants were asked whether, over their whole life, they had ever smoked more than 100 cigarettes or cigars (or equivalent use of pipes). Positive responses led to further questions regarding levels and duration of smoking. We estimated each participant's lifetime cumulative quantity of tobacco smoked (dose) in pack-years, derived by dividing the number of cigarettes smoked on a typical day by 20 and multiplying by the total number of years smoked. Participants were asked to report the frequency with which they consumed six classes of alcohol (reduced-alcohol beer, regular beer, white wine, red wine, port/sherry, and spirits/liqueur) during the age intervals of 20–29 years, 30–49 years, and 50 years and older, as applicable. For these analyses, total alcohol consumption was summed across all age groups and we then calculated the average number of standard drinks (10 g ethanol) consumed per week between age 20 years and current age. Participants were also asked if they had ever used, separately, aspirin or other non-steroidal anti-inflammatory drugs (NSAIDs) during the past 5 years and, if so, the frequency of use on a seven-point scale. Finally, participants were asked whether they had ever used H2-receptor antagonists and proton pump inhibitors (PPIs).
Serum availability
Non-fasting samples of whole blood were collected in plain tubes from 91% and 90% of participating simple BO and dysplastic BO patients, respectively, and from 85% of participating population controls. We had useable serum available for H pylori assays for 398 (62%) population controls, 217 (76%) patients with simple BO and 95 (88%) patients with dysplastic BO. Controls for whom serological data were available were older, on average, and more likely to have experienced symptoms of heartburn or acid reflux than controls without serological data. Cases (i.e., simple BO and dysplastic BO) with serological data were similar to cases without serological data.
Serologic methods
Blood samples were collected, transported overnight to the processing centre and serum samples were then stored at -80°C according to a common protocol. Serum immunoglobulin G antibodies to H pylori were measured using a commercially available enzyme-linked immunosorbent assay kit (Genesis Diagnostics Ltd, Littleport, Cambridge, UK), used according to the manufacturer's instructions. Briefly, diluted serum samples were incubated with partially purified H pylori antigens immobilised on microtiter wells. After washing away unbound serum components, rabbit anti-human immunoglobulin G conjugated to horseradish peroxidise was added to the wells. Unbound conjugate was removed by washing, and a solution containing 3, 3′, 5, 5′-tetramethylbenzidine and enzyme substrate was added to trace specific antibody binding. The optical densities of the standards, controls, and samples were measured using a microplate reader at 450 nm. Each rack of assays contained a mixed batch of case and control samples and analysts were blinded to participant status. For each participant, duplicate samples were run. In addition, test samples known to be positive or negative for H pylori antibodies were run in the batch. For these analyses, an index of <0.9 was considered ‘negative’, an index of ≥1.1 was considered ‘positive’, and values between 0.9 and <1.1 were ‘equivocal’.
Statistical analysis
Our primary aim was to measure the relative risk of BO associated with H pylori antibody status, and then to assess the effect of potential modifiers if associations were observed. We fitted multivariate logistic regression models to calculate the odds ratios (OR) and the 95% confidence intervals (95% CI) for the association between H pylori seropositivity and the two BO outcomes. Our approach was, first, to fit simple age- and sex-adjusted models for each exposure. We then additionally adjusted for those variables that were significantly associated with BO in our data set, namely education, cumulative smoking history (‘never smoker’, ‘<30 pack-years’, ‘≥30 pack-years’), average lifetime alcohol consumption (Never drinkers, <1, 1–6, 7–20, ≥21 drinks/week), frequency of aspirin/NSAID use (‘never’, ‘ever’), and BMI (<25.0, 25.0–29.9, ≥30 kg/m2). Fully adjusted models included the preceding variables as well as a term for frequency of GOR symptoms (‘never’, ‘less than weekly’, ‘at least weekly’). To explore whether any associations between H pylori antibody status and risk of BO were modified by exposure to known or suspected causal factors for BO, we repeated the above analyses within strata of sex, age (<60, ≥60 yrs), frequency of GOR symptoms, cumulative smoking history, BMI, use of H2-receptor antagonists and, separately, use of PPIs (‘never’, ‘ever’). To assess statistical significance of differences in associations across the strata of host characteristics, we assessed the P value for the type III analysis of effects for the interaction term.
We used imputation and sensitivity analysis to assess potential selection bias resulting from study participants (controls and cases) with missing serological data (termed ‘non-serology’ participants). For these analyses, we imputed the H pylori serostatus for ‘non-serology’ participants as per Pandeya et al.27 Briefly, probabilities for H pylori serostatus (positive, negative and equivocal) were derived from the distributions among participants with serological data for each stratum of age, sex and history of GOR symptoms. A random number from the uniform distribution U [0, 1] was then drawn for each ‘non-serology’ participant. ‘Non-serology’ participants with a random number less than or equal to the probability of H pylori seropositivity (π1) were assigned a ‘positive’ status. Those ‘non-serology’ participants not assigned ‘positive’ were then assigned a ‘negative’ status if a second random number was less than or equal to the probability of being negative (π2) excluding the positives (that is, π2/(1- π1)). Finally, ‘non-serology’ participants not assigned ‘positive’ or ‘negative’ were automatically assigned an ‘equivocal’ status. We then repeated our multivariate analyses by including all study participants (i.e., those who participated in the study for whom we had a measured serostatus and those for whom we had an ‘imputed’ serostatus).
Statistical significance was determined at α = 0.05, and all tests for statistical significance were two-sided. All analyses were performed by using SAS version 9.1 (SAS Institute, Inc, Cary, NC).
Results
Study population
Characteristics of cases and controls with serological data are presented in Table 1. The distributions of age and sex were similar among simple BO cases and controls due to the frequency matching. Dysplastic BO cases were older and more likely to be male than controls. Compared with controls, cases were generally more likely to have a lower education status and a lower income (not shown). Cases were also more likely to have smoked or experienced symptoms of heartburn or acid reflux and the prevalence of overweight and obesity was higher among cases than controls.
