

Abstract
There is increasing experimental and epidemiological evidence that fetal programming of genetic systems is a contributing factor in the recent increase in adult obesity and other components of metabolic syndrome. In particular, there is evidence that epigenetic changes associated with the use of manmade chemicals may interact with other factors that influence fetal and postnatal growth in contributing to the current obesity epidemic. The focus of this review is on the developmental effects of estrogenic endocrine disrupting chemicals (EDCs), and more specifically on effects of exposure to the estrogenic EDC bisphenol A (BPA), on adipocytes and their function, and the ultimate impact on adult obesity; BPA exposure also results in impaired reproductive capacity. We discuss the interaction of EDCs with other factors that impact growth during fetal and neonatal life, such as placental blood flow and nutrient transport to fetuses, and how these influence fetal growth and abnormalities in homeostatic control systems required to maintain normal body weight throughout life.
Keywords: Endocrine disrupting chemicals, Bisphenol A, Obesity, Metabolic syndrome
1. The importance of life stage
1.1 Effects of exposure to estrogens in adulthood on adipose tissue
Estrogens and other sex hormones regulate the functioning of tissues in addition to those involved in reproduction in adults. The effects that occur when a hormone is present often do not occur after the hormone is withdrawn. These are termed “activational” effects. The typical view is that increased plasma concentrations of estrogens are associated with a reduction in food intake and body weight in adults, and that the loss of ovarian estrogen secretion related to menopause in women results in weight gain [1]. However, evidence is accumulating that during critical periods in development, estrogenic chemicals can have unexpected effects on the differentiation of adipocytes as well as on postnatal growth [2,3]. The hypothesis that “programming” of obesity is related to exposure to environmental estrogens during critical periods in organogenesis [4] may seem counter-intuitive. There is considerable experimental evidence that in adult mice the most potent endogenous estrogen, estradiol-17β, acts via estrogen receptor α (ERα) to have an inhibitory effect on adipocyte number and lipogenesis, and removal of endogenous estrogens by ovariectomy or via a genetic mutation in the gene controlling the enzyme aromatase (CYP19), causes impaired glucose tolerance and insulin resistance in addition to increased fat mass [5–8]. Estrogens have central effects on food consumption and energy expenditure that also contribute to their overall inhibitory effects on adipose deposition in adults; these effects occur as a result of the interaction between hormones such as leptin produced by adipocytes with estrogens, and the interaction between these hormones also impacts reproductive processes [1]. However, a maxim in developmental biology and pediatric medicine is that it is inappropriate to use effects in adults to predict effects during development.
1. 2 The developmental origin of health and disease (DOHaD) hypothesis: Body weight and metabolic diseases
Estrogens and other hormones can cause permanent changes, termed “organizational” effects, by programming gene expression when exposure occurs during the time in development when cells are differentiating, referred to as “critical periods.” There is an extensive literature providing support for this going back over 50 years. In addition to epigenetic changes impacting gene expression, there are numerous mechanisms by which environmental endocrine disrupting chemicals (EDCs) can disrupt the development and subsequent functioning of tissues. For example, chemicals can disrupt the interaction between different cell types, such as mesenchyme and epithelium, during critical periods in the organization of tissues [9].
Considerable research is currently being directed at elucidating the mechanisms by which genes are programmed during cell differentiation under the influence of hormones such as estradiol, as well as estrogenic EDCs and other EDCs that operate through other mechanisms. The mechanisms that determine which genes in a cell can be transcribed, as well as the level at which transcription occurs, involve epigenetic modifications of DNA as well as the associated histone proteins [10,11]. There is now strong evidence indicating that estrogenic EDCs can program gene activity via epigenetic changes during critical periods in development, with long-term consequences that impact the health status of the individual throughout the remainder of life. One finding that serves to demonstrate this in a well characterized system is the change in both coat color and obesity in mice with a methylation sensitive promoter that is differentially methylated by genistein (increased methylation) and bisphenol A (BPA; decreased methylation). This finding revealed that two different estrogenic EDCs (genistein and BPA) can have opposite effects on epigenetic mechanisms and subsequent phenotype. Decreased methylation status caused by BPA predicted an adult phenotype of yellow coat color, diabetes, tumors and obesity [12].
The focus of this review will primarily be on developmental effects of the estrogenic EDC, BPA. The hypothesis that obesity and co-morbidities, such as impaired reproductive capacity, is related to events that occur during early development is known as the “developmental origin of health and disease” hypothesis, which has led to creation of a society and a journal devoted to studying this issue (Journal of Developmental Origin of Health and Disease; DOHaD) [4,13]. The incidence of metabolic syndrome, which includes obesity, type II diabetes, heart disease and hypertension, has increased dramatically over the last few decades in the USA and many other regions of the world [14,15]. The DOHaD hypothesis proposes that metabolic syndrome is related to factors that influence growth in utero as well as during postnatal development [16]. There is evidence that developmental exposure to elevated levels of estrogens is a factor in the development of metabolic syndrome and also results in abnormal development of the reproductive system; we have referred to these outcomes as being part of the “fetal estrogenization syndrome” [17].
There is increasing experimental and epidemiological evidence that fetal programming of genetic systems is a contributing factor in adult obesity [18]. This has led to the hypothesis that epigenetic changes associated with the increased use of manmade chemicals, such as chemicals used in the manufacture of plastic products, may interact with other factors that influence fetal and postnatal growth in contributing to the current obesity epidemic [4,19,20]. The hypothesis that epigenetic mechanisms are involved in the etiology of obesity relates to the general issue of developmental plasticity. The hypothesis is that individuals are adapted to function within a restricted range of potential responses throughout life as a consequence of their underlying genetic potential being acted on by environmental factors during the time in tissue differentiation when genetic programming occurs [21]. Thus, a fetus that develops in a uterine environment in which there is reduced placental blood flow and nutrient transport is thought to develop a “thrifty phenotype”, such that the mechanisms mediating weight homeostasis are permanently programmed for a lifetime of undernourishment. For example, when intrauterine growth restricted (IUGR) rats are subsequently exposed to a highly palatable diet (to mimic the modern fast food diet with excessive calories), these individuals are unable to regulate their body weight, resulting in weight gain [22]. The question being posed here is whether during fetal and neonatal life, environmental chemicals are playing a role in programming a phenotype that is prone to obesity regardless of postnatal nutritional factors or whether there is an interaction between environmental chemicals and both prenatal and postnatal nutritional factors? Our view is that the latter case (interactive effects) is the most likely scenario, and our ongoing research supports this prediction.
Restriction of placental blood flow is one cause of reduced fetal growth, and light-at-term human babies are at higher risk for subsequent obesity, type 2 diabetes and hypertension [23]. There is now convincing evidence in support of the hypothesis that during fetal life environmental factors that influence fetal growth interact with factors that increase the rate of postnatal growth, resulting in obesity and type 2 diabetes. The interaction between prenatal and postnatal factors is supported by findings that the best predictor of insulin resistance in 8-year-old children is the combination of being light at birth associated with a high postnatal growth velocity. The phenomenon of restricted intrauterine growth followed by accelerated postnatal growth shown in Figure 1 is referred to as “centile crossing” [15]. Recent evidence suggests that the age at which the rapid body weight increase occurs during postnatal life is a critical factor in the eventual health status of a person [13].
Figure 1.
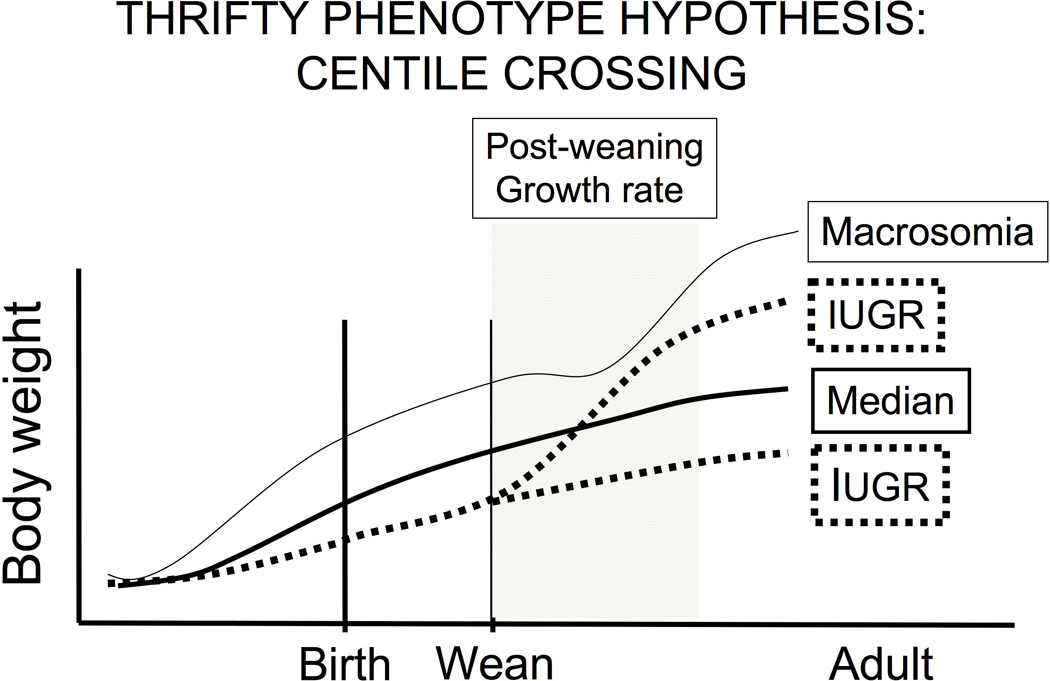
Schematic depicting the consequence of intrauterine growth restriction (IUGR) and macrosomia on postnatal growth. There are two trajectories for fetuses that experience IUGR; one sub-group may remain underweight throughout life, while another sub-group may experience overgrowth and obesity in adulthood. The IUGR fetuses differ from normal body weight at birth babies in their response to nutrients, and the postnatal growth trajectory of the different sub-groups of IUGR fetuses depends on factors during postnatal life, as predicted by the “thrifty phenotype” hypothesis. Research indicates that the mechanisms underlying obesity in macrosomic and IUGR individuals are different.
