

Summary
Polyploidization can precede the development of aneuploidy in cancer. Polyploidization in megakaryocytes (Mk), in contrast, is a highly controlled developmental process critical for efficient platelet production via unknown mechanisms. Using primary cells, we demonstrate that the guanine exchange factors GEF-H1 and ECT2, which are often overexpressed in cancer and are essential for RhoA activation during cytokinesis, must be downregulated for Mk polyploidization. The first (2N-to-4N) endomitotic cycle requires GEF-H1 downregulation while subsequent cycles (>4N) require ECT2 downregulation. Exogenous expression of both GEF-H1 and ECT2 prevents endomitosis, resulting in proliferation of 2N Mk. Furthermore, we have shown that the mechanism by which polyploidization is prevented in Mk lacking Mkl1, which is mutated in megakaryocytic leukemia, is via elevated GEF-H1 expression; shRNA-mediated GEF-H1 knockdown alone rescues this ploidy defect. These mechanistic insights enhance our understanding of normal versus malignant megakaryocytopoiesis, as well as aberrant mitosis in aneuploid cancers.
Introduction
Polyploidy resulting from cellular stress precedes aneuploidy, which can lead to tumors associated with transformation to malignancy and a poor prognosis (Nguyen and Ravid, 2006; Nguyen and Ravid, 2010). In contrast, polyploidy of megakaryocytes (Mk), the hematopoietic cells that give rise to platelets, is a tightly controlled normal differentiation process. Diploid megakaryoblasts differentiated from hematopoietic stem cells undergo a progressive increase in ploidy (up to 128N) due to repeated DNA replication without cell division, a process termed endomitosis, resulting in large multilobulated, polyploid nuclei (Battinelli et al., 2007). Polyploidization is essential for efficient platelet production. In megakaryoblastic leukemia, low ploidy megakaryoblasts predominate (Raslova et al., 2007).
Studies using time-lapse microscopy to observe endomitotic Mk suggest that the initial endomitotic cleavage event in which cells progress from 2N to 4N occurs due to failure at late cytokinesis with normal cleavage furrow ingression followed by furrow regression (Geddis et al., 2007; Papadantonakis et al., 2008; Lordier et al., 2008; Leysi-Derilou et al., 2010). These endomitotic Mk form an apparently intact midzone with normal localization of essential components including Survivin, Aurora B, INCENP, PRC1 (protein regulating cytokinesis 1), MKLP1 and 2 (mitotic kinesin-like protein), MgcRacGAP and microtubules (Geddis and Kaushansky, 2006; Lordier et al., 2008;). During cytokinesis, RhoA signaling is required to establish the actomyosin ring at the cleavage furrow, generating the contraction force for completion of cytokinesis (Bement et al., 2005; Narumiya and Yasuda, 2006; Melendez J et al., 2011). Activated RhoA and its effectors (ROCK, Citron, LIM and mDia) are localized to the cleavage furrow (Madaule et al., 1998; Yasui et al., 1998; Kosako et al., 2002; Tolliday et al., 2002). Dominant-negative Citron and ROCK inhibitors prevent normal cytokinesis (Madaule, 1998; Kosako et al., 2000). In contrast to normal cytokinesis, the contractile ring of Mk undergoing endomitosis lacks non-muscle myosin IIA and contains decreased levels of RhoA and actin at the 2N to 4N transition; in higher ploidy cells, RhoA is not detectable at the cleavage furrow during anaphase (Geddis and Kaushansky, 2006; Lordier et al., 2008).
Rho family small GTPases (e.g. RhoA, Rac1, and Cdc42) are molecular switches that regulate many cellular processes including actin cytoskeleton reorganization, microtubule dynamics, cell cycle progression and cytokinesis (Etienne-Manneville and Hall, 2002). Rho GTPase switching from the inactive GDP-bound state to the active GTP-bound state is facilitated by a group of proteins called Dbl family guanine nucleotide-exchange factors (GEFs), which have a tandem Dbl homology (DH) - Pleckstrin homology (PH) domain, in which the DH domain contains GDP/GTP exchange activity (Rossman et al., 2005). GEFs are involved in RhoA localization and activation during different stages of cytokinesis. Upon breakdown of the nuclear envelope during mitosis, the GEF ECT2 (Epithelial Cell Transforming Sequence 2) is dispersed from the nucleus to the cytoplasm, and recruited to the central spindle by the central spindlin complex (formed by MKlp1 and MgcRacGAP) during late anaphase for establishment of the cleavage furrow (Petronczki et al., 2007; Yuce et al., 2005). ECT2, required for cell cycle progression, is an oncogene that resides on chromosome 3q26, a region frequently targeted for chromosomal alterations in human tumors and overexpressed in many primary human tumors (Fields and Justilien, 2010; Iyoda et al., 2010). RNAi knock-down of ECT2 results in mitotic failure and binucleate cells due to the lack of cleavage furrow ingression (Birkenfeld et al., 2007). There are multiple studies suggested that ECT2 is important for RhoA localization and activation during cleavage furrow formation and ingression (Yuce et al., 2005; Nishimura and Yonemura, 2005; Yoshizaki et al., 2004), whereas some evidence suggested ECT2 may not be directly responsible for RhoA activation during furrow ingression. Without ECT2, RhoA still gets activated, but is mislocalized from the cleavage furrow (Chalamalasetty et al., 2006; Birkenfeld et al., 2007). An N-terminal fragment of ECT2 lacking the catalytic DH/PH domain can rescue the furrow ingression defect in ECT2 RNAi treated cells (Chalamalasetty et al., 2006). Thus, ECT2 recruits RhoA to the cleavage furrow, but may not directly catalyze its activation.
The microtubule associated protein GEF-H1, plays a critical role in cytokinesis by activating RhoA at the cleavage furrow (Birkenfeld et al., 2007). Association with polymerized microtubules inactivates GEF-H1 (Krendel et al., 2002). A truncated form of GEF-H1 lacking its microtubule-binding ability was discovered in the monocytic leukemia cell line U937 and is able to induce tumor formation in nude mice (Brecht et al., 2005). Also GEF-H1 is transcriptionally activated by mutant p53, and its expression is strongly induced in mutant p53 cell lines, leading to accelerated tumor cell proliferation (Mizuarai et al., 2006). During mitosis, GEF-H1 is first associated with the microtubule spindle, and later with the midbody (Birkenfeld et al., 2007). The same authors also show that GEF-H1 binding to microtubules is facilitated by phosphorylation at Ser885 and Ser959 by the mitotic kinases Aurora A/B and Cdk1, respectively. At the onset of cytokinesis, GEF-H1 is dephosphorylated and released from microtubules so that it can activate RhoA. In contrast to ECT2 RNAi, in which cytokinesis is blocked and there is no cleavage furrow formation, GEF-H1 knock down causes a defect at a later stage of cytokinesis - the cleavage furrow is induced normally, but the furrow fails to close completely resulting in binucleate cells (Birkenfeld et al., 2007).
Because of the importance of mitotic GEFs in RhoA localization and activation during cytokinesis, we hypothesized that decreased RhoA activation in Mk endomitosis may be caused by a decrease in mitotic GEFs. In the present work, we show that both GEF-H1 and ECT2 are downregulated at the mRNA and protein levels during Mk polyploidization. We show that MKL1-regulated GEF-H1 downregulation is required for endomitosis of 2N cells to become 4N, whereas ECT2 downregulation is required for polyploidization beyond the 4N stage. Together, these decreases in ECT2 and GEF-H1 are responsible for the decreased RhoA signaling that occurs in endomitosis, and downregulation of GEF-H1 represents one of the initiating events of endomitosis.