Table 1. Characteristics of controls and Barrett's oesophagus patients (with and without dysplasia).
| Variables | Categories | Controls (n = 398) n (%) |
Simple BO (n = 217) n (%) |
Dysplastic BO (n = 95) n (%) |
|---|---|---|---|---|
| Age, y | ||||
| ≤ 49 | 64 (16.1) | 41 (18.9) | 12 (12.7) | |
| 50 – 59 | 137 (34.4) | 71 (32.7) | 22 (23.2) | |
| 60 – 69 | 126 (31.7) | 68 (31.3) | 36 (37.9) | |
| 70 – 79 | 71 (17.8) | 37 (17.1) | 25 (26.3) | |
| Sex | ||||
| Female | 138 (34.7) | 74 (34.1) | 20 (21.1) | |
| Male | 260 (65.3) | 143 (65.9) | 75 (78.9) | |
| Highest level of education | ||||
| School only | 149 (37.4) | 103 (47.5) | 31 (32.6) | |
| Technical college/diploma | 162 (40.7) | 84 (38.7) | 52 (54.7) | |
| University | 87 (21.9) | 30 (13.8) | 12 (12.6) | |
| P value1 | .02 | .03 | ||
| BMI last year (kg/m2) | ||||
| < 25.0 | 142 (35.9) | 62 (29.1) | 27 (29.0) | |
| 25.0 – 29.9 | 171 (43.3) | 84 (39.4) | 44 (47.3) | |
| ≥ 30.0 | 82 (20.8) | 67 (31.5) | 22 (23.7) | |
| P value | .01 | .45 | ||
| Smoking history | ||||
| Never smoker | 212 (53.3) | 72 (33.2) | 23 (24.2) | |
| < 30 pack-years | 118 (29.6) | 92 (42.4) | 46 (48.4) | |
| ≥ 30 pack-years | 68 (17.1) | 53 (24.4) | 26 (27.4) | |
| P value | <.001 | <.001 | ||
| Mean alcohol consumption2 (standard drinks/week) | ||||
| Never drinkers | 47 (11.9) | 29 (13.6) | 3 (3.2) | |
| < 1 | 29 (7.4) | 16 (7.5) | 11 (11.8) | |
| 1 – 6.9 | 123 (31.2) | 63 (29.4) | 26 (28.0) | |
| 7 – 20.9 | 115 (29.2) | 58 (27.1) | 32 (34.4) | |
| ≥ 21 | 80 (20.3) | 48 (22.4) | 21 (22.6) | |
| P value | .91 | .07 | ||
| Frequency of symptoms of heartburn or reflux3 | ||||
| Never | 192 (48.9) | 31 (14.6) | 9 (9.7) | |
| Less than weekly | 168 (42.7) | 86 (40.4) | 41 (44.1) | |
| At least weekly | 33 (8.4) | 96 (45.1) | 43 (46.2) | |
| P value | <.001 | <.001 | ||
| Frequency of use of aspirin/NSAIDs past 5 years | ||||
| Never | 92 (23.1) | 56 (25.8) | 22 (23.2) | |
| Less than weekly | 219 (55.0) | 102 (47.0) | 43 (45.3) | |
| At least weekly | 87 (21.9) | 59 (27.2) | 30 (31.6) | |
| P value | .15 | .11 |
BMI, body mass index; Dysplastic BO, Barrett's oesophagus with dysplasia; NSAIDs, non-steroidal anti-inflammatory drugs; Simple BO, Barrett's oesophagus without dysplasia.
P value for χ2 test for heterogeneity for comparing group of cases to the controls for the distribution of each categorical variable.
One standard drink is equivalent to 10 g of ethanol.
Symptoms reported in the age interval 10 years prior to the reference age.
The overall prevalence of H pylori seropositivity among controls was 18% (95% CI: 15-22%). Among controls, there was a significant trend towards increasing prevalence with increasing age and increasing number of pack-years smoked (P trend both = .01) and there was some evidence of an association between seropositivity and low levels of education (P = .05). We found similar prevalence of seropositivity among controls by sex and across categories of BMI, alcohol consumption, frequency of GOR symptoms, and frequency of aspirin or other NSAID use. There was no difference in the prevalence of seropositivity among controls who were never (18%) versus ever (21%) users of PPIs (p=0.59). Among patients with simple BO, the prevalence of H pylori was higher among never users (15%) than ever users of PPIs (8%), although again, this difference was not statistically significant (p=0.14).
Risk estimates associated with Helicobacter pylori infection
The risk of BO was inversely associated with H pylori seropositivity (Table 2). The prevalences of H pylori infection were 12% (95% CI: 8-15%) and 3% (95% CI: 0-6%) among patients with simple BO and dysplastic BO, respectively. Patients with simple BO (OR=0.51, 95% CI: 0.30-0.86) and dysplastic BO (OR=0.10, 95% CI: 0.03-0.33) were significantly less likely than controls to have antibodies to H pylori. Adjustment for frequency of GOR symptoms attenuated the association between a positive H pylori antibody test and simple BO to OR = 0.66 (95% CI: 0.37-1.17), but made little difference to the risk estimate for dysplastic BO (Table 2).