2. Adipocytes
2.1 Production and response to hormones in adipocytes
Fat tissue is now recognized to produce hormones that are critical for regulating metabolism and other processes. Leptin is produced by fat cells and impacts body weight via effects on the hypothalamus, and also impacts reproductive processes via effects on gonadotropin releasing hormone (GnRH) [1,24]. Adiponectin is produced by adipocytes and plays a role in regulating glucose uptake into cells [25]. Interestingly, leptin and adiponectin are also produced by the human placenta [26]. While adipocytes produce these hormones, other hormones are major regulators of adipose tissue and are critical for adipocyte development and function [27]. An extensive array of hormones and growth factors modulate adipocyte development and activity, including growth hormone, thyroid hormone, catecholamines, glucagon, insulin and insulin-like growth factors, and glucocorticoids, as well as estradiol and thus chemicals (natural and synthetic) with estrogenic activity [17,28,29].
In abdominal fat, mitochondrial glycerol-3-phosphate acyltransferase (GPAT) catalyzes the initial step in glycerolipid synthesis [30]. Mice deficient in diglyceride acyltransferase (DGAT1) are resistant to diet-induced obesity and have increased insulin and leptin sensitivity [31]. The presence of the enzyme aromatase in adipocytes provides a source of intracellular estradiol via aromatization of testosterone. Aromatase activity in a variety of tissues is influenced by glucocorticoids, androgens, prostaglandins and estrogens, including the estrogenic chemical bisphenol A (BPA) [32–36], and the factors that impact intracellular estradiol levels in differentiating adipocytes are the subject of ongoing investigations [37]. In aromatase knockout mice, an increase in adipocyte volume and number was observed, suggesting a role for estrogen in the regulation of adipocyte proliferation and incorporation of lipids [8,38].
Lipoprotein lipase (LPL) is a key enzyme in regulating lipids. Adipose tissue with a lower level of endothelial LPL will not uptake lipid as rapidly. LPL is subject to regulation by estrogens [38]. Estradiol was reported to markedly decrease the amounts of lipoprotein lipase mRNA as well as triglyceride accumulation in 3T3-L1 adipocytes [39].
In humans, C/EBPα mRNA levels in adipocyte tissue are elevated in those with an obese relative to lean phenotype [40]. PPARγ, which is expressed in adult adipocytes, and C/EBP are both regulators of lipogenesis, and PPARγ activators result in increased fat deposition [41–43]. Although obesity typically involves adipocyte hypertrophy, adipocyte hyperplasia is also seen in certain types of human and rodent obesity [44,45]. A critical question to consider when postulating potential developmental effects on adipocyte number and subsequent function is whether such effects would be permanent or transitory and reversible once the stimulus inducing the change in adipocyte is removed.
2.2 The differentiation of adipose tissue
In humans, differentiation of preadipocytes into adipocytes begins prior to birth, but the majority of preadipocytes differentiate postnatally. Adipocyte number increases markedly between birth and 18 months of age, then continues to increase more slowly throughout early childhood [46]. The developmental sequence by which the adipocyte phenotype arises from mesenchymal stem cells is still being actively studied, although preadipocytes appear to differentiate from mesenchymal stem cells associated with blood vessels. Mesenchymal stem cells become committed to an adipogenic lineage and give rise to preadipocytes that express PPARγ, which can remain undifferentiated and quiescent, proliferate but remain undifferentiated, or differentiate as a postmitotic adipocyte [47]. Importantly, estrogens can block PPARγ ligand-induced differentiation into adipocytes [48].
While preadipocytes in mice do not begin to differentiate into adipocytes prior to birth [49], mouse preadipocytes develop from mesenchymal cells and proliferate during fetal life, and, as discussed further below, express estrogen receptors [2]. Mouse preadipocytes continue to proliferate rapidly at birth, but the majority then enters the differentiation pathway neonatally to give rise to post-mitotic adipocytes. By approximately 3 weeks of age, the basic adult number of adipocytes has been established in most mouse strains [45]. A preadipocyte population still remains in adults and can give rise to new adipocytes at any time during life in both mice and humans [50].
The genes critical for inducing adipocyte differentiation, as well as their temporal sequence of expression during adipocyte differentiation are being actively investigated [27,51]. Of central interest as a critical regulator of adipocyte differentiation is PPARγ [27,52]. There is evidence that CCAAT/enhancer-binding protein β (C/EBPβ) initially stimulates PPARγ, which together with C/EBPs plays a critical role in adipocyte differentiation from the preadipocyte to the fully functional, postmitotic adipocyte [41,52]. The transcription factor cAMP response element-binding protein (CREB) plays an important role in regulating these genes [43].
Estrogens are known to play an important role in regulating adipose deposition, and estrogens regulate key developmental events in adipogenesis [8]. Thus, while the factors regulating whether preadipocytes proliferate or differentiate are not well understood, estrogens appear to be a factor involved in their development. Adipose tissue expresses both ERα and ERβ. ERα is expressed in adipocytes, preadipocytes and stromal vascular cells, indicating that almost all cells in adipose tissue are potentially estrogen responsive [53,54]. Estrogens appear to play a crucial role in establishment of adult adipocyte number, although effects of estrogens on adipose tissue are complex, and may vary in adipocytes located in different parts of the body.
A number of studies have shown that estradiol increases adipogenesis using mouse 3T3-L1 fibroblasts that can differentiate into preadipocytes and eventually adipocytes, as well as human or rat preadipocytes in vitro [55–57]. Estradiol treatment of cultured preadipocytes induces increased release of mitogenic substances into the media [58]. However, these effects occur at supra-physiological doses of estradiol, and the relevance of these findings to in vivo effects of estradiol and low doses of environmental estrogens is thus of concern. The concern is based on the fact that low and high doses of hormones can result in opposite effects [59,60]. In this regard, it is interesting that, as discussed further below, in rats developmental exposure to a low dose of BPA, which has estrogenic activity, reduces adipocyte number but increases adipocyte volume, associated with an increase in body weight [29]). It is thus likely that developmental exposure to EDCs such as BPA influence adipocyte differentiation and adult body weight homeostasis via different molecular signaling systems than some other classes of EDCs, such as organotins [47].
In adults estrogens tend to preferentially promote differentiation of mesenchymal stem cells into osteoblasts over adipocytes [61]. While the precise role of estrogens during development on mesenchymal stem cells is not established, there is evidence that exposure to exogenous estrogens program fetal bone. Pharmacological doses of DES given pre- and postnatally to mice decreased femur length in adulthood [62–64]. As mentioned above and discussed further below, exposure to low versus high doses of estrogenic EDCs during development can have qualitatively different effects. This was recently observed for developmental programming of bone where exposure to low doses of DES (0.1 µg/kg), ethinylestradiol or BPA (10 µg/kg) increased femur length in adulthood [65], which was the opposite effect of that seen at higher pharmacologic doses. For example, developmental exposure to ethinylestradiol showed an inverted U dose response on femur length in mice, that is, low doses of 0.01 and 0.1 µg/kg increased femur length, while 1.0 µg/kg did not alter femur length relative to controls [65]. Since developmental exposure to BPA resulted in fewer (but larger) fat cells and increased bone length in adult mice, exposure to xenoestrogens like BPA may favor differentiation of mesenchymal stem cells into bone versus adipocytes during development as well as in adulthood, although a more detailed analysis is required to examine this hypothesis.
In summary, the fetal/neonatal period is a time in the mouse when changes in circulating estrogens (either exogenous or endogenous) could result in changes in adipose tissue function during later life, and changes in the methylation pattern of genes is one potential mechanism. This prediction is consistent with evidence from other systems that exposure to sex hormones and estrogenic EDCs during fetal and neonatal life can have latent effects on the functioning of tissues after birth [17,66,67].
3. Endocrine disrupting chemicals (EDCs): A paradigm shift in the study of environmental chemicals
A meeting held in 1991 [68] brought to the attention of the scientific community and the public that chemicals present in household products, such as building materials, plastics, cleaning fluids, cosmetics and pesticides, previously considered safe by regulatory agencies, could interfere with endocrine signaling systems in the body. This discovery [69] has led over the last two decades to an extensive amount of research on endocrine disrupting chemicals or EDCs. Labeling a chemical as an “endocrine disruptor” involves using the term to very broadly cover disruption of the synthesis and transport of all chemical messengers (autocrine, paracrine, endocrine, neurotransmitters), as well as their intracellular signaling pathways and receptor systems that regulate cell function and intercellular communication. Importantly, the effects of EDCs occur at doses far below those that had been examined in traditional toxicological studies that focused on acute toxic effects of chemicals in adults. This has led to a new term, “low-dose range”, being used to describe doses below those used in traditional toxicological studies or doses that are within the range of typical human exposure [60,70].
A paradigm shift in the approach to studying environmental chemicals has occurred as a result of the diversity of backgrounds of scientists who have chosen to examine the effects of EDCs, which when combined include studies of virtually every organ system. This has resulted in the application of information from virtually every field of biology to what was once a field that used prescribed approaches dating back to predictions about dose-response relationships initially formulated in the 16th century [59]. The result is that the core assumptions used in toxicological risk assessments have been shown to be invalid for EDCs [59,71]. As identified by Thomas Kuhn, the process of a paradigm shift in science is always a painful process for those who do not want to give up the firmly held beliefs that have governed their careers. One example of a central assumption in toxicology and chemical risk assessments is the 16th century proclamation that “the dose makes the poison”, which has led toxicologists to assume that testing very high doses of chemicals predicts effects at very low doses, based on the belief all dose-response relationships are monotonic [59]. However, non-monotonic dose-response curves are commonly observed in endocrinology, due to a variety of mechanisms, and have been observed for hormones, hormonally active drugs and environmental chemicals when tested at a wide range of doses [59,72,73]. Briefly, many of the effects that occur in response to low doses of hormones do not occur at much higher doses, and in many cases, opposite effects of low and high doses are observed. Thus, the dose used in both in vitro and in vivo experiments has to be taken into account in assessing the physiological relevance of the results.