Results
Establishment of in vitro models to study Mk endomitosis
We used primary murine Mk to study the involvement of Dbl family GEFs in Mk endomitosis. The first endomitotic event from 2N to 4N Mk occurs due to failure of cytokinesis at a late stage of cytokinesis with cleavage furrow regression. In contrast, there are different reports for when subsequent endomitotic events driving 4N to higher ploidy Mk interrupt the normal cytokinesis machinery. Some report significant cleavage furrow formation for higher ploidy Mk (Lordier et al., 2008; Leysi-Derilou et al., 2010), while others report little apparent cleavage furrow formation in high ploidy endomitosis (Geddis et al., 2007; Papadantonakis et al., 2008). Therefore, we first examined how primary mouse Mk undergo endomitosis at different stages using Mk from GFP-tagged Histone 2B (H2B-GFP) transgenic mice visualized by timelapse microscopy for changes in both chromatin and cell morphology during endomitosis. Mk progenitors (MkP, Kit+CD41+) sorted from BM were cultured in Tpo-only differentiation medium (DM) to promote Mk polyploidization. MkP endomitotic events were recorded by time-lapse microscopy, and 2N, 4N and higher ploidy MKs were distinguished based on division history as well as their cell size after mitosis or endomitosis, a method established previously (Levine et al., 1982; Leysi-Derilou et al., 2010; Tomer, 2004; Tomer et al., 1988). Of the 2N cells (24 events) whose DNA division events we observed, 80% of 2N DNA divisions resulted in endomitosis with clear cleavage furrow formation followed by regression and formation of 4N cells (Fig. 1a, Movie S1), 7% of 2N cells underwent endomitosis without significant cleavage furrow ingression, and 13% of 2N cells underwent normal mitosis with complete cytokinesis. In contrast, for division of cells that started out with ≥4N DNA (37 events), 86% of divisions were endomitotic without noticeable cleavage furrow ingression (Fig. 1b, Movie S2), and 14% were endomitotic with significant cleavage furrow ingression followed by regression. Our data confirm that there are two distinct phases of endomitosis in mouse primary Mk: for the 2N to 4N transition, the cleavage furrow forms but fails to complete cytokinesis; and for ≥4N endomitosis, there is usually, but not always, little to no cleavage furrow ingression.
Figure 1.
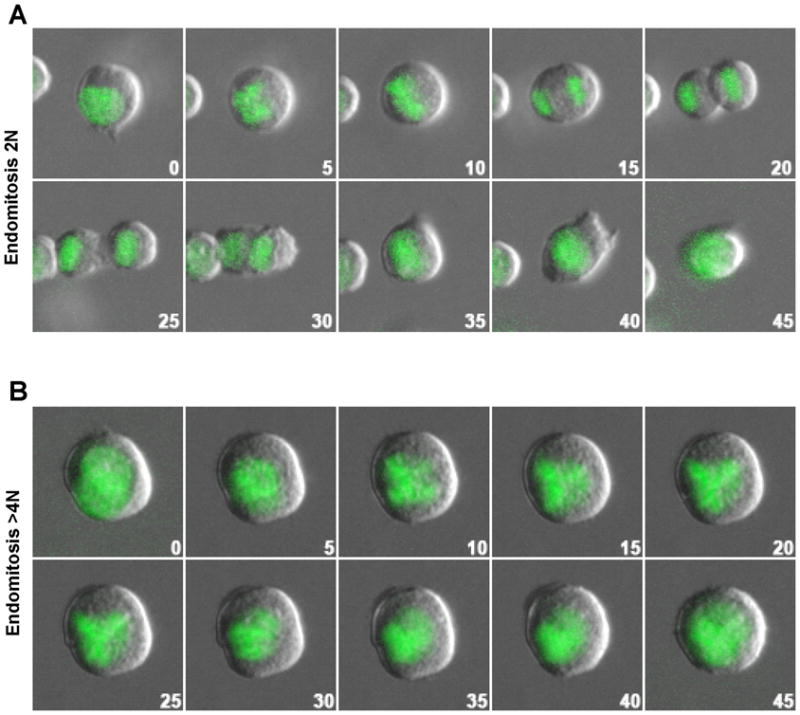
Endomitosis of MkP induced by Tpo. (A) 2N to 4N Mk endomitosis shows cleavage furrow ingression with subsequent regression, resulting in one 4N cell. (B) 4N to 8N Mk endomitosis showing no apparent cleavage furrow. The figures show overlays of DIC (gray) with green fluorescent H2B-GFP (green) images taken every 5 minutes. The time of each image relative to the first is indicated in each frame.
RhoA is not activated at the cleavage furrow during endomitosis
RhoA activity is important for cleavage furrow ingression during cytokinesis. To understand the mechanisms underlying the failure to complete cytokinesis in 2N to 4N Mk endomitosis, we assayed localization of active RhoA during endomitosis using a FRET-based RhoA biosensor. The biphenotypic Mk-erythroid progenitor population as defined by Akashi et al. and refined (Lin-Sca-Kit+CD41− CD150+CD105−) and renamed ‘PreMegE’ by Pronk et al. (Akashi et al., 2000; Pronk et al., 2007), was sorted from BM. To assess RhoA activation directly, a widely-used single molecule RhoA activation FRET probe (Birkenfeld et al., 2007; Pertz et al., 2006), pBabe-Puro-RhoA Biosensor, was transduced into primary mouse PreMegE that were then cultured in growth medium (Fig. 2a) or differentiation medium (Fig. 2b), and the FRET efficiencies (FRET/CFP ratio) were determined over time in cells undergoing anaphase using a Leica SP5 scanning microscope equipped with temperature and CO2 control. Total RhoA localization was assessed by analysis of the CFP signal from the RhoA biosensor, which reflects the endogenous RhoA localization during mitosis as reported previously (Birkenfeld et al., 2007; Pertz et al., 2006). PreMegE cultured in growth medium (GM) with SCF, TPO, IL-3 and Flt-3 serve as normal mitotic controls (Fig 2a). Under these conditions, 100% of 2N cells undergoing mitosis complete cytokinesis (data not shown). From early to late cytokinesis the RhoA biosensor is enriched at the equatorial cortex (Fig. 2ai,iii) as previously reported (Yuce et al., 2005). When MkP are cultured in DM, 80% of DNA divisions result in endomitosis, and during endomitotic anaphase and cytokinesis, RhoA is similarly present throughout the cytoplasm including at the equatorial region at early (Fig. 2bi,iii), which is consistent with previous reports that RhoA localizes correctly during 2N endomitosis by immunofluorescence of fixed Mk (Geddis and Kaushansky, 2006; Lordier et al., 2008). To determine whether RhoA protein levels change during Mk differentiation, RhoA protein from different stage of Mk differentiation as well as in platelets was analyzed by Western-Blotting. As shown in Figure S1, total RhoA protein was only slightly decreased (20%) during Mk differentiation, which is in agreement with the almost constant level of RhoA during Mk differentiation of CD34+ human blood cells (Chang et al. 2007).
Figure 2.

Active RhoA is absent from the cleavage furrow during the first endomitotic cleavage. Mouse primary PreMegE cells were transduced with the RhoA biosensor virus for 48 h before switching to fresh growth medium (A) or differentiation (B) medium. After 8 hours, the RhoA activation pattern throughout mitosis (A) or endomitosis (B) was assessed. The CFP channel indicates biosensor (and RhoA) localization (i, iii, v). The RhoA activation pattern was assessed by the RhoA biosensor’s FRET/CFP ratio (FRET). The red color in FRET images indicates high RhoA activation (ii, iv, vi). All images were processed identically. Elapsed time (minutes) with starting time set to 0 (8h post-Tpo administration) is indicated in each picture. Images are representative of at least 4 similar images for each condition.