Table 2. Relative risks of Barrett's oesophagus (with and without dysplasia) associated with H pylori seropositivity.
| H pylori serostatus | Controls | Cases | Model 11 | Model 22 | Model 33 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | (%) | n | (%) | OR | (95% CI) | OR | (95% CI) | OR | (95% CI) | ||
| Simple BO | Negative | 317 | (80) | 183 | (84) | 1.00 | (Referent) | 1.00 | (Referent) | 1.00 | (Referent) |
| Positive | 73 | (18) | 25 | (12) | 0.60 | (0.37–0.98) | 0.51 | (0.30–0.86) | 0.66 | (0.37–1.17) | |
| Dysplastic BO | Negative | 317 | (80) | 85 | (90) | 1.00 | (Referent) | 1.00 | (Referent) | 1.00 | (Referent) |
| Positive | 73 | (18) | 3 | (3) | 0.13 | (0.04–0.41) | 0.10 | (0.03–0.33) | 0.13 | (0.04–0.45) | |
| All cases4 | Negative | 317 | (80) | 268 | (86) | 1.00 | (Referent) | 1.00 | (Referent) | 1.00 | (Referent) |
| Positive | 73 | (18) | 28 | (9) | 0.44 | (0.27–0.70) | 0.37 | (0.22–0.61) | 0.47 | (0.27–0.81) | |
BMI, body mass index; CI, confidence interval; Dysplastic BO, Barrett's oesophagus with dysplasia; OR, odds ratio; Simple BO, Barrett's oesophagus without dysplasia.
NOTE: There were 8 (2%) controls, 9 (4%) simple BO and 7 (7%) dysplastic BO cases with equivocal index readings.
Adjusted for sex and age.
Adjusted for sex, age, education level, cumulative smoking history, BMI category, average lifetime alcohol consumption, and frequency of aspirin/NSAID use in past 5 years.
Additionally adjusted for frequency of reflux or heartburn symptoms in 10 years before study.
A single case group combining Simple BO + Dysplastic BO.
We additionally performed analysis combining the participants with simple and dysplastic BO in a single case group and there was a significant inverse association with H pylori status for the fully adjusted model (OR=0.47, 95% CI: 0.27-0.81).
Effect modification
The association between H pylori antibody status and BO was assessed within strata of known risk factors for BO (Table 3). We found consistently that the greatest risk reductions associated with H pylori seropositivity were observed among those who ever used PPIs (versus never used), and also among those who ever used H2-receptor antagonists (versus never used), although the interaction terms were uniformly nonsignificant. There was some evidence that the inverse association between H pylori seropositivity and BO was stronger among men than women, participants aged ≥ 60 years than those < 60 years, and participants with a history of GOR symptoms than those without. We observed strong inverse associations between H pylori seropositivity and BO among participants in the lowest and highest categories of cumulative smoking history and BMI, but not among those in the middle categories. These apparent differences in the magnitude of risks across strata of known causal factors were uniformly nonsignificant, and thus within the bounds of random variation. Stratified analyses could not be performed for dysplastic BO due to the small number of H pylori seropositive cases (n = 3).
Table 3. Relative risks of Barrett's oesophagus associated with H pylori seropositivity, stratified by other risk factors for Barrett's oesophagus.
| Controls | Simple BO | All cases1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. Pos | No. Neg | No. Pos | No. Neg | OR2 | (95% CI) | No. Pos | No. Neg | OR2 | (95% CI) | |
| Female | 18 | 115 | 8 | 59 | 1.21 | (0.43–3.41) | 8 | 76 | 0.93 | (0.34–2.58) |
| Male | 51 | 195 | 16 | 117 | 0.51 | (0.25–1.03) | 19 | 182 | 0.36 | (0.19–0.69) |
| P3 = .15 | P = .12 | |||||||||
| < 60 years of age | 25 | 164 | 11 | 93 | 1.06 | (0.43–2.58) | 11 | 122 | 0.83 | (0.34–2.02) |
| ≥ 60 years of age | 44 | 146 | 13 | 83 | 0.49 | (0.22–1.06) | 16 | 136 | 0.34 | (0.16–0.68) |
| P = .33 | P = .15 | |||||||||
| GOR never | 29 | 154 | 5 | 25 | 1.14 | (0.36–3.57) | 5 | 34 | 0.68 | (0.23–2.04) |
| GOR less than weekly4 | 37 | 128 | 14 | 68 | 0.53 | (0.25–1.13) | 16 | 104 | 0.39 | (0.19–0.78) |
| GOR at least weekly4 | 3 | 28 | 5 | 83 | 0.49 | (0.08–3.03) | 6 | 120 | 0.37 | (0.07–1.91) |
| P = .65 | P = .75 | |||||||||
| Never smoker | 30 | 175 | 5 | 63 | 0.67 | (0.22–2.06) | 6 | 83 | 0.53 | (0.19–1.51) |
| < 30 pack-years | 22 | 91 | 13 | 72 | 1.09 | (0.46–2.60) | 14 | 113 | 0.71 | (0.31–1.63) |
| ≥ 30 pack-years | 17 | 44 | 6 | 41 | 0.28 | (0.08–0.96) | 7 | 62 | 0.24 | (0.08–0.72) |
| P = .28 | P = .40 | |||||||||
| BMI < 25 kg/m2 | 26 | 110 | 7 | 48 | 0.52 | (0.18–1.49) | 8 | 72 | 0.42 | (0.15–1.14) |
| BMI 25 – 29.9 kg/m2 | 24 | 141 | 11 | 70 | 1.33 | (0.52–3.44) | 13 | 112 | 0.77 | (0.32–1.83) |
| BMI ≥ 30 kg/m2 | 19 | 59 | 6 | 58 | 0.28 | (0.08–0.94) | 6 | 74 | 0.20 | (0.06–0.67) |
| P = .28 | P = .40 | |||||||||
| Never used H2-receptor antagonists | 56 | 265 | 16 | 110 | 0.73 | (0.37–1.43) | 18 | 152 | 0.56 | (0.29–1.07) |
| Ever used H2-receptor antagonists | 13 | 43 | 8 | 65 | 0.47 | (0.13–1.66) | 9 | 104 | 0.24 | (0.07–0.77) |
| P = .55 | P = .29 | |||||||||
| Never used PPI | 59 | 267 | 18 | 106 | 0.79 | (0.41–1.54) | 18 | 125 | 0.66 | (0.35–1.28) |
| Ever used PPI | 10 | 41 | 6 | 69 | 0.33 | (0.08–1.32) | 9 | 131 | 0.22 | (0.06–0.73) |
| P = .41 | P = .19 | |||||||||
BMI, body mass index; CI, confidence interval; Dysplastic BO, Barrett's oesophagus with dysplasia; GOR, gastro-oesophageal reflux; OR, odds ratio; PPI, proton pump inhibitors; Simple BO, Barrett's oesophagus with no dysplasia.