The assumption that all dose-response relationships are monotonic, and other outdated assumptions in toxicology, are being challenged by the Endocrine Society and other scientific and medical societies (representing the fields of endocrinology, reproduction, genetics, and developmental biology) that are seeking a seat at the decision-making table regarding assessing the hazards of chemicals classified as endocrine disruptors [74,75]. Traditional toxicological testing involved examination of a narrow range of very high doses based on a dose that was found to be acutely toxic [71]. However, the disruption of endocrine signaling systems by low doses of EDCs is the issue that is central to the discussion regarding the potential for EDCs to disrupt metabolic processes and lead eventually to obesity at the relatively low internal doses encountered on a daily basis based on biomonitoring studies.
Of the different types of environmental chemicals that disrupt cell signaling, the most well known are those categorized as estrogenic EDCs. However, as we learn more about these chemicals, it is being discovered that similar to endogenous hormones, they often have the capacity to interact with multiple receptor systems as a function of dose [70]. Virtually all estrogenic EDCs have the capacity to bind to classical nuclear estrogen receptors (both the alpha and beta forms) and initiate or inhibit transcription of estrogen-responsive genes, but typically the gene-response profile is not identical to the response caused by an equal dose of estradiol [73]. In addition, specific responses caused by estrogenic EDCs and estradiol may vary between tissues in the same species. Estrogenic EDCs are thus categorized as selective estrogen receptor modulators (SERMs) [70,76]. In addition to classical estrogen receptors, estrogenic EDCs can bind to non-classical estrogen receptors associated with the cell membrane. The doses of estrogenic EDCs required to activate intracellular enzyme pathways that can rapidly change cell function as well as initiate transcription are much lower than doses of the same chemicals required to initiate transcription via binding to nuclear estrogen receptors; these rapid-response systems greatly amplify extracellular signals [77].
3.1 Endocrine disrupting chemicals and obesity: The obesogen hypothesis
In recent years the focus on obesity has involved the “big two” factors thought to be primarily responsible for the dramatic increase in obesity over the last two decades: reduced physical activity and over-consumption of high calorie “junk” food associated with food marketing practices [20]. However, it seems likely that environmental factors are also involved, since changes in nutrition and exercise do not explain the epidemic of obesity over the last few decades. While a positive energy balance can increase body weight, this simplistic view does not take into account that fetal programming events due to environmental stressors (including EDCs) can alter body weight homeostasis via effects on neuroendocrine, pancreatic, gut, adipocyte and other components of this complex integrated system [4]. The concept that EDCs can have obesogenic activity is beginning to stimulate interest in the research community [47]. However, despite efforts of physicians in the pediatrics community [78–80], there remains little attention to this issue in the general medical community, possibly due to the virtual absence of information about environmental medicine in the curriculum of most medical schools.
Although there are a large number of in vitro and experimental animal studies concerning the role of BPA and other “obesogens” in obesity and other co-morbidities associated with metabolic syndrome, only a few epidemiologic studies have examined the possible role of environmental endocrine disruptors, such as phthalates, that are used in a wide variety of products, and BPA in the human obesity epidemic [81–84]. Data from studies with experimental animals, as well as cell culture studies, indicate that more studies of EDCs and metabolic diseases in humans are needed, particularly those that focus on developmental effects [66,79,85].
3.2 Bisphenol A (BPA) and obesity
BPA (CAS #80-05-7) is one of the highest volume chemicals in worldwide production, with production capacity estimated at 10-billion pounds per year in 2011 for use in manufacturing polycarbonate plastic, the resin that lines metal cans, and as an additive in many other types of plastic [86]. All human fetuses that have been examined have measurable blood levels of BPA [87–90], and mean or median levels found in humans are higher than levels in fetal and neonatal mice in response to maternal doses that increase postnatal growth [90–93].
There are markedly different views regarding the potency of BPA, which varies in relation to estradiol as a function of both nuclear receptor subtype, ERα or ERβ [70], and the co-regulatory proteins that complex with these receptors and alter responses, resulting in an uncoupling of affinity for the receptor from potency in terms of specific responses [94]. In addition, BPA and estradiol are generally equally potent as activators of receptors associated with the cell membrane that initiate rapid signaling cascades at concentrations as low as 0.01 pM [77]. As indicated above, similar to other xenoestrogens, BPA is considered to be a SERM, since it has a variety of unique effects relative to estradiol [70,73,95].
Exposure during gestation and lactation to BPA has been shown to result in a wide range of effects observed during postnatal life in mice, rats and other vertebrate and invertebrate species, including disruption of all organs in the male and female reproductive system as well as neuroendocrine effects [96,97]. We initially reported [91], and other studies have confirmed that exposure to low doses of BPA during the perinatal period of development results in an increase in body weight [29,98–104]. It is thus not surprising that neonatal exposure to a low dose (1 µg/kg/day) of the estrogenic drug diethylstilbestrol (DES) also stimulated a subsequent increase in body weight and an increase in body fat in mice [66,105]. Interestingly, exposure to BPA just prior to puberty [106,107] has also been found to increase body weight. In addition to effects on abdominal fat (in rodents the gonadal and renal fat pads), prenatal exposure to a low dose of BPA (0.25 µg/kg/day) via the use of a continuous release pump resulted in advancement of differentiation of the adipocytes in the mammary gland in fetal female mice [108]. The earliest reported exposure to BPA that impacts subsequent growth is administration of a very low concentration of BPA (1 nM or 0.23 ng/ml) during the first two days after in vitro fertilization [109], which accelerated the rate of cell division of the fertilized oocyte. In a subsequent study, after implantation of embryos exposed in vitro for two days to 1 nM BPA into an untreated female mouse, the BPA exposure prior to the blastocyst stage led to accelerated postnatal growth of the offspring [110].
There are a number of genes that are likely to be involved in the control of effects of estrogenic chemicals such as BPA on adipocyte differentiation and subsequent function. The genes critical for inducing adipocyte differentiation, as well as their temporal sequence of expression during adipocyte differentiation are being actively investigated [27,51]. We have preliminary evidence that expression of a number of genes is permanently altered as a result of differential fetal growth, based on comparisons of intrauterine growth restricted (IUGR) and macrosomic male mice (macrosomia describes a newborn with an excessive birth weight, and we classified male mice as IUGR or macrosomic if they were at the bottom and top 5% for body weight at birth); expression of some of the same genes that differed in adult adipocytes based on body weight at birth were also permanently altered in adipocytes due to developmental exposure to BPA [29].
We are examining the expression of a number of genes that are implicated in adipocyte differentiation, function and obesity, such as PPAR, C/EBP, LPL, GLUT4, Cyp19 (aromatase), GPAT and DGAT. In rats, developmental exposure to approximately 70-µg/kg/day BPA resulted in up-regulation of a number of genes in abdominal adipocytes in adulthood, including PPAR, C/EBP and LPL [29]. In mouse 3T3-L1 cells BPA increased lipoprotein lipase (LPL) activity and triacylglycerol accumulation; BPA resulted in the presence of larger lipid droplets in the differentiated cells [111]. Insulin and BPA interacted synergistically to further accelerate these processes. BPA also stimulated an increase in the glucose transporter GLUT4 and glucose uptake into 3T3-F442A adipocytes in cell culture [112]. In a separate study up-regulation of GLUT4 increased basal and insulin-induced glucose uptake into adipocytes [113], however these in vitro effects required doses considerably higher than concentrations found in human tissues [90].
Aspects of metabolic disease are influenced by adult exposure to BPA. Low doses of BPA stimulated rapid secretion of insulin in mouse pancreatic β cells in primary culture through a non-classical, non-genomic estrogen-response system, and the magnitude of the response was the same at equal doses of BPA and estradiol. In contrast, prolonged exposure to a low oral dose of BPA (10 µg/kg/day) resulted in stimulation of insulin secretion in adult mice that was mediated by the classical nuclear estrogen receptors; the prolonged hypersecretion of insulin was followed by insulin resistance [114]. In a subsequent study [115] these investigators reported that mice exposed to a low dose of BPA (10 µg/kg/day) during fetal life were heavier at birth relative to controls, and at 6 months of age, males prenatally exposed to BPA displayed glucose intolerance, insulin resistance and altered insulin release from pancreatic cells compared with control mice. Pregnant mice exposed to this same dose of BPA displayed glucose intolerance relative to untreated controls, and at a higher dose (100 µg/kg/day) a trend toward altered insulin sensitivity. Furthermore, 4 months after delivery, the mice treated during pregnancy with BPA were heavier than control unexposed pregnant mice and had decreased insulin sensitivity and glucose intolerance. As mentioned previously, this interesting finding stands in contrast to the typical assumption that exposure to chemicals such as BPA in adulthood alter metabolic systems during the time of exposure (activational effects), but that the consequences of exposure are not permanent. This observation in mice suggests that BPA exposure during pregnancy in women could also affect body weight and glucose metabolism later in life. Consistent with the prediction that fetuses are more sensitive to environmental chemicals than adults, the permanent effects of BPA on offspring’s metabolic systems occurred at a dose 10-fold lower than the dose required to cause subsequent effects in the adult mother [115].
An interesting comparison is the finding that subcutaneous (sc) injection of pregnant CD-1 (ICR) mice with low doses of BPA (2 and 20 µg/kg/day) and DES (0.02, 0.2 and 2 µg/kg/day) accelerated puberty (a common finding), but reduced body weight at puberty in the female offspring [116]. In contrast, a similar prenatal BPA dose (2.4 µg/kg/day) fed to pregnant CF-1 mice both accelerated puberty and increased body weight at puberty in female offspring [91]. An important difference, aside from the route of administration, is that in the Howdeshell study, fetuses were delivered by cesarean section and raised by untreated foster mothers. In a separate study, Palanza and colleagues reported a decrease in maternal nursing behavior in CD-1 mouse mothers fed 10 µg/kg/day BPA during the last five days of pregnancy [117]; similar disruption of nursing behavior has also been reported in rats exposed to a low dose of BPA throughout pregnancy and lactation [118].