In PreMegE undergoing cytokinesis, there is a high FRET signal at the cleavage furrow (Fig. 2aii,iv) indicating high RhoA activity in this region, in agreement with published data on RhoA activation during cytokinesis in NIH3T3 cells (Yoshizaki et al., 2003). However, in Mk undergoing endomitosis, there was no significant FRET signal in this region (Fig. 2bii,iv), indicating lack of RhoA activation at the cleavage furrow. Our data provide direct evidence that during 2N to 4N endomitosis, there is a failure of RhoA activation at the cleavage furrow, even though RhoA is localized correctly to this region.
GEF-H1 and ECT2 levels decrease during Mk polyploidization
To investigate the mechanisms underlying the failure of RhoA activation in Mk during endomitosis of 2N cells, and the absence of RhoA protein at the cleavage furrow region during endomitosis of higher ploidy cells, we assessed whether the mitosis-associated GEFs, ECT2 and GEF-H1, are differentially expressed during Mk differentiation. WT PreMegE and MkP were cultured in DM, and RNA expression assessed by quantitative RT-PCR over time. As shown in Figure 3(a, b), MkPs have lower levels of both GEF-H1 and ECT2 mRNA compared with PreMegE. Tpo treatment of PreMegE decreases GEF-H1 and ECT2 mRNA levels by 67% and 78%, respectively. GEF-H1 mRNA levels subsequently increased after prolonged Tpo exposure when high ploidy Mk were forming, whereas ECT2 mRNA levels remained low. GEF-H1 and ECT2 protein levels also decreased (Fig. 3c), but with slightly different kinetics than the mRNA: GEF-H1 protein reached a nadir after 1 day of culture of PreMegE in Tpo medium, and then began to increase; in contrast, ECT2 protein levels were significantly decreased after two days, and then remained low.
Figure 3.

GEF-H1 and ECT2 are down-regulated during Mk differentiation. Data shown for freshly sorted (d0) PreMegE and MkP cells from WT mice, as well as PreMegE cultured in DM for the time indicated. (A) Relative levels of GEF-H1 mRNA are reduced during Mk differentiation. ***, P<0.005, versus value of PreMegE day 0. (B) Relative levels of ECT2 mRNA were also decreased. ***, P<0.005, versus value of PreMegE day 0. (C) The protein levels of GEF-H1 and ECT2 are reduced with different kinetics as shown by Western-blotting. Anti-α-tubulin was used as the loading control. Relative protein level of each sample after normalization to tubulin and setting PreMegE level as 1 are indicated above each band.
GEF-H1 and ECT2 levels during endomitosis
ECT2 expression is induced by growth factors that promote cell cycle entry, and cells in G0 have little ECT2 (Saito et al., 2003). However, it is unlikely that decreased GEF-H1 and ECT2 levels during Tpo-induced Mk differentiation are caused by cell cycle exit. First, Tpo induces Mk progenitor cell cycling (Drayer, 2006). Second, the level of Anillin protein, an indicator of mitotic activity, which peaks in mitosis and decreases dramatically upon mitotic exit (Zhao and Fang, 2005), increased dramatically upon Mk Tpo treatment (Fig. S1), indicating increased cell cycling after Tpo treatment. To directly confirm that Mk undergoing the cell cycle have decreased GEF-H1 and ECT2 levels, we examined GEF-H1 and ECT2 protein in endomitotic Mk by immunofluorescence. In mitotic control cells (PreMegEs in GM for one day), the microtubule spindle is normal, and GEF-H1 co-localizes with the microtubule spindle as reported previously (Birkenfeld et al., 2007) (Fig. 4ai–iii). In contrast, in endomitotic MkP cultured in DM for one day, the GEF-H1 protein level is low with little fluorescence signal detected above background (Fig. 4aiv–vi). After 2 days in DM, in polyploid (>4N) cells undergoing anaphase, the spindle appeared normal, and GEF-H1 protein was again easily detected with localization to the microtubule spindle (Fig. 4avii–ix). The relative increase in GEF-H1 was consistent with the changes in protein levels detected by Western blotting (Fig. 3c). In PreMegE mitosis controls, ECT2 localized to the central spindle (Fig. 4bi–iii) as published previously (Tatsumoto et al., 1999; Yuce et al., 2005). During 2N to 4N endomitosis of MkP, ECT2 was easily detected and localized to the central spindle (Fig. 4biv–vi). The immunofluorescence signal between these two samples did not show any apparent differences (Fig. 4bi, 4biv), although the ECT2 protein level in Mkp is about 30% lower than that of PreMegE by Western-Blot (Fig. 3c). In high ploidy cells, ECT2 protein levels were very low (Fig. 4b, vii–ix), which is in agreement with the Western Blot data (Fig. 3c, S1). These data not only confirm that in endomitotic Mk cells, GEF-H1 and ECT2 levels decreases (albeit with different kinetics), but also suggest that during Mk differentiation, loss of GEF-H1 predominates in low ploidy Mk, whereas loss of ECT2 occurs later and predominates in higher ploidy endomitosis.
Figure 4.

GEF-H1 and ECT2 are reduced in endomitotic Mk. PreMegE were grown in GM and MkP in DM. (A) Cells were stained with anti-GEF-H1 (red), anti-α-tubulin (green) and DAPI. Examples of normal mitosis (i–iii, PreMegE cultured in growth medium), 2N to 4N endomitosis (iv–vi), and ≥ 4N endomitosis (vii–ix) are shown. GEF-H1 protein level is reduced at the 2N to 4N stage of endomitosis and increases at later stages (2 days) of endomitosis. (B) ECT2 protein, stained with anti-ECT2 antibody (red), is clearly detected in PreMegE undergoing mitosis (i – iii), and at the 2N to 4N stage of endomitosis (iv–vi), but ECT2 levels are reduced at later stages (2 days) of endomitosis (vii–ix).
Ectopic expression of GEF-H1 or ECT2 causes low ploidy Mk in vitro
To test whether the decreases in GEF-H1 and/or ECT2 cause the sequential stages of endomitosis, we expressed exogenous GEF-H1 or ECT2 by retroviral transduction of PreMegE cells, which were then induced to differentiate with DM in which 80% of mitotic events are endomitotic. After 3 days in DM, the ploidy of GFP positive (control vector vs. ECT2 overexpression vs. GEF-H1 overexpression) CD41+ Mk was assessed by flow cytometry. Enforced expression of ECT2 or GEF-H1 did not affect the percentage of cells that were CD41+ (Fig. S2), suggesting that exogenous GEF-H1 or ECT2 does not affect PreMegE differentiation down the Mk lineage. Cells transduced with empty viral vector had a normal Mk ploidy distribution. Although enforced expression of either GEF-H1 or ECT2 resulted in significantly decreased mean ploidy (Fig. 5a,b), the ploidy profiles were significantly different. GEF-H 1 caused an increase in 2N cells, although Mk that became 4N were still able to become highly polyploid (Fig. 5a, middle). In contrast, ECT2 expression led to accumulation at the 2N, 4N and 8N stage with far fewer cells having >8N (Figure 5b, right). We believe that the 8N peak in cells overexpressing ECT2 represents the G2/M peak of cycling 4N cells. Expression of GEF-H1 and ECT2 was confirmed by Western blot (figure 5d). To evaluate whether enforced expression of GEF-H1 plus ECT2 completely prevents endomitosis, we transduced PreMegE cells with both GEF-H1 (with IRES-RFP) and ECT2 (with IRES-GFP) retroviruses. After 3 days in Tpo, the ploidy of CD41+ RFP+ and GFP+ (double positive) cells was determined. Compared with cells only expressing GEF-H1 or ECT2, double positive cells have more 2N cells, and far fewer >4N cells (Fig. 5c, far right, upper panel). These data indicate that exogenous expression of GEF-H1 prevents the progression of cells from 2N to 4N in the first endomitotic event, whereas exogenous expression of ECT2 prevents the subsequent >4N endomitotic events to form highly polyploid Mk. Since RhoA acts downstream of ECT2 and GEF-H1, we next tested whether the dominant-negative RhoA mutant RhoA N19 can block the effect on Mk ploidy caused by enforced expression of ECT2 or GEF-H1 in Mk. As shown in Fig. 5c (lower panel), the ability to make high ploidy Mk was restored in cells expressing both GEF-H1 and RhoA N19 or ECT2 and RhoA N19 compared with only expressing GEF-H1 or ECT2 (Fig. 5c, upper panel).