A single case group combining Simple BO + Dysplastic BO.
Adjusted for sex, age, education level, cumulative smoking history, BMI category, average lifetime alcohol consumption, frequency of aspirin/NSAID use in past 5 years, and frequency of reflux or heartburn symptoms in 10 years before study.
P value for the type III analysis of effects for the addition of the interaction term to the saturated model.
Symptoms reported in the age interval 10 years prior to the reference age.
Sensitivity analysis
We used sensitivity analysis to assess the robustness of our results to possible misclassification of measured H pylori serostatus, and also to examine the effects of missing serology information among study participants. Thus, we first considered the impact of reclassifying all participants with a measured ‘equivocal’ reading as either ‘negative’ or ‘positive’. We observed essentially the same patterns for the risk estimates of simple BO and dysplastic BO as above (data not shown). Secondly, we used an imputation technique to assess sensitivity to missing serostatus information among case and control participants.27 Given the relationship between reduced acid secretion and H pylori infection, exclusion of ‘non-serology’ control participants may bias any inverse associations towards the null (i.e., given the characteristics of the ‘non-serology’ control participants, we would expect them to have a higher prevalence of H pylori seropositivity than that observed for controls with a measured serostatus). All sensitivity analyses were adjusted for potential confounders, including frequency of GOR symptoms. Under the first imputation model, H pylori serostatus among ‘non-serology’ participants was imputed assuming the same distribution as that for participants with a measured serostatus. Compared with the original analysis, the risks of simple BO (OR=0.66, 95% CI: 0.39-1.12) and dysplastic BO (OR=0.11, 95% CI: 0.03-0.40) were essentially the same under this model. The next model assumed that the prevalence of H pylori seropositivity among ‘non-serology’ participants was two-fold higher than we observed among participants with a measured serostatus. Relative risk estimates were strengthened for both simple BO (OR=0.52, 95% CI: 0.30-0.90) and dysplastic BO (OR=0.08, 95% CI: 0.02-0.30). Finally, we assumed that the prevalence of H pylori seropositivity among ‘non-serology’ participants was half that observed among participants with a measured serostatus. Under this extreme scenario, the risk estimates for both simple BO (OR=0.68, 95% CI: 0.38-1.21) and dysplastic BO (OR=0.14, 95% CI: 0.04-0.52) were again similar to our original analyses.
Discussion
In this case-control study, we found a strong inverse association between serological evidence of a past infection with H pylori and the risk of BO. The magnitude of the risk reduction was larger for BO with dysplasia than for BO without dysplasia. Notably, the association between H pylori and BO without dysplasia was attenuated after adjustment for the frequency of GOR symptoms. These findings suggest that H pylori infection is inversely associated with the risk for BO and this association may be at least partly mediated through the suppression of GOR by H pylori infection.
Of note in our study was the very low prevalence of H pylori seropositivity. At 18% among controls, the prevalence was considerably less than that observed in a comparable population-based study of BO in Ireland,25 and moderately less than the prevalence observed in a population-based study of BO conducted in northern California.24 Prevalence estimates for H pylori seropositivity in Australia vary with the sampling frame, but are typically lower than in the United States and other developed countries.28
The magnitude of the inverse association we observed between H pylori seropositivity and simple BO was similar to that reported in the recent Californian study,24 but weaker than that reported in the Irish study.25 In keeping with the findings of both of those prior studies, we observed that the association between H pylori and BO was attenuated after adjusting for GOR symptoms. Our results are also in keeping with a meta-analysis of 9 studies, that showed H pylori infection was significantly associated with reduced risks of BO in those studies that compared BO cases to endoscopically normal controls.23
This study aimed to examine the possibility that exposure to other risk factors known to be strongly associated with risk of BO may confound or modify the association between H pylori and BO. We found consistently greater risk reductions among those who had used PPIs. As PPIs are used in combination with antibiotics to eradicate H pylori infection, the observed associations may be due to confounding by indication since BO patients are more likely to have used PPIs than controls, and use of PPIs is associated with treatment for infection. While not conclusive, our study found that BO patients were less likely than population controls to have positive H pylori serology, regardless of PPI use. We did not collect detailed information regarding duration and dose of PPIs, and thus we cannot definitely exclude PPI use as a possible explanation of the inverse associations. However, these results combined with the previously reported inverse association between H pylori and risks of OAC,12, 13 suggests that confounding by PPI use is unlikely to fully explain the inverse association between H pylori and BO. Although not statistically significant, the strength of association between H pylori seropositivity and BO also varied by sex, age and GOR symptoms. Additionally, there was variation in the risk estimates across strata of BMI and cumulative smoking history. Pooled analyses of published data are needed to establish definitively whether any of these factors modified the associations.
There are several potential mechanisms through which H pylori infection could be associated with reduced BO risk. Firstly, it is generally recognised that the presence of H pylori decreases gastric acid secretion and increases the risk of gastric atrophy.17, 29-32 Therefore, H pylori colonisation reduces the risk of GOR and this, in turn, directly reduces the risk of BO. In our study, adjustment for the frequency of GOR symptoms did not entirely eliminate the associations between H pylori and BO and thus our results suggest that decreased acid load is not the only mechanism underlying the H pylori protective effect. Secondly, H pylori also appears to suppress levels of ghrelin,33-35 a hormone known to increase gastric emptying and potentially decrease GOR. Thus, if H pylori infection suppresses ghrelin, it might impair emptying and enhance GOR. However, there is no evidence that H pylori infection affects gastric emptying.36, 37 Ghrelin is also a potent appetite stimulant and potential contributor to obesity.38 As obesity is consistently observed to increase the risk of GOR, BO and OAC,39-42 it is therefore feasible that H pylori colonisation may decrease the risks of these diseases. However, the findings from a recently published prospective study do not provide support for this hypothesis,43 reporting that, independent of H pylori infection, high ghrelin levels (rather than low) were associated with reduced risks of OAC. Our study also found no statistical evidence to support this hypothesis, as adjustment for BMI made little difference to the effect estimates.