3.3 Controversies regarding low-dose studies showing effects of BPA
There are a number of controversies regarding reports of research in laboratory animals of effects of low doses of BPA on diseases that are increasing in frequency in humans [96,119,120]. The assumption that using sc injection of a dose of BPA that was thousands of time below the presumed “no effect dose” would lead to extremely high internal doses relative to oral administration [discussed in [93,121]], has been used to declare studies that did not use oral bolus administration as essentially meaningless. Importantly, there is no argument that in adults, orally absorbed BPA is subject to greater first pass metabolism in the liver relative to BPA administered by non-oral routes [122]. However, the argument that studies are of no value if they used a very low dose of BPA that was administered by a non-oral route is flawed for two reasons: first, in adult rodents, the difference in serum unconjugated BPA based on route of administration is primarily in the maximum value reached after a bolus administration, while the average exposure over the 24 hr after administration (in particular between 12 – 24 hr after administration) does not differ based on route of administration [122]. Thus, the average internal concentration of BPA over the 24 hr after administration via oral or sc route is actually not very different, which dramatically contradicts assumptions made by the European Food Safety Agency [123]. Second, in newborn rodents, route of administration is even less of a factor than the relatively small effect it has in adults [93], and the same adverse outcome has been observed in male rats in terms of adult prostate disease as a result of neonatal exposure to the same low 10 µg/kg/day dose of BPA as a result of oral or sc administration [124]. Finally, an important recent observation is that pharmacokinetics of BPA does not differ between mice, rhesus monkeys and humans [121].
A major controversy has been that regulatory agencies in the USA and Europe have focused on a very narrow set of BPA studies that followed regulatory guidelines and used “good laboratory practices (GLP)” protocols. However, this name does not imply “good science”, since GLP was instituted as a result of fraud in record keeping by commercial chemical testing laboratories [125]. Thus, lack of use of GLP protocols, which greatly increase the cost of the research, has been identified by scientists [125,126], and science review boards in the USA [127] and in Europe [128], as an inappropriate basis for rejecting studies for inclusion in assessing the health hazards of BPA.
While a large number of studies (reviewed above) have reported an increase in body weight as a result of exposure during development to low doses of BPA, not all studies involving developmental exposure to BPA have reported an increase in body weight. In fact, in our initial study in which CF-1 male mice were exposed via feeding the pregnant mother 2 or 20 µg/kg/day BPA, male offspring exposed to the 2 µg/kg/day dose of BPA were slightly (9%), but significantly, lighter than controls in adulthood [129]. Other studies have also reported that developmental exposure to low doses of BPA decreased subsequent body weight in mice [116] and rats [130]. Some studies have reported no significant effect of BPA exposure during development on body weight, for example [131–133]. Finally, an interesting feature of the literature on the effects of developmental exposure to BPA on obesity and other aspects of metabolic disease is that significant effects are often observed on either males or females, but there is not consistency between studies regarding which sex is most affected [29,115]. What is interesting about this issue is that a consistent finding regarding the effects of low doses of BPA on neurobehavioral development is that the effects in rats and mice of BPA are different in males and females [134]. A similar gender difference in the association between maternal BPA levels during pregnancy and subsequent behavior in children has been reported [135,136].
How can all of these different results be reconciled? First, body fat, not body weight, needs to be examined in rodents if one wants to draw conclusions about BPA influencing obesity. The rodent does not have the extensive amount of subcutaneous fat found in humans, particularly women, although there are subcutaneous adipocytes that contribute to an increase in body weight in mice on a high fat diet [137]. We have found that even dramatic differences in abdominal body fat (associated with the kidneys and gonads) in mice result in a very small percent change in body weight [17].
Second, since estrogens function at extremely low concentrations, investigators should be concerned about background levels of estrogenic chemical contamination in control animals. For example, contamination can occur through water. In order to avoid this in our laboratory, we have a reverse osmosis system, copper pipes and a series of carbon filters that we use to provide water to mice in glass bottles. Using a very sensitive assay (HPLC with CoulArray detection; limit of detection = 2 parts per trillion) we do not detect any BPA in water provided to our mice. Another source of contamination to be considered is BPA exposure due to the use of polycarbonate bottles and animal cages; we use polypropylene cages that do not contain BPA [138]. Polycarbonate bottles and cages are known to leach amounts of BPA that can interfere with finding effects of low doses of BPA [138–140]. Investigators who are examining effects of low doses of BPA during development on subsequent obesity (for example a study that examined 0.25 µg/kg/day BPA) need to be aware of the potential for contamination due to housing animals in polycarbonate cages and providing water in polycarbonate bottles [141].
Third, the issue of the appropriate feed to use in studies involving EDCs such as BPA is complicated, and in some cases findings have been quite unexpected. One prediction that seemed reasonable was that it would be best to avoid the use of soy-containing feed in studies involving estrogenic EDCs, since the phytoestrogen content is known to be highly variable from batch to batch of the same feed [142]; this was the subject of a NIEHS meeting on variability in estrogenic activity in feeds used experimental animal research [143].
The potential effects on EDC experiments of components of feed, such as soy phytoestrogens or casein, animal fat vs. vegetable fat, etc. are complex. For example, the complete absence of phytoestrogens in the diet of mice has the surprising effect of resulting in gross adult obesity and other aspects of metabolic syndrome, as well as adverse effects on reproductive organs [17,144]. The surprising finding here is that in laboratory rodents there is an important role for phytoestrogens in preventing obesity as well as preventing abnormal development of the reproductive organs, which may be the consequence of selection over thousands of generations for mice that could withstand high levels of phytoestrogens in the soy-based feeds typically used by commercial breeding facilities because it is the least expensive feed. Since the amount of phytoestrogens in soy-based feed can vary dramatically while the amount of protein is held constant, controlling the amount of phytoestrogens in soy-based feed is required for consistency of results in rodent studies, while completely eliminating phytoestrogens from the feed, which might seem to be a reasonable solution, could actually dramatically interfere with the ability to study environmental factors involved in obesity in rodent models [17,143]
Given the strange effects that various feeds have on both the endocrine and metabolic control systems involved in regulating body fat, glucose homeostasis and reproductive processes in experimental animals [17,29,145], it is not difficult to imagine that differences in feed (even though the feed used might have the same name) due to batch-to-batch variation, could contribute to variability in outcome of studies examining the effects of EDCs on obesity [143]. To make matters worse, variability in phytoestrogen content of feed has a greater effect on some strains of laboratory animals than it does on other strains [146]. An issue that is clearly of great interest, but at this time remains controversial, is the interaction between BPA and the nutritional components of feed in terms of specific outcomes associated with obesity and other aspects of metabolic disease.
4. The adult phenotype due to intrauterine growth restriction (IUGR) is similar to developmental exposure to BPA
There is extensive epidemiological evidence showing that babies with intrauterine growth restriction (IUGR) who then experience a rapid “catch-up” growth spurt during childhood are at high risk for adult obesity and type 2 diabetes (as well as other aspects of metabolic syndrome), consistent with the DOHaD hypothesis [4,18]. Thus, fetal growth rate interacts with childhood-adolescence growth rate in terms of whether IUGR leads to adult obesity and other metabolic diseases (Figure 1). We have developed a novel crowded uterine horn mouse model that results in siblings that range from growth restricted to macrosomic due to differences in placental blood flow based on location in the crowded uterus (Figure 2). Importantly, the IUGR mice experience a rapid period of catch-up growth: IUGR mice experience about a 90% increase in body weight, while macrosomic males have about a 30% increase in body weight during the first week after weaning (Coe et al. in preparation). Adult IUGR male mice show marked similarities to IUGR humans in terms of glucose intolerance and elevated insulin as well as an increase in total abdominal fat weight [147]. Importantly, while both IUGR and macrosomic males remained significantly heavier than male mice with a median body weight at birth, we have found significant differences in adipocyte gene expression and number in adult male gonadal fat (Coe et al. in preparation). There is thus a markedly different etiology of obesity in these two overweight sub-populations of mice relating to differences in both fetal and post-weaning growth.
Figure 2.

Blood flow (indicated by arrows) into the loop uterine artery is bi-directional from both the ovarian (O) and cervical (C) ends of the uterine horn, which leads to greater placental blood flow at the ends relative to the middle (M) of each uterine horn. The data shown are placental blood flow measurements taken from a hemi-ovariectomized pregnant female CD-1 mouse on gestation day 18; the female was injected with radiolabelled microspheres to measure blood flow [150]. Removing the left ovary prior to pregnancy results in double the number of oocytes being ovulated from the remaining right ovary and crowding in the right uterine horn, which is separate from the left uterine horn in mice and rats.
As noted above, there are some interesting parallels between the effect of perinatal exposure to BPA and the consequence of a mouse pup being IUGR; BPA-exposed developing mouse pups appear to have traits similar to those of IUGR pups. This is interesting in that a recent occupational study from China reported data suggesting that maternal exposure to BPA was related to IUGR [148]. In addition, there are similarities in gonadal adipocyte gene expression in comparisons of IUGR vs. macrosomic animals and BPA-exposed vs. control animals; similar to IUGR, BPA elevates PPAR, C/EBP and LPL in abdominal adipocytes [29].
It is interesting that different EDCs identified as obesogens can lead to adult obesity with underlying differences in phenotype. For example, developmental exposure to a low dose of BPA results in an increase in adult body weight associated with a decrease in abdominal adipocyte number, but an increase in adipocyte size (hypertrophy) [29]; we have similar findings in male CD-1 mice (Coe et al. in preparation). The effects of BPA are similar to the effect of developmental exposure to nicotine [149], but different from effects of organotins, which result in obesity characterized by adipocyte hyperplasia [47].
5. Conclusions
At this time there is limited human data relating obesity with environmental chemicals, and specifically environmental chemicals that are estrogenic or otherwise disrupt estrogen homeostasis. However, there has been an increase in experimental animal research relating environmental chemical exposure to obesity, insulin and glucose dysregulation, and type 2 diabetes. Findings from these studies have led to an increased awareness that energy expenditure and components of diet, while important, likely do not explain the rate of increase in obesity that has been documented over the last two decades [4,19,20]. While the contribution of environmental chemicals to the obesity epidemic remains a largely unexamined issue, the dramatic increase in the incidence of obesity has occurred in parallel with a dramatic increase in the use of plastic and other products containing EDCs [for example personal care products that contain phthalates [84]]. Plastics also contain other EDCs in addition to BPA. The animal experiments showing a relationship between accelerated postnatal growth, altered insulin secretion and glucose sensitivity due to developmental exposure to daily doses of BPA within the range of human exposure provide a strong argument for further research into the possibility that developmental exposure to BPA as well as other EDCs are contributing to the development of obesity later in life.
-
➢
Estrogenic EDC exposure may lead to obesity via genetic programming.
-
➢
Estrogens and EDCs directly regulate adipocyte function.