Figure 5.

Differential effects of overexpressing GEF-H1 or ECT2 on polyploidization of Mk. (A–C) PreMegE cells in growth medium were transduced with retroviral vectors (control and expressing individual GEFs) and cultured for two days, then transferred to differentiation medium for 3 days before the ploidy of each sample was assessed. (A) Shown is the effect of overexpressing control (GFP) virus, GEF-H1 or ECT2 encoding virus. Ploidy of GFP+ cells in each condition is shown. The percentages of Mk in 2N, 4N, and ≥ 8N ploidy are indicated. (B) The average ploidy of Mk expressing GFP only (control), GEF-H1 or ECT2 is compared. ***, P<0.005, versus control. (C) In this experiment, cells were transduced with 2 retroviral vectors (one encoding RFP and the other GFP). Controls received vectors encoding only RFP and GFP. GEF-H1 (upper second panel) received GFP control plus GEF-H1-ires-RFP vectors, ECT2 (upper third panel) received ECT2-ires-GFP plus control RFP vectors, the upper fourth panel shows cells transduced with ECT2-ires-GFP plus GEF-H1-ires-RFP. Samples in lower panel were transduced with RhoAN19-ires-GFP together with RFP control (left), GEF-H1-ires-RFP (middle), or ECT2-ires-RFP (right). Ploidy is shown for GFP+RFP+ (double positive) cells. (D) Western blot of HEL cells transduced with the indicated virus validate expression vectors. (E) Overexpression of GEF-H1 also decreases polyploidization of Mk in vivo. CD45.1 BM cells were transduced with control (GFP only) or GEF-H1–ires-GFP virus, and transplanted into lethally irradiated CD45.2 mice. After 6 weeks, the ploidy of GFP positive Mk was analyzed. Representative ploidy profiles from GFP+ Mk expressing empty virus or GEF-H1 are shown. (F) Average ploidy of GFP positive Mk recovered 6 weeks post-transplant. *, P<0.05, versus the value of control.
To assess whether downregulation of GEF-H1 plays a role in polyploidization of Mk in vivo, GEF-H1 transduced CD45.1 BM cells were transplanted into lethally irradiated WT CD45.2 mice. After 6 weeks, the ploidy of GFP positive (control vector vs. GEF-H1 transduced) Mk was assessed. In agreement with in vitro differentiation, ectopic expression of GEF-H1 resulted in more 2N Mk (27.8%) compared with empty vector (13.1%), leading to a decrease in the average ploidy (Fig. 5e, f). However, as seen in vitro, those GEF-H1 transduced cells that did become 4N were able to undergo subsequent further polyploidization. These data demonstrate that enforced GEF-H1 decreases the likelihood of 2N to 4N endomitotic events in Mk in vivo.
Down-regulation of GEF-H1 during Mk differentiation is mediated by MKL1
Our lab previously demonstrated that knockout of the transcriptional cofactor Megakaryoblastic leukemia 1 gene (Mkl1) leads to reduced Mk ploidy in vivo (Cheng et al., 2009). To study whether the down-regulation of GEF-H1 and/or ECT2 during Mk differentiation is Mkl1 dependent, we analyzed GEF-H1 and ECT2 mRNA levels during Mk differentiation of PreMegE and MkP from Mkl1−/− and WT mice. GEF-H1 levels in both PreMegE and MkP are more than 15 times higher in Mkl1−/− mice than in WT mice (Fig. 6a). Unlike WT cells, there is little decrease of GEF-H1 mRNA in Mkl1−/− cells differentiated in vitro. In contrast, ECT2 mRNA levels are the same in Mkl1−/− vs. WT cells (Fig. 6b).
Figure 6.

Down-regulation of GEF-H1 is MKL1- dependent. (A) The relative mRNA level of GEF-H1 is significantly increased in Mkl1−/− Mk, and shows very little decrease with Mk differentiation. WT or Mkl1−/− PreMegE or MkP were cultured in DM as indicated. ***, P<0.005, versus the corresponding WT control. (B) Relative ECT2 mRNA levels are the same for WT and Mkl1−/− cells. In both, ECT2 mRNA decreases during Mk differentiation. P>0.1 for the values of Mkl1−/− versus the corresponding WT control, (C) PreMegE cultured in growth medium (mitotic controls, i–iii and vii–ix) and MkP cultured in Tpo-only medium, which induces endomitosis, were immunostained as indicated. Unlike WT MkP (iv–vi), Mkl1−/− MkP do not show a loss of GEF-H1 protein level in response to TPO induction (x–xii). (D and E) 293FT cells were transduced with empty vector (−); WT Mkl1 (WT); constitutively active (CA) Mkl1, which lacks the actin binding domain; or dominant negative (DN) Mkl1, which lacks the transcriptional activation domain, but can still heterodimerize with endogenous Mkl1. Overexpression of CA MKL1 significantly reduces endogenous GEF-H1 mRNA (D) and protein (E) levels compared to cells transduced with empty vector (−), wild type Mkl1 (WT) and dominant negative (DN) Mkl1. GAPDH was used as a loading control in (E). ***, P<0.005, and **, P<0.01, versus the value of empty vector (−). (F) Quantitative RT-PCR reveals much higher levels of GEF-H1 and ECT2 mRNA in the 6133 cell line compared to WT PreMegE with PreMegE value set to 1. Values normalized to 18S RNA. ***, P<0.005.
We next assessed GEF-H1 protein expression and localization in Mkl1−/− vs. WT PreMegE cultured for 1 day in DM. GEF-H1expression is much higher in Mkl1−/− Mk than WT, although it is localized to both the cell periphery and the microtubule spindle compared with predominantly spindle localization in WT cells during normal mitosis (Fig. 6c). No obvious differences between WT and Mkl1−/− cells were found in ECT2 detection or localization (data not shown). As expected with higher GEF-H1 levels in Mkl1−/− PreMegE and Mk, GTP-bound active RhoA levels in PreMegE with or without 24h exposure to Tpo are higher than wild type (Fig. S3). However the RhoA activity in Mkl1−/− cells is able to decrease after one day in Tpo similar to wild type cells (Fig. S3). To further confirm that GEF-H1 down-regulation is MKL1-dependent, we assessed the GEF-H1 mRNA levels in nonhematopoietic 293FT cells transfected with control vector, wild type, constitutive active, or dominant negative MKL1 constructs. Wild type MKL1 slightly decreased GEF-H1, constitutively active MKL1 further decreased GEF-H1 levels, and dominant negative MKL1 actually increased GEF-H1 levels (Fig. 6d,e). MKL1 is part of the t(1;22)(p13;q13) chromosomal translocation, associated with acute megakaryoblastic leukemia (Mercher, 2001; Ma 2001). To test whether GEF-H1 is dysregulated in acute megakaryoblastic leukemia containing the MKL1 translocation, we analyzed the levels of GEF-H1 and ECT2 in 6133 cells, a megakaryoblastic leukemia cell line derived from a transgenic mouse in which MKL1 was knocked in downstream of exon 1 of RBM15 to allow expression of the OTT-MKL1 fusion product (Mercher et al., 2009), compared with wild type PreMegE. As shown (Fig. 6f), 6133 cells express elevated GEF-H1 and ECT2 compared to normal Mk progenitor PreMegE. In summary, GEF-H1 is down-regulated by MKL1 during Mk differentiation, whereas the ECT2 decrease is MKL1-independent, and acute megakaryoblastic leukemia cells have increased GEF-H1 expression.