Strengths of the present study include the prospective, population-based recruitment of patients with newly diagnosed BO and the use of population controls. The relatively large sample size and the collection of information on a wide range of potential confounders enhanced the accuracy of risk estimates. We adopted strict and consistent criteria throughout the study ascertainment period, using standardised histologic and endoscopic definitions to make the formal diagnosis of BO and dysplasia.44 The possibility of recall bias, associated with long-standing awareness of a BO diagnosis and reflecting on possible causes, was minimised by recruiting incident cases soon after diagnosis. Also, if present at all, recall bias would be limited to self-reported exposures (i.e., confounders) and would not apply to the main exposure (H pylori antibody status). Assays were performed blinded to participant status, and there is no reason to suspect that our findings were the result of systematic differences in laboratory analysis.
A limitation of these analyses was the relatively low rate of participation, raising concerns about possible biased selection of cases and controls. The distribution of BMI and the prevalence of GOR symptoms among our controls were similar to population data from the Australian National Health Survey 2007,45 and to another Australian survey,46 respectively. In contrast, the prevalence of current smokers in our controls was lower than the population average, which suggests our estimates might be under-estimates of the true effect as H pylori infection is positively associated with smoking. The most notable limitation was the absence of serological data for a proportion of study participants. This may have introduced bias as controls with serological data were older but also more likely to have experienced symptoms of heartburn or acid reflux than those controls missing serostatus information. As H pylori infection is more common among older than younger people, but less common among those with frequent symptoms of GOR, the likely direction of any bias due to missing serostatus is difficult to predict. We took account of these possible contrasting influences in our sensitivity analyses and found that when the H pylori antibody status among ‘non-serology’ participants was imputed assuming the same distribution as that among participants with a measured serostatus, the magnitude of the inverse association was unchanged. When we modelled higher prevalences of H pylori seropositivity among ‘non-serology’ participants, the inverse associations were strengthened. We can therefore conclude that missing serology status is unlikely to account for our findings; indeed the observed effect may actually underestimate the true effect.
As the majority of BO cases remain undiagnosed in the general population,47-49 our study may be subject to selection bias if, for example, patients diagnosed differ from those who remain undiagnosed. However, this is the same for all studies of BO. Moreover, if the prevalence of undiagnosed BO in the general population is high, this would attenuate the observed inverse associations since our control group would very likely include some people with undiagnosed BO. Misclassification of H pylori exposure cannot be ruled out, especially false negative tests, since the patients with dysplasia were older than population controls. It is possible that some such cases may have been infected at young ages but then seroreverted over time. While our study collected self-reported data on prior H pylori diagnosis and treatment, it was not of sufficiently high quality to merit further analysis. This would be unlikely to account for the overall strength of the inverse association however. The inverse association between H pylori and BO may be partly mediated by CagA (cytotoxin-associated gene product A) status.24 Although we did not test for antibodies to the CagA protein, had we measured CagA status, it is likely that the inverse association with BO would have been strengthened. Finally, we attempted to control for known confounders, however it is possible that unknown or unmeasured variables related to H pylori infection and BO might have influenced our results.
In summary, we found a reduced risk of BO associated with serological evidence of infection with H pylori. Understanding the mechanisms through which H pylori mediates its effect on oesophageal epithelium remains the focus of ongoing research.
Figure 1.
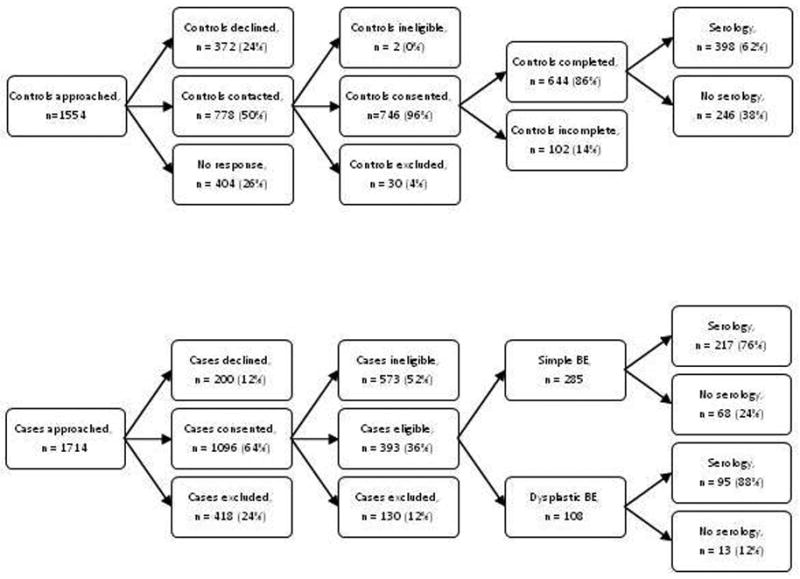
Flow chart of recruitment and serum availability.
Background
Barrett's oesophagus, a metaplastic change of the oesophageal lining, is the only known precursor to oesophageal adenocarcinoma.
While Helicobacter pylori infection is causally associated with gastric cancers, infection with this organism is associated with reduced risks of oesophageal adenocarcinoma.
It is not known when the apparent protective effect of H pylori occurs in the metaplasia-dysplasia-carcinoma sequence.