-
➢
Developmental EDC exposure influences adult body weight and metabolism.
-
➢
Effects of EDCs on body weight and on glucose homeostasis are seen at low doses.
Acknowledgments
Role of the funding source.
Support during the preparation of this chapter was provided by grants to FSvS (ES018764) and to SCN (HD056441). The study sponsors had no further part in the study or in the decision to submit the paper for publication.
Abbreviations
- BPA
bisphenol A
- Cyp19
aromatase
- DGAT1
diglyceride acyltransferase
- DOHaD
developmental origin of health and disease
- ERα
estrogen receptor α
- ERβ
estrogen receptor β
- C/EBP
CCAAT/enhancer-binding protein
- C/EBPβ
CCAAT/enhancer-binding protein β
- GLP
good laboratory practices
- GLUT4
glucose transporter type 4
- GnRH
gonadotropin releasing hormone
- GPAT
glycerol-3-phosphate acyltransferase
- HPLC
high performance liquid chromatography
- IUGR
intrauterine growth restriction
- LPL
lipoprotein lipase
- PPARγ
peroxisome proliferator-activated receptor gamma
- SERMs
selective estrogen receptor modulators
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement.
SCN, JAT, BLC and BMA have nothing to disclose. FSvS has been a consultant for attorneys involved in product labeling litigation.
References
- 1.Gao Q, Horvath TL. Cross-talk between estrogen and leptin signaling in the hypothalamus. Am J Physiol Endocrinol Metab. 2008;294:E817–E826. doi: 10.1152/ajpendo.00733.2007. [DOI] [PubMed] [Google Scholar]
- 2.Cooke PS, Naaz A. The role of estrogens in adipocyte development and function. Exp. Biol. Med. 2004;229:1127–1135. doi: 10.1177/153537020422901107. [DOI] [PubMed] [Google Scholar]
- 3.Newbold RR, Padilla-Banks E, Snyder RJ, Phillips TM, Jefferson WN. Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod Toxicol. 2007;23:290–296. doi: 10.1016/j.reprotox.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heindel JJ, vom Saal FS. Overview of obesity and the role of developmental nutrition and environmental chemical exposures. Mol. Cell Endocrinol. 2009;304:90–96. doi: 10.1016/j.mce.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 5.Cooke PS, Heine PA, Taylor JA, Lubahn DB. The role of estrogen and estrogen receptor-alpha in male adipose tissue. Mol. Cell Endocrinol. 2001;178:147–154. doi: 10.1016/s0303-7207(01)00414-2. [DOI] [PubMed] [Google Scholar]
- 6.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc. Natl. Acad. Sci. 2000;97:12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones ME, Thorburn AW, Britt KL, Hewitt KN, Misso ML, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice accumulate excess adipose tissue. J. Steroid Biochem. Mol. Biol. 2001;79:3–9. doi: 10.1016/s0960-0760(01)00136-4. [DOI] [PubMed] [Google Scholar]
- 9.Marker PC, Donjacour AA, Dahiya R, Cunha GR. Hormonal, cellular, and molecular control of prostatic development. Dev Biol. 2003;253:165–174. doi: 10.1016/s0012-1606(02)00031-3. [DOI] [PubMed] [Google Scholar]
- 10.Choudhuri S, Cui Y, Klaassen CD. Molecular targets of epigenetic regulation and effectors of environmental influences. Toxicol Appl Pharmacol. 2010;245:378–393. doi: 10.1016/j.taap.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab. 2010;21:214–222. doi: 10.1016/j.tem.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wild SH, Byrne CD. Evidence for fetal programming of obesity with a focus on putative mechanisms. Nutrition Research Reviews. 2004;17:153–162. doi: 10.1079/NRR200487. [DOI] [PubMed] [Google Scholar]
- 14.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal K. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 15.Yajnik CS. Fetal origins of adult disease: Where do we stand? Int. J. Diab. Dev. Countries. 2001;21:42–50. [Google Scholar]
- 16.Barker DJ. The developmental origins of adult disease. Journal of the American College of Nutrition. 2004;23:588S–595S. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- 17.Ruhlen RL, Howdeshell KL, Mao J, Taylor JA, Bronson FH, Newbold RR, Welshons WV, vom Saal FS. Low phytoestrogen levels in feed increase fetal serum estradiol resulting in the "fetal estrogenization syndrome" and obesity in CD-1 mice. Environ Health Perspect. 2008;116:322–328. doi: 10.1289/ehp.10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oken E, Gillman MW. Fetal origins of obesity. Obes. Res. 2003;11:496–506. doi: 10.1038/oby.2003.69. [DOI] [PubMed] [Google Scholar]
- 19.Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J. Altern. Complement. Med. 2002;8:185–192. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- 20.Keith SW, Redden DT, Katzmarzyk PT, Boggiano MM, Hanlon EC, Benca RM, Ruden D, Pietrobelli A, Barger JL, Fontaine KR, Wang C, Aronne LJ, Wright SM, Baskin M, Dhurandhar NV, Lijoi MC, Grilo CM, Deluca M, Westfall AO, Allison DB. Putative contributors to the secular increase in obesity: exploring the roads less traveled. Int J Obes (Lond) 2006 doi: 10.1038/sj.ijo.0803326. Online June 27. [DOI] [PubMed] [Google Scholar]
- 21.Gluckman PD, Hanson MA, Morton SM, Pinal CS. Life-long echoes--a critical analysis of the developmental origins of adult disease model. Biol Neonate. 2005;87:127–139. doi: 10.1159/000082311. [DOI] [PubMed] [Google Scholar]
- 22.Stocker CJ, Arch JR, Cawthorne MA. Fetal origins of insulin resistance and obesity. Proc Nutr Soc. 2005;64:143–151. doi: 10.1079/pns2005417. [DOI] [PubMed] [Google Scholar]
- 23.Adair LS, Cole TJ. Rapid child growth raises blood pressure in adolescent boys who were thin at birth. Hypertension. 2003;41:451–456. doi: 10.1161/01.HYP.0000054212.23528.B2. [DOI] [PubMed] [Google Scholar]
- 24.Donato J, Jr, Cravo RM, Frazao R, Elias CF. Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology. 2011;93:9–18. doi: 10.1159/000322472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben-Jonathan N, Hugo ER, Brandebourg TD. Effects of bisphenol A on adipokine release from human adipose tissue: Implications for the metabolic syndrome. Mol Cell Endocrinol. 2009;304:49–54. doi: 10.1016/j.mce.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lappas M, Yee K, Permezel M, Rice GE. Release and regulation of leptin, resistin and adiponectin from human placenta, fetal membranes, and maternal adipose tissue and skeletal muscle from normal and gestational diabetes mellitus-complicated pregnancies. J Endocrinol. 2005;186:457–465. doi: 10.1677/joe.1.06227. [DOI] [PubMed] [Google Scholar]
- 27.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 28.Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Developmental exposure to estrogenic compounds and obesity. Birth Defects Res A Clin Mol Teratol. 2005;73:478–480. doi: 10.1002/bdra.20147. [DOI] [PubMed] [Google Scholar]
- 29.Somm E, Schwitzgebel VM, Toulotte A, Cederroth CR, Combescure C, Nef S, Aubert ML, Huppi PS. Perinatal exposure to bisphenol a alters early adipogenesis in the rat. Environ Health Perspect. 2009;117:1549–1555. doi: 10.1289/ehp.11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 31.Chen HC, Smith SJ, Ladha Z, Jensen DR, Ferreira LD, Pulawa LK, McGuire JG, Pitas RE, Eckel RH, Farese RV., Jr Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J Clin Invest. 2002;109:1049–1055. doi: 10.1172/JCI14672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdelgadir SE, Resko JA, Ojeda SR, Lephart ED, McPhaul MJ, Roselli CE. Androgens regulate aromatase cytochrome P450 messenger ribonucleic acid in rat brain. Endocrinology. 1994;135:395–401. doi: 10.1210/endo.135.1.8013375. [DOI] [PubMed] [Google Scholar]
- 33.Arase S, Ishii K, Igarashi K, Aisaki K, Yoshio Y, Matsushima A, Shimohigashi Y, Arima K, Kanno J, Sugimura Y. Endocrine disrupter bisphenol a increases in situ estrogen production in the mouse urogenital sinus. Biol Reprod. 2011;84:734–742. doi: 10.1095/biolreprod.110.087502. [DOI] [PubMed] [Google Scholar]
- 34.Blouin K, Nadeau M, Mailloux J, Daris M, Lebel S, Luu-The V, Tchernof A. Pathways of adipose tissue androgen metabolism in women: depot differences and modulation by adipogenesis. Am J Physiol Endocrinol Metab. 2009;296:E244–E255. doi: 10.1152/ajpendo.00039.2008. [DOI] [PubMed] [Google Scholar]
- 35.Nativelle-Serpentini C, Richard S, Seralini GE, Sourdaine P. Aromatase activity modulation by lindane and bisphenol-A in human placental JEG-3 and transfected kidney E293 cells. Toxicol In Vitro. 2003;17:413–422. doi: 10.1016/s0887-2333(03)00046-8. [DOI] [PubMed] [Google Scholar]
- 36.Subbaramaiah K, Hudis CA, Dannenberg AJ. The prostaglandin transporter regulates adipogenesis and aromatase transcription. Cancer Prev Res (Phila) 2011;4:194–206. doi: 10.1158/1940-6207.CAPR-10-0367. [DOI] [PubMed] [Google Scholar]
- 37.Pino AM, Rodriguez JM, Rios S, Astudillo P, Leiva L, Seitz G, Fernandez M, Rodriguez JP. Aromatase activity of human mesenchymal stem cells is stimulated by early differentiation, vitamin D and leptin. J Endocrinol. 2006;191:715–725. doi: 10.1677/joe.1.07026. [DOI] [PubMed] [Google Scholar]
- 38.Misso ML, Murata Y, Boon WC, Jones ME, Britt KL, Simpson ER. Cellular and molecular characterization of the adipose phenotype of the aromatase-deficient mouse. Endocrinology. 2003;144:1474–1480. doi: 10.1210/en.2002-221123. [DOI] [PubMed] [Google Scholar]
- 39.Homma H, Kurachi H, Nishio Y, Takeda T, Yamamoto T, Adachi K, Morishige K, Ohmichi M, Matsuzawa Y, Murata Y. Estrogen suppresses transcription of lipoprotein lipase gene. Existence of a unique estrogen response element on the lipoprotein lipase promoter. J. Biol. Chem. 2000;275:11404–11411. doi: 10.1074/jbc.275.15.11404. [DOI] [PubMed] [Google Scholar]