Knock-down of GEF-H1 rescues the ploidy defect in Mkl1−/− Mk
In order to test whether downregulation of GEF-H1 alone can restore polyploidization in Mkl1−/− Mk, we decreased GEF-H1 by shRNA in WT and Mkl1−/− PreMegE, that were then differentiated in DM. As expected, GEF-H1 shRNA had no effect on WT Mk ploidy compared with a control shRNA targeting luciferase (Fig. 7a). Mk differentiated from Mkl1−/− cells transduced with control shRNA had lower ploidy than WT cells (Fig. 7a). However, transduction of Mkl1−/− Mk with GEF-H1 shRNA led to much higher ploidy (Fig. 7a, far right). The average ploidy for each condition (Fig. 7b) demonstrates that shRNA against GEF-H1 restores the ploidy level of Mkl1−/− cells to that of WT cells. The GEF-H1 shRNA construct was validated in mouse NIH3T3 cells, in which there was approximately 70% knock-down (Fig. 7c).
Figure 7.

Knockdown of GEF-H1 restores polyploidy in Mkl1−/− Mk in vitro. (A) WT or Mkl1−/− PreMegE were transduced with retrovirus encoding either shRNA targeting luciferase or GEF-H1, as indicated. After Tpo induced differentiation, ploidy was assessed. A representative ploidy plot for each condition is shown. The percentages of Mk in 2N, 4N, 8N and ≥16N ploidy are indicated. (B) Average ploidy of these samples. *** P <0.005. (C) Validation of shRNA mediated knockdown of GEF-H1 protein in NIH3T3 cells transduced with the indicated constructs. GFP positive shRNA expressing cells were sorted, and analyzed by Western Blot. GAPDH was used as the loading control.
Discussion
Our data confirm that there are two distinct stages of Mk endomitosis. For the 2N to 4N transition, cleavage furrow ingression occurs followed by furrow regression, whereas in endomitosis associated with ≥ 4N cells, there is little cleavage furrow ingression. Our data provide evidence that RhoA activation is absent from the cleavage furrow during the first of endomitotic cleavage, and that two mitotic GEFs, GEF-H1 and ECT2, are down-regulated during Mk differentiation. Overexpression of GEF-H1 in differentiating Mk leads to more 2N MKs, but for those cells that do fail to complete cytokinesis, high levels of polyploidization are still achievable. In contrast, exogenous ECT2 inhibits 4N polyploid cells undergoing endomitosis but does not affect proliferation of 2N and 4N cells. Enforced overexpression of both GEF-H1 and ECT2 completely prevents polyploidization of primary Mk.
We conclude that decrease of GEF-H1 during megakaryocytopoiesis is mediated by the transcriptional cofactor MKL1, based on the markedly elevated GEF-H1 expression in Mkl1−/− Mk, and the downregulation of GEF-H1 in 293FT cells overexpressing constitutively active MKL1. The block of polyploidization in Mkl1−/− Mk can be restored by shRNA-mediated downregulation of GEF-H1, further confirming that GEF-H1 is downstream of MKL1.
Using primary murine PreMegE and MkP differentiated in vitro down the Mk lineage with TPO, we observed late cytokinesis defects in the first round of endomitosis whereas for later rounds of endomitosis (>=4N), the majority of endomitosis occurred without apparent cleavage furrow ingression. These data differ from reports on endomitosis of human CD34+ derived Mk, especially cord blood CD34+, in which only about 30% of 2N cells undergo endomitosis, and the majority of high ploidy cells showed significant furrow ingression, and some high ploidy cells were able to complete cytokinesis and divide to two high ploidy cells (Lordier et al., 2008; Leysi-Derilou et al., 2010). The low endomitosis ratio at the 2N stage of the human CD34+ derived Mk may be due to contamination with other cell types, and the majority of immunofluorescence images of those cells may represent normal mitotic divisions. Alternatively, there may be intrinsic differences between murine and human megakaryopoiesis, especially that of cord blood, in which Mk ploidy is lower than in adults indicating fewer endomitotic cleavage events. While the absence of cleavage furrow formation in high ploidy endomitosis has been reported previously as being associated with a lack of accumulation of RhoA (Lordier et al., 2008, Geddis et al., 2006), the mechanism of cytokinesis failure in the early rounds of endomitosis was previously unknown since RhoA was correctly localized to the cleavage furrow at the 2N stage. Here we show that this correctly localized RhoA is not activated in cells undergoing endomitosis with a FRET-based RhoA activity biosensor in live Mk.
We show that the mRNA and protein levels of two RhoA GEFs involved in cytokinesis are decreased with different kinetics during Mk differentiation: the reduction of GEF-H1 occurs at the 2N to 4N stage, while the significant reduction of ECT2 protein occurs at 4N and higher stages. Forced expressing GEF-H1 results in a higher percentage of Mk that are 2N, although high ploidy Mk can still form, whereas forced expression of ECT2 results in accumulation of 2N to 8N Mk, and a lack of higher ploidy Mk. Taken together, these data suggest that loss of GEF-H1 and ECT2 play distinct roles in Mk polyploidization: in low ploidy cells, with correct ECT2 localization at the central spindle, RhoA is recruited to the equatorial region, but due to the lack of GEF-H1 protein, most of this RhoA is inactive. In high ploidy cells, despite restored levels of GEF-H1, significantly decreased levels of ECT2 lead to failure of RhoA recruitment to the equatorial region. GEF-H1 levels decrease again in platelets (Fig. S1). As we and others reported previously (Halene et al., 2010; Patel et al., 2005), the extensive microtubule network and actin cytoskeleton are essential for Mk maturation, platelet formation and function. It is thus possible that the reappearance of GEF-H1, a unique microtubule-associated RhoA GEF, in higher ploidy Mk may be essential for subsequent Mk maturation but not for platelet function.
Our data from Mkl1−/− mice and overexpression of constitutively active (nuclear) MKL1 in 293FT cells suggest that MKL1 down-regulates GEF-H1 mRNA, but not ECT2. MKL1 is a transcriptional cofactor for SRF. MKL1 is up-regulated and required for Mk maturation and the formation of highly polyploid Mk (Cheng et al., 2009). There is a confirmed SRF binding site close to the GEF-H1 promoter region (Cooper et al., 2007). The mechanism by which MKL1 causes reduction of GEF-H1 during Mk differentiation requires further study.
Acute megakaryoblastic leukemia is characterized by a high percentage of immature, low ploidy megakaryoblasts. We showed that GEF-H1 and ECT2 levels are dramatically increased in a megakaryoblastic leukemia cell line derived from a mouse AMKL model in which the t(1;22) fusion protein is encoded from the endogenous murine RBM15 locus. How and whether the RBM15-MKL1 fusion protein regulates GEF-H1 and/or ECT2 expression remains to be shown. One possibility is that RBM15-MKL1 inhibits MKL1 activity, but this would not explain the increase in ECT2 levels. Future studies will need to explore the levels of GEF-H1 and ECT2 in AMKL patient samples, and whether targeting of either of these GEFs could be used to promote Mk polyploidy and maturation.
Furthermore, these mechanistic insights into the normal polyploidization process of Mk differentiation have important implications for aneuploid cells, and their malignant transformation. In normal Mk, although there are high levels of cyclin E (Eliades et al., 2010) and D (Muntean et al., 2007) activity, which are required to overcome the cell cycle checkpoints in high ploidy cells undergoing endomitosis, proliferation is controlled due to the lack of GEF-H1 and ECT2, which promotes polyploidy and stops expansion. In contrast, abnormal polyploid cells formed under stress conditions, with highly active GEF-H1 and ECT2, can divide, expand and transform to malignant aneuploid cells. Therefore GEF-H1 and ECT2 are potential therapeutic targets for cancer cell proliferation and expansion.