Novelty of the paper
In a population with low prevalence of H pylori infection, serological evidence of a past infection with H pylori still had a strong protective effect on Barrett's oesophagus.
The inverse association between H pylori infection and Barrett's oesophagus was greater among users of PPIs or H2-receptor antagonists than among never users of these medications.
The inverse association remained following adjustment for reflux symptoms, suggesting that acid load is not the only mechanism underlying the H pylori protective effect.
Impact of the paper
Systematic eradication of H pylori infection may, in the future, contribute to even higher rates of Barrett's oesophagus and oesophageal adenocarcinoma.
Acknowledgments
We gratefully acknowledge the cooperation of the following institutions: Sullivan and Nicolaides Pathology (Brisbane); Queensland Medical Laboratory (Brisbane); Queensland Health Pathology Services (Brisbane). We also acknowledge the contribution of the study nurses and research assistants and would like to thank all of the people who participated in the study.
Grant sponsor: National Cancer Institute; Grant number: 5 RO1 CA 001833-02. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute. APT is supported by an Australian Postgraduate Award (University of Queensland) and the Cancer Council NSW STREP grant 08-04. NP, NKH and PMW are supported by Fellowships from the National Health and Medical Research Council of Australia. DCW is supported by a Future Fellowship from the Australian Research Council.
Abbreviations used
- BMI
body mass index
- BO
Barrett's oesophagus
- CI
confidence interval
- GOR
gastro-oesophageal reflux
- OAC
oesophageal adenocarcinoma
- OR
odds ratio
- PPI
proton pump inhibitors
Footnotes
The authors disclose no conflicts of interest.
Study of Digestive Health Investigators:
Queensland Institute of Medical Research, Brisbane, Australia: David C Whiteman MBBS, PhD; Adele C Green MBBS, PhD; Nicholas K Hayward PhD; Peter G Parsons PhD; Sandra J Pavey PhD, David M Purdie PhD; Penelope M Webb DPhil.
University of Queensland, Brisbane, Australia: David Gotley FRACS; Mark Smithers FRACS.
The University of Adelaide, Adelaide, Australia: Glyn G Jamieson FRACS.
Flinders University, Adelaide, Australia: Paul Drew PhD; David I Watson FRACS.
Envoi Pathology, Brisbane, Australia: Andrew Clouston PhD, FRCPA.
Study of Digestive Health Research Staff:
Project Manager: Suzanne O'Brien (QIMR); Data Manager: Troy Sadkowsky (QIMR); Research Nurses: Andrea McMurtrie, Linda Terry, Michael Connard, Lea Jackman, Susan Perry, Marcia Davis; Clinical Collaborators: Ian Brown (S&N Pathology), Neal Walker (QML Pathology).
Author contributions: APT performed the statistical analysis and wrote the first draft of the manuscript. DCW, PMW, NKH and ACG designed the original study, obtained funding and provided overall supervision. KJS assisted in data collection, cleaning and derivation of variables. NP assisted in statistical analysis and in preparing the manuscript. All authors read and approved the final version of the manuscript.
References
- 1.Falk GW. Barrett's esophagus. Gastroenterology. 2002;122:1569–91. doi: 10.1053/gast.2002.33427. [DOI] [PubMed] [Google Scholar]
- 2.Bosetti C, Levi F, Ferlay J, Garavello W, Lucchini F, Bertuccio P, Negri E, La Vecchia C. Trends in oesophageal cancer incidence and mortality in Europe. Int J Cancer. 2008;122:1118–29. doi: 10.1002/ijc.23232. [DOI] [PubMed] [Google Scholar]
- 3.Pohl H, Sirovich B, Welch HG. Esophageal adenocarcinoma incidence: are we reaching the peak? Cancer Epidemiol Biomarkers Prev. 2010;19:1468–70. doi: 10.1158/1055-9965.EPI-10-0012. [DOI] [PubMed] [Google Scholar]
- 4.Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. 2007;17:2–9. doi: 10.1016/j.semradonc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Lord RVN, Law MG, Ward RL, Giles GG, Thomas RJS, Thursfield V. Rising incidence of oesophageal adenocarcinoma in men in Australia. J Gastroenterol Hepatol. 1998;13:356–62. doi: 10.1111/j.1440-1746.1998.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 6.Brown LM, Devesa SS, Chow WH. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age. J Natl Cancer Inst. 2008;100:1184–87. doi: 10.1093/jnci/djn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron AJ, Ott BJ, Payne WS. The incidence of adenocarcinoma in columnar-lined (Barrett's) esophagus. N Engl J Med. 1985;313:857–59. doi: 10.1056/NEJM198510033131404. [DOI] [PubMed] [Google Scholar]
- 8.Cook MB, Wild CP, Everett SM, Hardie LJ, Bani-Hani KE, Martin IG, Forman D. Risk of mortality and cancer incidence in Barrett's esophagus. Cancer Epidemiol Biomarkers Prev. 2007;16:2090–96. doi: 10.1158/1055-9965.EPI-07-0432. [DOI] [PubMed] [Google Scholar]
- 9.Blaser MJ. Disappearing microbiota: Helicobacter pylori protection against esophageal adenocarcinoma. Cancer Prev Res (Phila Pa) 2008;1:308–11. doi: 10.1158/1940-6207.CAPR-08-0170. [DOI] [PubMed] [Google Scholar]