- 40.Krempler F, Breban D, Oberkofler H, Esterbauer H, Hell E, Paulweber B, Patsch W. Leptin, peroxisome proliferator-activated receptor-gamma, and CCAAT/enhancer binding protein-alpha mRNA expression in adipose tissue of humans and their relation to cardiovascular risk factors. Arterioscler Thromb Vasc Biol. 2000;20:443–449. doi: 10.1161/01.atv.20.2.443. [DOI] [PubMed] [Google Scholar]
- 41.Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev. 1998;78:783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 42.Yamauchi T, Kamon J, Waki H, Murakami K, Motojima K, Komeda K, Ide T, Kubota N, Terauchi Y, Tobe K, Miki H, Tsuchida A, Akanuma Y, Nagai R, Kimura S, Kadowaki T. The mechanisms by which both heterozygous peroxisome proliferator-activated receptor gamma (PPARgamma) deficiency and PPARgamma agonist improve insulin resistance. J Biol Chem. 2001;276:41245–41254. doi: 10.1074/jbc.M103241200. [DOI] [PubMed] [Google Scholar]
- 43.Zhang JW, Klemm DJ, Vinson C, Lane MD. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J Biol Chem. 2004;279:4471–4478. doi: 10.1074/jbc.M311327200. [DOI] [PubMed] [Google Scholar]
- 44.Gregoire FM. Adipocyte differentiation: From fibroblast to endocrine cell. Exp. Biol. Med. 2001;226:997–1002. doi: 10.1177/153537020122601106. [DOI] [PubMed] [Google Scholar]
- 45.Johnson PR, Hirsch J. Cellularity of adipose depots in six strains of genetically obese mice. J. Lipid Res. 1972;13:2–11. [PubMed] [Google Scholar]
- 46.Hager A, Sjostrm L, Arvidsson B, Bjorntorp P, Smith U. Body fat and adipose tissue cellularity in infants: a longitudinal study. Metabolism. 1977;26:607–614. doi: 10.1016/0026-0495(77)90082-8. [DOI] [PubMed] [Google Scholar]
- 47.Janesick A, Blumberg B. Endocrine disrupting chemicals and the developmental programming of adipogenesis and obesity. Birth Defects Res C Embryo Today. 2011;93:34–50. doi: 10.1002/bdrc.20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benvenuti S, Cellai I, Luciani P, Deledda C, Saccardi R, Mazzanti B, Dal Pozzo S, Serio M, Peri A. Androgens and Estrogens Prevent Rosiglitazone-Induced Adipogenesis in Human Mesenchymal Stem Cells. J Endocrinol Invest. 2011 doi: 10.3275/7739. online May 19. [DOI] [PubMed] [Google Scholar]
- 49.Ailhaud G, Grimaldi P, Negrel R. Cellular and molecular aspects of adipose tissue development. Annu. Rev. Nutr. 1992;12:207–233. doi: 10.1146/annurev.nu.12.070192.001231. [DOI] [PubMed] [Google Scholar]
- 50.Larsen TM, Toubro S, Astrup A. PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int. J Obes. Relat. Metab. Disord. 2003;27:147–161. doi: 10.1038/sj.ijo.802223. [DOI] [PubMed] [Google Scholar]
- 51.Tong Q, Hotamisligil GS. Molecular mechanisms of adipocyte differentiation. Rev. Endocr. Metab. Disord. 2001;2:349–355. doi: 10.1023/a:1011863414321. [DOI] [PubMed] [Google Scholar]
- 52.Janesick A, Blumberg B. Minireview: PPARgamma as the target of obesogens. J Steroid Biochem Mol Biol. 2011;147:4–8. doi: 10.1016/j.jsbmb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joyner JM, Hutley LJ, Cameron DP. Estrogen receptors in human preadipocytes. Endocrine. 2001;15:225–230. doi: 10.1385/ENDO:15:2:225. [DOI] [PubMed] [Google Scholar]
- 54.Naaz A, Zakroczymski M, Heine P, Taylor J, Saunders P, Lubahn D, Cooke PS. Effect of ovariectomy on adipose tissue of mice in the absence of estrogen receptor alpha (ERalpha): a potential role for estrogen receptor beta (ERbeta) Horm. Metab. Res. 2002;34:758–763. doi: 10.1055/s-2002-38259. [DOI] [PubMed] [Google Scholar]
- 55.Anderson LA, McTernan PG, Barnett AH, Kumar S. The effects of androgens and estrogens on preadipocyte proliferation in human adipose tissue: influence of gender and site. J. Clin. Endocrinol. Metab. 2001;86:5045–5051. doi: 10.1210/jcem.86.10.7955. [DOI] [PubMed] [Google Scholar]
- 56.Dieudonne MN, Pecquery R, Leneveu MC, Giudicelli Y. Opposite effects of androgens and estrogens on adipogenesis in rat preadipocytes: evidence for sex and site-related specificities and possible involvement of insulin-like growth factor 1 receptor and peroxisome proliferator-activated receptor gamma2. Endocrinology. 2000;141:649–656. doi: 10.1210/endo.141.2.7293. [DOI] [PubMed] [Google Scholar]
- 57.Lea-Currie YR, Monroe D, McIntosh MK. Dehydroepiandrosterone and related steroids alter 3T3-L1 preadipocyte proliferation and differentiation. Comp. Biochem. Physiol. C. Pharmacol. Toxicol. Endocrinol. 1999;123:17–25. doi: 10.1016/s0742-8413(99)00003-1. [DOI] [PubMed] [Google Scholar]
- 58.Cooper SC, Roncari DA. 17-beta-estradiol increases mitogenic activity of medium from cultured preadipocytes of massively obese persons. J. Clin. Invest. 1989;83:1925–1929. doi: 10.1172/JCI114099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Myers JP, Zoeller TJ, vom Saal FS. A clash of old and new scientific concepts in toxicity, with important implications for public health. Environ. Health Perspect. 2009a;117:1652–1655. doi: 10.1289/ehp.0900887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Welshons WV, Thayer KS, Taylor J, Judy B, vom Saal FS. Large effects from small exposures: I. Mechanisms for endocrine-disrupting chemicals with estrogenic activity. Environ. Health Perspect. 2003;111:904–106. doi: 10.1289/ehp.5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao JW, Gao ZL, Mei H, Li YL, Wang Y. Differentiation of human mesenchymal stem cells: the potential mechanism for estrogen-induced preferential osteoblast versus adipocyte differentiation. Am J Med Sci. 2011;341:460–468. doi: 10.1097/MAJ.0b013e31820865d5. [DOI] [PubMed] [Google Scholar]
- 62.Migliaccio S, Newbold RR, McLachlan JA, Korach KS. Alterations in estrogen levels during development affects the skeleton: use of an animal model. Environ Health Perspect. 1995;103 Suppl 7:95–97. doi: 10.1289/ehp.95103s795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Migliaccio S, Newbold RR, Bullock BC, Jefferson WJ, Sutton FG, Jr, McLachlan JA, Korach KS. Alterations of maternal estrogen levels during gestation affect the skeleton of female offspring. Endocrinology. 1996;137:2118–2125. doi: 10.1210/endo.137.5.8612556. [DOI] [PubMed] [Google Scholar]
- 64.Migliaccio S, Newbold RR, Teti A, Jefferson WJ, Toverud SU, Taranta A, Bullock BC, Suggs CA, Spera G, Korach KS. Transient estrogen exposure of female mice during early development permanently affects osteoclastogenesis in adulthood. Bone. 2000;27:47–52. doi: 10.1016/s8756-3282(00)00286-6. [DOI] [PubMed] [Google Scholar]
- 65.Pelch KE, Carleton SM, Phillips CL, Nagel SC. Developmental Exposure to Low Dose Xenoestrogens Alters Femur Length and Tensile Strength in Adult Mice. Biol Reprod. 2011 doi: 10.1095/biolreprod.111.096545. Online: Nov 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Newbold RR, Padilla-Banks E, Jefferson WN, Heindel JJ. Effects of endocrine disruptors on obesity. Int J Androl. 2008;31:201–208. doi: 10.1111/j.1365-2605.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- 67.vom Saal FS. Sexual differentiation in litter-bearing mammals: influence of sex of adjacent fetuses in utero. J Anim Sci. 1989;67:1824–1840. doi: 10.2527/jas1989.6771824x. [DOI] [PubMed] [Google Scholar]
- 68.Colborn T, Clement C. Chemically-Induced alterations in sexual and functional development: The wildlife/human connection. In: Mehlman MA, editor. Advances in Modern Environmental Toxicology. Princeton: Princeton Scientific Publishing C., Inc.; 1992. p. 403. [Google Scholar]
- 69.Colborn T, vom Saal FS, Soto AM. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environmental Health Perspectives. 1993;101:378–384. doi: 10.1289/ehp.93101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology. 2006;147:S56–S69. doi: 10.1210/en.2005-1159. [DOI] [PubMed] [Google Scholar]
- 71.vom Saal FS, Sheehan DM. Challenging risk assessment. Forum for Applied Research and Public Policy. 1998;13:11–18. [Google Scholar]
- 72.Medlock KL, Forrester TM, Sheehan DM. Short-term effects of physiological and pharmacological doses of estradiol on estrogen receptor and uterine growth. Journal of Receptor Research. 1991;11:743–756. doi: 10.3109/10799899109064677. [DOI] [PubMed] [Google Scholar]
- 73.Shioda T, Chesnes J, Coser KR, Zou L, Hur J, Dean KL, Sonnenschein C, Soto AM, Isselbacher KJ. Importance of dosage standardization for interpreting transcriptomal signature profiles: evidence from studies of xenoestrogens. Proc. Natl. Acad. Sci. 2006;103:12033–12038. doi: 10.1073/pnas.0605341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hunt PA. Assessing chemical risk: Societies offer expertise. Science. 2011;331:1136. doi: 10.1126/science.331.6021.1136-a. [DOI] [PubMed] [Google Scholar]
- 76.Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, Watson CS, Zoeller RT, Belcher SM. In vitro molecular mechanisms of bisphenol A action. Reproductive Toxicology. 2007;24:178–198. doi: 10.1016/j.reprotox.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 77.Watson CS, Jeng YJ, Kochukov MY. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci. 2010;115:1–11. doi: 10.1093/toxsci/kfp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goldman L, Falk H, Landrigan PJ, Balk SJ, Reigart JR, Etzel RA. Environmental pediatrics and its impact on government health policy. Pediatrics. 2004;113:1146–1157. [PubMed] [Google Scholar]