Experimental Procedures
Cell sorting
All mouse procedures were performed according to Yale University Animal Care and Use Committee–approved protocols and complied with federal laws. Murine bone marrow (BM) cells were obtained by crushing hips, femurs and tibias in cold PBS with 1% FBS. After lineage depletion with the BD IMag™ Mouse Hematopoietic Progenitor (Stem) Cell Enrichment kit (BD Biosciences, San Jose, CA), the remaining cells were stained with FITC anti-CD41, PE anti-lineage markers, PE-Cy5 anti-CD150, PE-Cy7 anti-CD105, Alexa 647 anti-Sca1, and APC-Hy7 anti-CD117 antibodies (eBioscience, San Diego, CA; Biolegend, San Diego, CA). The PreMegE population, defined as the Lin−Sca−Kit+CD41−CD105−CD150+, and MkP population, defined as Lin−Sca−Kit+CD41+, were separated using a FACSAria sorter. For sorting MkP from H2B-GFP transgenic mice (Jackson Laboratory, Bar Harbor, ME), eFluoro-450 labeled anti-CD41 antibody (eBioscience) was used.
Cell culture, Mk differentiation, and Western blotting
For expansion, sorted PreMegE were cultured at 1 x106 cells/mL in Growth Medium containing StemSpan (StemCell Technologies, Vancouver, Canada) supplemented with 30% BIT9500 (StemCell Technologies), 100 ng/mL murine SCF, 50 ng/mL murine Flt3-L, 10 ng/mL murine IL-3 (all purchased from PeproTech, Rocky Hill, NJ), 50 ng/mL murine Tpo, (from ConnStem, Cheshire, CT) and penicillin/streptomycin. For differentiation, PreMegE and MkP were cultured in Differentiation Medium similar to the Growth Medium but with only a single cytokine, 20 ng/mL murine Tpo. 6133 cells, a gift from Thomas Mercher (Villejuif, France), were grown as described (Mercher et al., 2009). For Western-blotting, about 1×105 cells per sample were washed with cold PBS, lysed in RIPA buffer for 15 minutes, and centrifuged at 18,000g for 15 minutes. Platelet lysate was prepared as described (Halene et al., 2009), and 20 μg of lysate per sample was analyzed by Western blot with: mouse anti-α-tubulin antibody (1:2000, Sigma-Aldrich), rabbit anti-GEF-H1 antibody (1:1000, Upstate), and rabbit anti-ECT2 antibody (1:500, Santa Cruz), and the corresponding secondary antibody. The density of protein bands was analyzed with ImageJ.
Time-lapse video microscopy
Primary murine MkP from H2B-GFP transgenic mice were sorted and cultured in 35mm glass bottom dishes (MatTeK, Ashland, MA) in differentiation medium. To maintain non-adherent cells within the field of view for time-lapse microscopy, 1% methylcellulose was added to increase the viscosity. Live cell imaging was performed using a Vivaview system (Olympus, Japan) with a 20x objective. DIC and green fluorescence images were taken every 5 minutes for two days with an Orca-R2 camera (Hamamatsu, Japan). Imaging data were analyzed, compiled and exported into Quick Time video with Metamorph for Vivaview software.
Immunofluorescent staining and confocal analysis
PreMegE and MkP cells cultured as indicated, were cytospun onto glass slides, fixed with 3.7% formaldehyde for 15 minutes, and permeabilized with 0.1% Triton-X-100 (Sigma-Aldrich) for 15 minutes. After blocking (PBS plus 3% BSA), slides were incubated with mouse anti-α-tubulin plus rabbit anti-GEF-H1 antibody (1:200, Upstate) or rabbit anti-ECT2 antibody (1:100, Santa Cruz). Bound antibody was detected using Alexa-488 labeled donkey anti-mouse secondary antibody and Alexa-555 donkey anti-rabbit secondary antibody, or Alexa-488 labeled anti-rabbit secondary antibody and Alexa-555 anti-mouse secondary antibody (Invitrogen). Fluorescent images were obtained on a Leica SP5 confocal microscope (Leica microsystem, Wetzlar, Germany).
Plasmids, Virus production and PreMegE transduction
Details regarding plasmids can be found in the online supplemental information. For retrovirus production, in a T-175 flask, 90% confluent HEK293 cells constitutively expressing Gag/Pol were transfected with 40 μg pBabe-Puro-RhoA Biosensor, MigR1 empty vector control, MigR1-GEF-H1, MigR1-ECT2 or Mieg3 RhoA N19 and 14 μg of VSVG packaging plasmid by Lipofectamine 2000 (Invitrogen) per manufacturer’s instructions. Supernatant collected at 48 and 72 hours was combined, spun at 3700g through Amicon filters (Millipore, Billerica, MA) to concentrate the virus to approximately 1×108 per ml, and aliquots stored at −80°C. Freshly sorted 5×104 PreMegE cells in growth medium were infected with indicated viruses in the presence of polybrene (8 ug/mL) by spinfection at 900g for 1 hour at 30°C. After two days in growth medium, some cells were switched to differentiation medium for 8 hours before FRET analysis or another 3 days for ploidy analysis.
FRET analysis of RhoA activation in endomitotic cells
RhoA activity during endomitosis was visualized using a widely-used RhoA biosensor FRET probe (Pertz et al., 2006). Two days after viral transduction, DAN of live PreMegE cells was stained with Hoechst red 33342 for 15 minutes, then the cells were washed and placed in differentiation medium or growth medium in a 35 mm glass bottom dish. After 8 hours, dividing cells in prometaphase expressing the RhoA biosensor were identified by YFP-fluorescence and chromosome morphology by UV excitation on a Leica SP5 confocal microscope equipped with temperature and CO2 control. CFP, FRET, YFP and DIC images were acquired using a 63× NA 1.4 objective. The fluorescence emission resulted from excitation with 458 (for CFP and FRET) and 514 (for YFP) nm laser lines from an argon laser through acousto-optical filter was detected with a prism spectrophotometer detection system, at a 490–500 nm spectral band width setting for CFP and a 520–590 nm spectral band width setting for FRET and YFP. Image analysis was performed using ImageJ essentially as described by Hodgson et al., 2010. Briefly, after shading correction and background subtraction, each CFP and the FRET image was multiplied with a binary threshold based mask to eliminate noise outside of the cell. Then, the FRET ratio image was generated by dividing the raw FRET image with the CFP image.
Quantitative RT-PCR
Total RNA from 5×104 cells was isolated using the RNAqueous-Micro Kit (Applied Biosystems, Foster City, CA), and treated with RNase-free DNase I. First strand cDNA was produced with Superscript II Reverse Transcriptase (Invitrogen) and random primers (Invitrogen) with 20 ng RNA from each sample. Gene expression levels were quantified on an iCycler iQ RT machine (Bio-Rad, Hercules, CA) with 2 μl of cDNA product from each sample using TaqMan probes (Applied Biosystems) as follows: murine GEF-H1: Mm00434757_m1; murine ECT2: Mm01289559_g1; and Eukaryotic18S rRNA: Hs99999901_s1. Relative gene levels were calculated from standard curves and normalized to 18s levels.
Flow cytometric analysis of DNA content and surface markers
In vitro differentiated Mk were stained with APC-conjugated anti-CD41 antibody, then fixed and permeabilized using BD cytofix/cytoperm on ice for 30 minutes. After incubation with RNAase at 37°C for 30 min, nuclear DNA was stained with 1 g/mL propidium iodide and analyzed using a FACS Calibur cytometer (BD Biosciences) and FlowJo software (TreeStar, Ashland, OR). In experiments using MigR1 RFP vectors, the nuclear DNA was stained with DAPI, and analyzed on a BD LSRII cytometer. To assay the Mk ploidy in transplanted mice, BM was collected, red cells lysed with PharmLyse (BD Biosciences), and ploidy analyzed as described above.