- 10.Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology. 1998;114:1169–79. doi: 10.1016/s0016-5085(98)70422-6. [DOI] [PubMed] [Google Scholar]
- 11.Ye WM, Held M, Lagergren J, Engstrand L, Blot WJ, McLaughlin JK, Nyren O. Helicobacter pylori infection and gastric atrophy: risk of adenocarcinoma and squamous-cell carcinoma of the esophagus and adenocarcinoma of the gastric cardia. J Natl Cancer Inst. 2004;96:388–96. doi: 10.1093/jnci/djh057. [DOI] [PubMed] [Google Scholar]
- 12.Whiteman DC, Parmar P, Fahey P, Moore SP, Stark M, Zhao ZZ, Montgomery GW, Green AC, Hayward NK, Webb PM Australian Cancer Study. Association of Helicobacter pylori infection with reduced risk for esophageal cancer is independent of environmental and genetic modifiers. Gastroenterology. 2010;139:73–83. doi: 10.1053/j.gastro.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 13.Islami F, Kamangar F. Helicobacter pylori and Esophageal Cancer Risk: A Meta-analysis. Cancer Prev Res (Phila Pa) 2008;1:329–38. doi: 10.1158/1940-6207.CAPR-08-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Martel C, Llosa AE, Farr SM, Friedman GD, Vogelman JH, Orentreich N, Corley DA, Parsonnet J. Helicobacter pylori infection and the risk of development of esophageal adenocarcinoma. J Infect Dis. 2005;191:761–67. doi: 10.1086/427659. [DOI] [PubMed] [Google Scholar]
- 15.Chow WH, Blaser MJ, Blot WJ, Gammon MD, Vaughan TL, Risch HA, Perez-Perez GI, Schoenberg JB, Stanford JL, Rotterdam H, West AB, Fraumeni JF. An inverse relation between cagA+ strains of Helicobacter pylori infection and risk of esophageal and gastric cardia adenocarcinoma. Cancer Res. 1998;58:588–90. [PubMed] [Google Scholar]
- 16.Henrik Siman J, Forsgren A, Berglund G, Floren CH. Helicobacter pylori infection is associated with a decreased risk of developing oesophageal neoplasms. Helicobacter. 2001;6:310–6. doi: 10.1046/j.1523-5378.2001.00041.x. [DOI] [PubMed] [Google Scholar]
- 17.Jones AD, Bacon KD, Jobe BA, Sheppard BC, Deveney CW, Rutten MJ. Helicobacter pylori induces apoptosis in Barrett's-derived esophageal adenocarcinoma cells. J Gastrointest Surg. 2003;7:68–76. doi: 10.1016/S1091-255X(02)00129-4. [DOI] [PubMed] [Google Scholar]
- 18.McColl KE, El-Omar E, Gillen D. Helicobacter pylori gastritis and gastric physiology. Gastroenterol Clin North Am. 2000;29:687–703. doi: 10.1016/s0889-8553(05)70138-2. [DOI] [PubMed] [Google Scholar]
- 19.El-Omar EM, Carrington M, Chow WH, McColl KEL, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 20.McColl KE. Helicobacter pylori and oesophageal cancer - not always protective. Gut. 2007;56:457–59. doi: 10.1136/gut.2006.111385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Omar EM, Penman ID, Ardill JES, Chittajallu RS, Howie C, McColl KEL. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology. 1995;109:681–91. doi: 10.1016/0016-5085(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 22.Rokkas T, Pistiolas D, Sechopoulos P, Robotis I, Margantinis G. Relationship between Helicobacter pylori infection and esophageal neoplasia: a meta-analysis. Clin Gastroenterol Hepatol. 2007;5:1413–17. doi: 10.1016/j.cgh.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Wang CC, Yuan YH, Hunt RH. Helicobacter pylori Infection and Barrett's esophagus: a systematic review and meta-analysis. Am J Gastroenterol. 2009;104:492–500. doi: 10.1038/ajg.2008.37. [DOI] [PubMed] [Google Scholar]
- 24.Corley DA, Kubo A, Levin TR, Block G, Habel L, Zhao W, Leighton P, Rumore G, Quesenberry C, Buffler P, Parsonnet J. Helicobacter pylori infection and the risk of Barrett's oesophagus: a community-based study. Gut. 2008;57:727–33. doi: 10.1136/gut.2007.132068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson LA, Murphy SJ, Johnston BT, Watson RGP, Ferguson HR, Bamford KB, Ghazy A, McCarron P, McGuigan J, Reynolds JV, Comber H, Murray LJ. Relationship between Helicobacter pylori infection and gastric atrophy and the stages of the oesophageal inflammation, metaplasia, adenocarcinoma sequence: results from the FINBAR case-control study. Gut. 2008;57:734–39. doi: 10.1136/gut.2007.132662. [DOI] [PubMed] [Google Scholar]
- 26.Smith KJ, O'Brien SM, Green AC, Webb PM, Whiteman DC Study of Digestive Health. Current and past smoking significantly increase risk for Barrett's esophagus. Clin Gastroenterol Hepatol. 2009;7:840–48. doi: 10.1016/j.cgh.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 27.Pandeya N, Williams GM, Green AC, Webb PM, Whiteman DC. Do low control response rates always affect the findings? Assessments of smoking and obesity in two Australian case-control studies of cancer. Aust N Z J Public Health. 2009;33:312–19. doi: 10.1111/j.1753-6405.2009.00401.x. [DOI] [PubMed] [Google Scholar]
- 28.Go MF. Review article: natural history and epidemiology of Helicobacter pylori injection. Aliment Pharmacol Ther. 2002;16(Suppl. 1):3–15. doi: 10.1046/j.1365-2036.2002.0160s1003.x. [DOI] [PubMed] [Google Scholar]
- 29.El-Omar EM, Oien K, ElNujumi A, Gillen D, Wirz A, Dahill S, Williams C, Ardill JES, McColl KEL. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15–24. doi: 10.1016/s0016-5085(97)70075-1. [DOI] [PubMed] [Google Scholar]