- 79.Landrigan P, Garg A, Droller DB. Assessing the effects of endocrine disruptors in the National Children's Study. Environ Health Perspect. 2003;111:1678–1682. doi: 10.1289/ehp.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Landrigan PJ. Children's health and the environment--the first Herbert L. Needleman Award Lecture. Matern Child Health J. 1997;1:61–64. doi: 10.1023/a:1026280520459. [DOI] [PubMed] [Google Scholar]
- 81.Hatch EE, Nelson JW, Stahlhut RW, Webster TF. Association of endocrine disruptors and obesity: perspectives from epidemiological studies. Int J Androl. 2010;33:324–332. doi: 10.1111/j.1365-2605.2009.01035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, Melzer D. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA. 2008;300:1303–1310. doi: 10.1001/jama.300.11.1303. [DOI] [PubMed] [Google Scholar]
- 83.Melzer D, Rice NE, Lewis C, Henley WE, Galloway TS. Association of urinary bisphenol a concentration with heart disease: evidence from NHANES 2003/06. PLoS One. 2010;5:e8673. doi: 10.1371/journal.pone.0008673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stahlhut RW, van Wijngaarden E, Dye TD, Cook S, Swan SH. Concentrations of urinary phthalate metabolites are associated with increased waist circumference and insulin resistance in adult U.S. males. Environ Health Perspect. 2007;115:876–882. doi: 10.1289/ehp.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blumberg B. Obesogens, stem cells and the maternal programming of obesity. Journal of Developmental Origins of Health and Disease. 2011;2:3–8. doi: 10.1017/S2040174410000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bailin PD, Byrne M, Lewis S, Liroff R. Public awareness drives market for safer alternatives: bisphenol A market analysis report. [Access date: April 20, 2011];2008 http://www.iehn.org/publications.reports.bpa.php. [Google Scholar]
- 87.Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in human maternal-fetal-placental unit. Environ. Health Perspect. 2002;110:A703–A707. doi: 10.1289/ehp.110-1241091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Human Reproduction. 2002;17:2839–2841. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- 89.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA) Reproductive Toxicology. 2007;24:139–177. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 90.Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ Health Perspect. 2010;118:1055–1070. doi: 10.1289/ehp.0901716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature. 1999;401:763–764. doi: 10.1038/44517. [DOI] [PubMed] [Google Scholar]
- 92.Zalko D, Soto AM, Dolo L, Dorio C, Rathahao E, Debrauwer L, Faure R, Cravedi JP. Biotransformations of bisphenol A in a mammalian model: answers and new questions raised by low-dose metabolic fate studies in pregnant CD-1 mice. Environ. Health Perspect. 2002;111:309–319. doi: 10.1289/ehp.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Taylor JA, Welshons WV, Vom Saal FS. No effect of route of exposure (oral; subcutaneous injection) on plasma bisphenol A throughout 24h after administration in neonatal female mice. Reprod Toxicol. 2008;25:169–176. doi: 10.1016/j.reprotox.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Routhledge EJ, White R, Parker MG, Sumpter JP. Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) α and ERββ. J. Biol. Chem. 2000;46:35986–35993. doi: 10.1074/jbc.M006777200. [DOI] [PubMed] [Google Scholar]
- 95.Nagel SC, Hagelbarger JL, McDonnell DP. Development of an ER action indicator mouse for the study of estrogens, selective ER modulators (SERMs), and Xenobiotics. Endocrinology. 2001;142:4721–4728. doi: 10.1210/endo.142.11.8471. [DOI] [PubMed] [Google Scholar]
- 96.vom Saal FS, Hughes C. An extensive new literature concerning low-dose effects of bisphenol A shows the need for a new risk assessment. Environ Health Perspect. 2005;113:926–933. doi: 10.1289/ehp.7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, Vandenbergh JG, Walser-Kuntz DR, vom Saal FS. In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol. 2007;24:199–224. doi: 10.1016/j.reprotox.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ashby J, Tinwell H, Haseman J. Lack of effects for low dose levels of bisphenol A (BPA) and diethylstilbestrol (DES) on the prostate gland of CF1 mice exposed in utero. Reg Tox Pharm. 1999;30:156–166. doi: 10.1006/rtph.1999.1317. [DOI] [PubMed] [Google Scholar]
- 99.Markey CM, Coombs MA, Sonnenschein C, Soto AM. Mammalian development in a changing environment: exposure to endocrine disruptors reveals the developmental plasticity of steroid-hormone target organs. Evol Dev. 2003;5:67–75. doi: 10.1046/j.1525-142x.2003.03011.x. [DOI] [PubMed] [Google Scholar]
- 100.Miyawaki J, Sakayama K, Kato H, Yamamoto H, Masuno H. Perinatal and postnatal exposure to bisphenol a increases adipose tissue mass and serum cholesterol level in mice. Journal of atherosclerosis and thrombosis. 2007;14:245–252. doi: 10.5551/jat.e486. [DOI] [PubMed] [Google Scholar]
- 101.Newbold RR, Jefferson WN, Padilla-Banks E. Long-term adverse effects of neonatal exposure to bisphenol A on the murine female reproductive tract. Reprod Toxicol. 2007;24:253–258. doi: 10.1016/j.reprotox.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nikaido Y, Yoshizawa K, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. Effects of maternal xenoestrogen exposure on development of the reproductive tract and mammary gland in female CD-1 mouse offspring. Reprod Toxicol. 2004;18:803–811. doi: 10.1016/j.reprotox.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 103.Patisaul HB, Bateman HL. Neonatal exposure to endocrine active compounds or an ERbeta agonist increases adult anxiety and aggression in gonadally intact male rats. Horm Behav. 2008;53:580–588. doi: 10.1016/j.yhbeh.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 104.Rubin BS, Murray MK, Bamassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ. Health Perspect. 2001;109:657–680. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Newbold RR, Jefferson WN, Padilla-Banks E, Haseman J. Developmental exposure to diethylstilbestrol (DES) alters uterine response to estrogens in prepubescent mice: low versus high dose effects. Reproductive Toxicology. 2004;18:399–406. doi: 10.1016/j.reprotox.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 106.Akingbemi BT, Sottas CM, Koulova AI, Klinefelter GR, Hardy MP. Inhibition of testicular steroidogenesis by the xenoestrogen bisphenol A is associated with reduced pituitary luteinizing hormone secretion and decreased steroidogenic enzyme gene expression in rat Leydig cells. Endocrinology. 2004;145:592–603. doi: 10.1210/en.2003-1174. [DOI] [PubMed] [Google Scholar]
- 107.Markey CM, Michaelson CL, Veson EC, Sonnenschein C, Soto AM. The mouse uterotrophic assay: a reevaluation of its validity in assessing the estrogenicity of bisphenol A. Environ Health Perspect. 2001;109:55–60. doi: 10.1289/ehp.0110955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology. 2007;148:116–127. doi: 10.1210/en.2006-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Takai Y, Tsutsumi O, Ikezuki Y, Hiroi H, Osuga Y, Momoeda M, Yano T, Taketani Y. Estrogen receptor-mediated effects of a xenoestrogen, bisphenol A on preimplantation mouse embryos. Biochem Biophys Res Commun. 2000a;270:918–921. doi: 10.1006/bbrc.2000.2548. [DOI] [PubMed] [Google Scholar]
- 110.Takai Y, Tsutsumi O, Ikezuki Y, Kamei Y, Osuga Y, Yano T, Taketan Y. Preimplantation exposure to bisphenol A advances postnatal development. Reproductive Toxicology. 2000b;15:71–74. doi: 10.1016/s0890-6238(00)00119-2. [DOI] [PubMed] [Google Scholar]
- 111.Masuno H, Kidani T, Sekiya K, Sakayama K, Shiosaka T, Yamamoto H, Honda K. Bisphenol A in combination with insulin can accelerate the conversion of 3T3-L1 fibroblasts to adipocytes. Journal of Lipid Research. 2002;43:676–684. [PubMed] [Google Scholar]
- 112.Sakurai K, Kawazuma M, Adachi T, Harigaya T, Saito Y, Hashimoto N, Mori C. Bisphenol A affects glucose transport in mouse 3T3-F442A adipocytes. Br J Pharmacol. 2004;141:209–14. doi: 10.1038/sj.bjp.0705520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deems RO, Evans JL, Deacon RW, Honer CM, Chu DT, Burki K, Fillers WS, Cohen DK, Young DA. Expression of human GLUT4 in mice results in increased insulin action. Diabetologia. 1994;37:1097–1104. doi: 10.1007/BF00418373. [DOI] [PubMed] [Google Scholar]