Murine bone marrow transplant
WT CD45.1 (B6.SJL-PtprcaPep3b/BoyJ mice) and CD45.2 (C57Bl6) mice were purchased from the Jackson Laboratory. After treatment with 150 mg/kg 5FU for 4 days, red cell depleted BM from 4–6 week old CD45.1 donor mice was transduced with empty vector or MigR1 GEF-H1 retrovirus by spinfection in Stemspan medium with 30% BIT9500, supplemented with 100 ng/ml SCF, 50 ng/ml TPO, 50 ng/ml Flt3 ligand, and 10 ng/ml IL-3. Lethally irradiated recipient C57Bl6/J mice were transplanted with 1 million cells 24 hours after transduction. Transplant efficiencies were monitored by detecting the CD45.1 ratio in the peripheral blood 4 wks post-transplantation. GFP positive Mk ploidy from recipient mice was analyzed 6 wks post-transplant.
RNA interference
The lentivirus pGIPZ vector containing shRNA targeting GEF-H1 (RHS4430-101128431) was obtained from Open Biosystems. The mir30 shRNA GEF-H1 sequence was cut with restriction enzymes Hpa1 and Bsut1, and ligated into the CMV-YFP-shRNA retroviral vector (gift from D. Wu, Yale University) digested with Hpa1, and retrovirus produced as described above. CMV-YFP vector containing shRNA targeting luciferase was used as a control. Sorted PreMegE cells from WT or Mkl1−/− mice (kind gift from Stephan Morris, Memphis, TN) were transduced with the shRNA viruses, and incubated for 3 days in growth medium followed by 3 days in differentiation medium, and the ploidy of YFP positive cells then assessed. To confirm the GEF-H1 shRNA efficiency, NIH3T3 cells were also transduced. After three days, YFP cells were sorted, and GEF-H1 protein levels detected by Western Blot.
Statistical analysis
Data are represented as means ± s.e.m of at least three independent experiments. Statistical significance was calculated with Student’s t-test. *P<0.05, **P<0.01, ***P<0.005.
Supplementary Material
Highlights.
Endomitotic megakaryocytes (Mk) do not activate equatorial RhoA and do not divide
RhoA GEFs, GEF-H1 and ECT2, must be downregulated for Mk polyploidization
GEF-H1 downregulation in Mk requires MKL1
Mk leukemia with the RBM15-MKL fusion expresses high levels of GEF-H1
Acknowledgments
We thank Susannah Kassmer and Stephanie Donaldson for assistance with mice, Ping-Xia Zhang for expert technical assistance, and Emanuela Bruscia and Shangqin Guo for helpful discussions. This work was supported by the R01DK086267, the Yale Center of Excellence in Molecular Hematology (P30DK072442), the State of Connecticut Stem Cell Research Fund, and the Yale Cancer Center (P30CA016359). The authors declare that they have no competing financial interests. Y.G. designed and performed the study and wrote the manuscript. E.S. designed and created plasmids, performed experiments, modified manuscript and provided intellectual input. E.K. and P.C. performed some time-lapse experiments. E.C. produced the stable HEL cell clones. S.Z. performed experiments, and provided a figure. S.L. helped with mouse transplantation and other mouse work. L.W. provided assistance with plasmid and retroviral vectors. S.H. provided reagents and intellectual input. D.S.K. oversaw the studies and wrote the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- Battinelli EM, Hartwig JH, Italiano JE., Jr Delivering new insight into the biology of megakaryopoiesis and thrombopoiesis. Curr Opin Hematol. 2007;14:419–426. doi: 10.1097/MOH.0b013e3282bad151. [DOI] [PubMed] [Google Scholar]
- Bement WM, Benink HA, von Dassow G. A microtubule-dependent zone of active RhoA during cleavage plane specification. J Cell Biol. 2005;170:91–101. doi: 10.1083/jcb.200501131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht M, Steenvoorden AC, Collard JG, Luf S, Erz D, Bartram CR, Janssen JW. Activation of gef-h1, a guanine nucleotide exchange factor for RhoA, by DNA transfection. Int J Cancer. 2005;113:533–540. doi: 10.1002/ijc.20626. [DOI] [PubMed] [Google Scholar]
- Chang Y, Auradé F, Larbret F, Zhang Y, Le Couedic JP, Momeux L, Larghero J, Bertoglio J, Louache F, Cramer E, Vainchenker W, Debili N. Proplatelet formation is regulated by the Rho/ROCK pathway. Blood. 2007;109:4229–4236. doi: 10.1182/blood-2006-04-020024. [DOI] [PubMed] [Google Scholar]
- Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, Prywes R. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol. 2003;23:6597–6608. doi: 10.1128/MCB.23.18.6597-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalamalasetty RB, Hummer S, Nigg EA, Sillje HH. Influence of human Ect2 depletion and overexpression on cleavage furrow formation and abscission. J Cell Sci. 2006;119:3008–3019. doi: 10.1242/jcs.03032. [DOI] [PubMed] [Google Scholar]
- Cheng EC, Luo Q, Bruscia EM, Renda MJ, Troy JA, Massaro SA, Tuck D, Schulz V, Mane SM, Berliner N, et al. Role for MKL1 in megakaryocytic maturation. Blood. 2009;113:2826–2834. doi: 10.1182/blood-2008-09-180596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SJ, Trinklein ND, Nguyen L, Myers RM. Serum response factor binding sites differ in three human cell types. Genome Res. 2007;17:136–144. doi: 10.1101/gr.5875007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayer AL, Olthof SG, Vellenga E. Mammalian target of rapamycin is required for thrombopoietin-induced proliferation of Mk progenitors. Stem Cells. 2006;24:105–114. doi: 10.1634/stemcells.2005-0062. [DOI] [PubMed] [Google Scholar]
- Eliades A, Papadantonakis N, Ravid K. New roles for cyclin E in megakaryocytic polyploidization. J Biol Chem. 2010;285:18909–18917. doi: 10.1074/jbc.M110.102145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fields AP, Justilien V. The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv Enzyme Regul. 2010;50:190–200. doi: 10.1016/j.advenzreg.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddis AE, Fox NE, Tkachenko E, Kaushansky K. Endomitotic Mk that form a bipolar spindle exhibit cleavage furrow ingression followed by furrow regression. Cell Cycle. 2007;6:455–460. doi: 10.4161/cc.6.4.3836. [DOI] [PubMed] [Google Scholar]
- Geddis AE, Kaushansky K. Endomitotic Mk form a midzone in anaphase but have a deficiency in cleavage furrow formation. Cell Cycle. 2006;5:538–545. doi: 10.4161/cc.5.5.2537. [DOI] [PubMed] [Google Scholar]
- Halene S, Gao Y, Hahn K, Massaro S, Italiano JE, Jr, Schulz V, Lin S, Kupfer GM, Krause DS. Serum response factor is an essential transcription factor in megakaryocytic maturation. Blood. 2010;116:1942–1950. doi: 10.1182/blood-2010-01-261743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson L, Shen F, Hahn K. Biosensors for characterizing dynamics of Rho family GTPases in living cells. Curr Prot Cell Biol. 2010;46:14.11.1–14.11.26. doi: 10.1002/0471143030.cb1411s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyoda M, Kasamatsu A, Ishigami T, Nakashima D, Endo-Sakamoto Y, Ogawara K, Shiiba M, Tanzawa H, Uzawa K. Epithelial cell transforming sequence 2 in human oral cancer. PLoS One. 2010;5:e14082. doi: 10.1371/journal.pone.0014082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosako H, Yoshida T, Matsumura F, Ishizaki T, Narumiya S, Inagaki M. Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene. 2000;19:6059–6064. doi: 10.1038/sj.onc.1203987. [DOI] [PubMed] [Google Scholar]
- Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol. 2002;4:294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]
- Levine RF, Hazzard KC, Lamberg JD. The significance of Mk size. Blood. 1982;60:1122–1131. [PubMed] [Google Scholar]
- Leysi-Derilou Y, Robert A, Duchesne C, Garnier A, Boyer L, Pineault N. Polyploid Mk can complete cytokinesis. Cell Cycle. 2010:9. doi: 10.4161/cc.9.13.12078. [DOI] [PubMed] [Google Scholar]
- Lordier L, Jalil A, Aurade F, Larbret F, Larghero J, Debili N, Vainchenker W, Chang Y. Megakaryocyte endomitosis is a failure of late cytokinesis related to defects in the contractile ring and Rho/Rock signaling. Blood. 2008;112:3164–3174. doi: 10.1182/blood-2008-03-144956. [DOI] [PubMed] [Google Scholar]
- Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T, Narumiya S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature. 1998;394:491–494. doi: 10.1038/28873. [DOI] [PubMed] [Google Scholar]
- Ma Z, Morris SW, Valentine V, Li M, Herbrick JA, Cui X, Bouman D, Li Y, Mehta PK, Nizetic D, Kaneko Y, Chan GC, Chan LC, Squire J, Scherer SW, Hitzler JK. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet. 2001;28:220–221. doi: 10.1038/90054. [DOI] [PubMed] [Google Scholar]
- Melendez J, Stengel K, Zhou X, Chauhan BK, Debidda M, Andreassen P, Lang RA, Zheng Y. RhoA GTPase is dispensable for actomyosin regulation but is essential for mitosis in primary mouse embryonic fibroblasts. J Biol Chem. 2011;286:15132–15137. doi: 10.1074/jbc.C111.229336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercher T, Coniat MB, Monni R, Mauchauffé M, Khac FN, Gressin L, Mugneret F, Leblanc T, Dastugue N, Berger R, Bernard OA. Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A. 2001;98:5776–5779. doi: 10.1073/pnas.101001498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercher T, Raffel GD, Moore SA, Cornejo MG, Baudry-Bluteau D, Cagnard N, Jesneck JL, Pikman Y, Cullen D, Williams IR, Akashi K, Shigematsu H, Bourquin JP, Giovannini M, Vainchenker W, Levine RL, Lee BH, Bernard OA, Gilliland DG. The OTT-MAL fusion oncogene activates RBPJ-mediated transcription and induces acute megakaryoblastic leukemia in a knockin mouse model. J Clin Invest. 2009;119:852–864. doi: 10.1172/JCI35901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuarai S, Yamanaka K, Kotani H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells. Cancer Res. 2006;66:6319–6326. doi: 10.1158/0008-5472.CAN-05-4629. [DOI] [PubMed] [Google Scholar]
- Muntean AG, Pang L, Poncz M, Dowdy SF, Blobel GA, Crispino JD. Cyclin D-Cdk4 is regulated by GATA-1 and required for Mk growth and polyploidization. Blood. 2007;109:5199–5207. doi: 10.1182/blood-2006-11-059378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S, Yasuda S. Rho GTPases in animal cell mitosis. Curr Opin Cell Biol. 2006;18:199–205. doi: 10.1016/j.ceb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Nguyen HG, Ravid K. Tetraploidy/aneuploidy and stem cells in cancer promotion: The role of chromosome passenger proteins. Journal of Cellular Physiology. 2006;208:12–22. doi: 10.1002/jcp.20565. [DOI] [PubMed] [Google Scholar]
- Nguyen HG, Ravid K. Polyploidy: mechanisms and cancer promotion in hematopoietic and other cells. Adv Exp Med Biol. 2010;676:105–122. doi: 10.1007/978-1-4419-6199-0_7. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Yonemura S. Centralspindlin regulates ECT2 and RhoA accumulation at the equatorial cortex during cytokinesis. J Cell Sci. 2006;19:104–114. doi: 10.1242/jcs.02737. [DOI] [PubMed] [Google Scholar]
- Papadantonakis N, Makitalo M, McCrann DJ, Liu K, Nguyen HG, Martin G, Patel-Hett S, Italiano JE, Ravid K. Direct visualization of the endomitotic cell cycle in living Mk: differential patterns in low and high ploidy cells. Cell Cycle. 2008;7:2352–2356. doi: 10.4161/cc.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SR, Richardson JL, Schulze H, Kahle E, Galjart N, Drabek K, Shivdasani RA, Hartwig JH, Italiano JE., Jr Differential roles of microtubule assembly and sliding in proplatelet formation by Mk. Blood. 2005;106:4076–4085. doi: 10.1182/blood-2005-06-2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Glotzer M, Kraut N, Peters JM. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell. 2007;12:713–725. doi: 10.1016/j.devcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Pronk CJ, Rossi DJ, Mansson R, Attema JL, Norddahl GL, Chan CK, Sigvardsson M, Weissman IL, Bryder D. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell. 2007;1:428–442. doi: 10.1016/j.stem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Raslova H, Kauffmann A, Sekkai D, Ripoche H, Larbret F, Robert T, Le Roux DT, Kroemer G, Debili N, Dessen P, et al. Interrelation between polyploidization and Mk differentiation: a gene profiling approach. Blood. 2007;109:3225–3234. doi: 10.1182/blood-2006-07-037838. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Saito S, Tatsumoto T, Lorenzi MV, Chedid M, Kapoor V, Sakata H, Rubin J, Miki T. Rho exchange factor ECT2 is induced by growth factors and regulates cytokinesis through the N-terminal cell cycle regulator-related domains. J Cell Biochem. 2003;90:819–836. doi: 10.1002/jcb.10688. [DOI] [PubMed] [Google Scholar]
- Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol. 1999;147:921–928. doi: 10.1083/jcb.147.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer A. Human marrow Mk differentiation: multiparameter correlative analysis identifies von Willebrand factor as a sensitive and distinctive marker for early (2N and 4N) Mk. Blood. 2004;104:2722–2727. doi: 10.1182/blood-2004-02-0769. [DOI] [PubMed] [Google Scholar]
- Tomer A, Harker LA, Burstein SA. Flow cytometric analysis of normal human Mk. Blood. 1988;71:1244–1252. [PubMed] [Google Scholar]
- Tolliday N, VerPlank L, Li R. Rho1 directs formin-mediated actin ring assembly during budding yeast cytokinesis. Curr Biol. 2002;12:1864–1870. doi: 10.1016/s0960-9822(02)01238-1. [DOI] [PubMed] [Google Scholar]
- Yasui Y, Amano M, Nagata K, Inagaki N, Nakamura H, Saya H, Kaibuchi K, Inagaki M. Roles of Rho-associated kinase in cytokinesis; mutations in Rho-associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J Cell Biol. 1998;143:1249–1258. doi: 10.1083/jcb.143.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki H, Ohba Y, Parrini MC, Dulyaninova NG, Bresnick AR, Mochizuki N, Matsuda M. Cell type-specific regulation of RhoA activity during cytokinesis. J Biol Chem. 2004;279:44756–44762. doi: 10.1074/jbc.M402292200. [DOI] [PubMed] [Google Scholar]
- Yoshizaki H, Ohba Y, Kurokawa K, Itoh RE, Nakamura T, Mochizuki N, Nagashima K, Matsuda M. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J cell Biol. 2003;162:223–232. doi: 10.1083/jcb.200212049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuce O, Piekny A, Glotzer M. An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol. 2005;170:571–582. doi: 10.1083/jcb.200501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WM, Fang G. Anillin is a substrate of anaphase-promoting complex/cyclosome (APC/C) that controls spatial contractility of myosin during late cytokinesis. J Biol Chem. 2005;280:33516–33524. doi: 10.1074/jbc.M504657200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.