- 30.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–33. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oksanen A, Sipponen P, Karttunen R, Miettinen A, Veijola L, Sarna S, Rautelin H. Atrophic gastritis and Helicobacter pylori infection in outpatients referred for gastroscopy. Gut. 2000;46:460–63. doi: 10.1136/gut.46.4.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuipers EJ, Uyterlinde AM, Pena AS, Roosendaal R, Pals G, Nelis GF, Festen HPM, Meuwissen SGM. Long-term sequelae of helicobacter pylori gastritis. Lancet. 1995;345:1525–28. doi: 10.1016/s0140-6736(95)91084-0. [DOI] [PubMed] [Google Scholar]
- 33.Tatsuguchi A, Miyake K, Gudis K, Futagami S, Tsukui T, Wada K, Kishida T, Fukuda Y, Sugisaki Y, Sakamoto C. Effect of Helicobacter pylori infection on ghrelin expression in human gastric mucosa. Am J Gastroenterol. 2004;99:2121–27. doi: 10.1111/j.1572-0241.2004.30291.x. [DOI] [PubMed] [Google Scholar]
- 34.Jang EJ, Park SW, Park JS, Park SJ, Hahm KB, Paik SY, Sin MK, Lee ES, Oh SW, Park CY, Baik HW. The influence of the eradication of Helicobacter pylori on gastric ghrelin, appetite, and body mass index in patients with peptic ulcer disease. J Gastroenterol Hepatol. 2008;23(Suppl. 2):S278–S85. doi: 10.1111/j.1440-1746.2008.05415.x. [DOI] [PubMed] [Google Scholar]
- 35.Nwokolo CU, Freshwater DA, O'Hare P, Randeva HS. Plasma ghrelin following cure of Helicobacter pylori. Gut. 2003;52:637–40. doi: 10.1136/gut.52.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parente F, Maconi G, Sangaletti O, Minguzzi M, Vago L, Porro GB. Behavior of acid secretion, gastrin release, serum pepsinogen I, and gastric emptying of liquids over six months from eradication of Helicobacter pylori in duodenal ulcer patients. A controlled study Gut. 1995;37:210–15. doi: 10.1136/gut.37.2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perri F, Clemente R, Festa V, Annese V, Quitadamo M, Rutgeerts P, Andriulli A. Patterns of symptoms in functional dyspepsia: role of Helicobacter pylori infection and delayed gastric emptying. Am J Gastroenterol. 1998;93:2082–88. doi: 10.1111/j.1572-0241.1998.00597.x. [DOI] [PubMed] [Google Scholar]
- 38.Azuma T, Suto H, Ito Y, Muramatsu A, Ohtani M, Dojo M, Yamazaki Y, Kuriyama M, Kato T. Eradication of Helicobacter pylori infection induces an increase in body mass index. Aliment Pharmacol Ther. 2002;16(Suppl. 2):240–44. doi: 10.1046/j.1365-2036.16.s2.31.x. [DOI] [PubMed] [Google Scholar]
- 39.Kubo A, Corley DA. Body mass index and adenocarcinomas of the esophagus or gastric cardia: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:872–78. doi: 10.1158/1055-9965.EPI-05-0860. [DOI] [PubMed] [Google Scholar]
- 40.Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143:199–211. doi: 10.7326/0003-4819-143-3-200508020-00006. [DOI] [PubMed] [Google Scholar]
- 41.Whiteman DC, Sadeghi S, Pandeya N, Smithers BM, Gotley DC, Bain CJ, Webb PM, Green AC Australian Cancer Study. Combined effects of obesity, acid reflux and smoking on the risk of adenocarcinomas of the oesophagus. Gut. 2008;57:173–80. doi: 10.1136/gut.2007.131375. [DOI] [PubMed] [Google Scholar]
- 42.Cook MB, Greenwood DC, Hardie LJ, Wild CR, Forman D. A systematic review and meta-analysis of the risk of increasing adiposity on Barrett's esophagus. Am J Gastroenterol. 2008;103:292–300. doi: 10.1111/j.1572-0241.2007.01621.x. [DOI] [PubMed] [Google Scholar]
- 43.de Martel C, Haggerty TD, Corley DA, Vogelman JH, Orentreich N, Parsonnet J. Serum ghrelin levels and risk of subsequent adenocarcinoma of the esophagus. Am J Gastroenterol. 2007;102:1166–72. doi: 10.1111/j.1572-0241.2007.01116.x. [DOI] [PubMed] [Google Scholar]
- 44.Sampliner RE. Updated guidelines for the diagnosis, surveillance, and therapy of Barrett's esophagus. Am J Gastroenterol. 2002;97:1888–95. doi: 10.1111/j.1572-0241.2002.05910.x. [DOI] [PubMed] [Google Scholar]
- 45.Australian Bureau of Statistics. National Health Survey: Summary of results 2007-2008. Australian Bureau of Statistics; Canberra: 2009. Vol cat no. 4364.0. Released 25 August 2009. [Google Scholar]
- 46.Watson DI, Lally CJ. Prevalence of symptoms and use of medication for gastroesophageal reflux in an Australian community. World J Surg. 2009;33:88–94. doi: 10.1007/s00268-008-9780-9. [DOI] [PubMed] [Google Scholar]
- 47.Cameron AJ, Lomboy CT. Barrett's esophagus: age, prevalence, and extent of columnar epithelium. Gastroenterology. 1992;103:1241–45. doi: 10.1016/0016-5085(92)91510-b. [DOI] [PubMed] [Google Scholar]
- 48.Cameron AJ, Zinsmeister AR, Ballard DJ, Carney JA. Prevalence of columnar-lined (Barrett's) esophagus. Comparison of population-based clinical and autopsy findings. Gastroenterology. 1990;99:918–22. doi: 10.1016/0016-5085(90)90607-3. [DOI] [PubMed] [Google Scholar]
- 49.Gerson LB, Shetler K, Triadafilopoulos G. Prevalence of Barrett's esophagus in asymptomatic individuals. Gastroenterology. 2002;123:461–67. doi: 10.1053/gast.2002.34748. [DOI] [PubMed] [Google Scholar]