- 114.Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ Health Perspect. 2006;114:106–112. doi: 10.1289/ehp.8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Alonso-Magdalena P, Vieira E, Soriano S, Menes L, Burks D, Quesada I, Nadal A. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. 2010;118:1243–1250. doi: 10.1289/ehp.1001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Honma S, Suzuki A, Buchanan DL, Katsu Y, Watanabe H, Iguchi T. Low dose effect of in utero exposure to bisphenol A and diethylstilbestrol on female mouse reproduction. Reprod. Toxicol. 2002;16:117–122. doi: 10.1016/s0890-6238(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 117.Palanza P, Howdeshell KL, Parmigiani S, vom Saal FS. Exposure to a low dose of bisphenol A during fetal life or in adulthood alters maternal behavior in mice. Environ. Health Perspect. 2002;110:415–422. doi: 10.1289/ehp.02110s3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Della Seta D, Minder I, Dessi-Fulgheri F, Farabollini F. Bisphenol-A exposure during pregnancy and lactation affects maternal behavior in rats. Brain Res Bull. 2005;65:255–260. doi: 10.1016/j.brainresbull.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 119.Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr Rev. 2009;30:75–95. doi: 10.1210/er.2008-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vandenberg LN, Chahoud I, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Biomonitoring studies should be used by regulatory agencies to assess human exposure levels and safety of bisphenol a. Environ Health Perspect. 2010b;118:1051–1054. doi: 10.1289/ehp.0901717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Taylor JA, Vom Saal FS, Welshons WV, Drury B, Rottinghaus G, Hunt PA, Toutain PL, Laffont CM, Vandevoort CA. Similarity of bisphenol a pharmacokinetics in rhesus monkeys and mice: relevance for human exposure. Environ Health Perspect. 2011;119:422–430. doi: 10.1289/ehp.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pottenger LH, Domoradzki JY, Markham DA, Hansen SC, Cagen SZ, Waechter JM. The relative bioavailability and metabolism of bisphenol A in rats is dependent upon the route of administration. Toxicological Sciences. 2000;54:3–18. doi: 10.1093/toxsci/54.1.3. [DOI] [PubMed] [Google Scholar]
- 123.EFSA. Authority EFS, editor. Toxicokinetics of Bisphenol A - Scientific Opinion of the Panel on Food additives Flavourings Processing aids and Materials in Contact with Food (AFC) [Access date: May 30, 2010];2008 doi: 10.2903/j.efsa.2008.759. http://www.efsa.europa.eu/EFSA/efsa_locale1178620753812_1211902017492.htm. [DOI] [PMC free article] [PubMed]
- 124.Prins GS, Ye SH, Birch L, Ho SM, Kannan K. Serum bisphenol A pharmacokinetics and prostate neoplastic responses following oral and subcutaneous exposures in neonatal Sprague-Dawley rats. Reprod Toxicol. 2011;31:1–9. doi: 10.1016/j.reprotox.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Myers JP, vom Saal FS, Akingbemi BT, Arizono K, Belcher S, Colborn T, Chahoud I, Crain DA, Farabollini F, Guillette LJ, Jr, Hassold T, Ho SM, Hunt PA, Iguchi T, Jobling S, Kanno J, Laufer H, Marcus M, McLachlan JA, Nadal A, Oehlmann J, Olea N, Palanza P, Parmigiani S, Rubin BS, Schoenfelder G, Sonnenschein C, Soto AM, Talsness CE, Taylor JA, Vandenberg LN, Vandenbergh JG, Vogel S, Watson CS, Welshons WV, Zoeller RT. Why public health agencies cannot depend on good laboratory practices as a criterion for selecting data: the case of bisphenol A. Environ Health Perspect. 2009b;117:309–315. doi: 10.1289/ehp.0800173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tweedale AC. Uses of 'Good Laboratory Practices' by regulated industry and agencies, and the safety of bisphenol A. J Epidemiol Community Health. 2011;65:475–476. doi: 10.1136/jech.2010.127761. [DOI] [PubMed] [Google Scholar]
- 127.FDA. FDA Science Board Subcommittee Report on Bisphenol A. [Accessed October 4, 2011];2008 http://www.fda.gov/Food/FoodIngredientsPackaging/ucm166145.htm.
- 128.Gies A, Heinzow B, Dieter HH, Heindel J. Bisphenol a workshop of the german federal environment agency - March 30–31, 2009 work group report: public health issues of bisphenol a. Int J Hyg Environ Health. 2009;212:693–696. doi: 10.1016/j.ijheh.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 129.Nagel SC, vom Saal FS, Thayer KA, Dhar MG, Boechler M, Welshons WV. Relative binding affinity-serum modified access (RBA-SMA) assay predicts the relative in vivo bioactivity of the xenoestrogens bisphenol A and octylphenol. Environmental Health Perspectives. 1997;105:70–76. doi: 10.1289/ehp.9710570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Talsness C, Fialkowski O, Gericke C, Merker H-J, Chahoud I. The effects of low and high doses of bisphenol A on the reproductive system of female and male rat offspring. Congenital Anomalies. 2000;40:S94–S107. [Google Scholar]
- 131.Ishido M, Masuo Y, Kunimoto M, Oka S, Morita M. Bisphenol A causes hyperactivity in the rat concomitantly with impairment of tyrosine hydroxylase immunoreactivity. J Neurosci Res. 2004;76:423–33. doi: 10.1002/jnr.20050. [DOI] [PubMed] [Google Scholar]
- 132.Kabuto H, Amakawa M, Shishibori T. Exposure to bisphenol A during embryonic/fetal life and infancy increases oxidative injury and causes underdevelopment of the brain and testis in mice. Life Sci. 2004;74:2931–2940. doi: 10.1016/j.lfs.2003.07.060. [DOI] [PubMed] [Google Scholar]
- 133.Ryan BC, Vandenbergh JG. Developmental exposure to environmental estrogens alters anxiety and spatial memory in female mice. Horm Behav. 2006;50:85–93. doi: 10.1016/j.yhbeh.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 134.Palanza P, Gioiosa L, vom Saal FS, Parmigiani S. Effects of developmental exposure to bisphenol A on brain and behavior in mice. Environ Res. 2008;108:150–157. doi: 10.1016/j.envres.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 135.Braun JM, Hauser R. Bisphenol A and children's health. Curr Opin Pediatr. 2011;23:233–239. doi: 10.1097/MOP.0b013e3283445675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Braun JM, Yolton K, Dietrich KN, Hornung R, Ye X, Calafat AM, Lanphear BP. Prenatal bisphenol A exposure and early childhood behavior. Environ Health Perspect. 2009;117:1945–1952. doi: 10.1289/ehp.0900979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ezure T, Amano S. Increased subcutaneous adipose tissue impairs dermal function in diet-induced obese mice. Exp Dermatol. 2010;19:878–882. doi: 10.1111/j.1600-0625.2009.00970.x. [DOI] [PubMed] [Google Scholar]
- 138.Howdeshell KL, Peterman PH, Judy BM, Taylor JA, Orazio CE, Ruhlen RL, vom Saal FS, Welshons WV. Bisphenol A is released from used polycarbonate animal cages into water at room temperature. Environ. Health Perspect. 2003;111:1180–1187. doi: 10.1289/ehp.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Hagan A, Voigt RC, Thomas S, Thomas BF, Hassold TJ. Bisphenol A causes meiotic aneuploidy in the female mouse. Current Biology. 2003;13:546–553. doi: 10.1016/s0960-9822(03)00189-1. [DOI] [PubMed] [Google Scholar]
- 140.Koehler KE, Voigt RC, Thomas S, Lamb B, Urban C, Hassold TJ, Hunt PA. When disaster strikes: Rethinking caging materials. Lab Animal. 2003;32:32–35. doi: 10.1038/laban0403-24. [DOI] [PubMed] [Google Scholar]
- 141.Ryan KK, Haller AM, Sorrell JE, Woods SC, Jandacek RJ, Seeley RJ. Perinatal exposure to bisphenol-a and the development of metabolic syndrome in CD-1 mice. Endocrinology. 2010;151:2603–2612. doi: 10.1210/en.2009-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thigpen JE, Setchell KD, Saunders HE, Haseman JK, Grant MG, Forsythe DB. Selecting the appropriate rodent diet for endocrine disruptor research and testing studies. ILAR J. 2004;45:401–416. doi: 10.1093/ilar.45.4.401. [DOI] [PubMed] [Google Scholar]
- 143.Heindel JJ, vom Saal FS. Meeting report: batch-to-batch variability in estrogenic activity in commercial animal diets--importance and approaches for laboratory animal research. Environ Health Perspect. 2008;116:389–393. doi: 10.1289/ehp.10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cederroth CR, Vinciguerra M, Kühne F, Madani R, Klein M, James RW, Doerge DR, Visser TJ, Foti M, Rohner-Jeanrenaud F, Vassalli J-D, Nef S. A phytoestrogen-rich diet increases energy expenditure and decreases adiposity in mice. Environ. Health Perspect. 2007;115:1467–1473. doi: 10.1289/ehp.10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ruhlen RL, Taylor JA, Mao J, Kirkpatrick J, Welshons WV, vomSaal FS. Choice of animal feed can alter fetal steroid levels and mask developmental effects of endocrine disrupting chemicals. Journal of Developmental Origins of Health and Disease. 2011;2:36–48. [Google Scholar]
- 146.Thigpen JE, Setchell KD, Padilla-Banks E, Haseman JK, Saunders HE, Caviness GF, Kissling GE, Grant MG, Forsythe DB. Variations in phytoestrogen content between different mill dates of the same diet produces significant differences in the time of vaginal opening in CD-1 mice and F344 rats but not in CD Sprague-Dawley rats. Environ Health Perspect. 2007;115:1717–1726. doi: 10.1289/ehp.10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Budge H, Gnanalingham MG, Gardner DS, Mostyn A, Stephenson T, Symonds ME. Maternal nutritional programming of fetal adipose tissue development: long-term consequences for later obesity. Birth Defects Res C Embryo Today. 2005;75:193–199. doi: 10.1002/bdrc.20044. [DOI] [PubMed] [Google Scholar]
- 148.Xi W, Lee CK, Yeung WS, Giesy JP, Wong MH, Zhang X, Hecker M, Wong CK. Effect of perinatal and postnatal bisphenol A exposure to the regulatory circuits at the hypothalamus-pituitary-gonadal axis of CD-1 mice. Reprod Toxicol. 2011;31:410–417. doi: 10.1016/j.reprotox.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 149.Somm E, Schwitzgebel VM, Vauthay DM, Camm EJ, Chen CY, Giacobino JP, Sizonenko SV, Aubert ML, Huppi PS. Prenatal nicotine exposure alters early pancreatic islet and adipose tissue development with consequences on the control of body weight and glucose metabolism later in life. Endocrinology. 2008;149:6289–6299. doi: 10.1210/en.2008-0361. [DOI] [PubMed] [Google Scholar]
- 150.Coe BL, Kirkpatrick JR, Taylor JA, vom Saal FS. A new 'crowded uterine horn' mouse model for examining the relationship between foetal growth and adult obesity. Basic & clinical pharmacology & toxicology. 2008;102:162–167. doi: 10.1111/j.1742-7843.2007.00195.x. [DOI] [PubMed] [Google Scholar]