

Abstract
Malignant astrocytomas are highly aggressive brain tumours with poor prognosis. While a number of structural genomic changes and dysregulation of signalling pathways in gliomas have been described, the identification of biomarkers and druggable targets remains an important task for novel diagnostic and therapeutic approaches. Here, we show that the Wnt-specific secretory protein Evi (also known as GPR177/Wntless/Sprinter) is overexpressed in astrocytic gliomas. Evi/Wls is a core Wnt signalling component and a specific regulator of pan-Wnt protein secretion, affecting both canonical and non-canonical signalling. We demonstrate that its depletion in glioma and glioma-derived stem-like cells led to decreased cell proliferation and apoptosis. Furthermore, Evi/Wls silencing in glioma cells reduced cell migration and the capacity to form tumours in vivo. We further show that Evi/Wls overexpression is sufficient to promote downstream Wnt signalling. Taken together, our study identifies Evi/Wls as an essential regulator of glioma tumourigenesis, identifying a pathway-specific protein trafficking factor as an oncogene and offering novel therapeutic options to interfere with the aberrant regulation of growth factors at the site of production.
Keywords: cancer research, glioma, RNAi, Wnt secretion, Wnt signalling
INTRODUCTION
Malignant astrocytomas are the largest group of primary brain tumours. Glioblastoma, the most common and most aggressive form, is characterized by marked cellular heterogeneity, high proliferative activity, aberrant microvascular proliferation, presence of necrosis and highly invasive growth (Riemenschneider & Reifenberger, 2009). They most commonly arise de novo (‘primary glioblastoma’) or develop by progression from pre-existing lower grade tumours (‘secondary glioblastoma’) (Ohgaki & Kleihues, 2007; Wettenhall & Smyth, 2004). Glioblastomas are characterized by complex genetic and epigenetic aberrations that differ between primary and secondary glioblastomas but affect a similar set of pathways, in particular receptor tyrosine kinase/Ras, phosphoinositol 3-kinase, p53 and pRb signalling (TCGA, 2008). Despite highly aggressive multimodal therapy, including surgical resection followed by combined radio- and chemotherapy, the median survival of glioblastoma patients has remained as low as 12–14 months throughout the past decade (Furnari et al, 2007; Holland, 2001).
Aberrant Wnt signalling is molecularly linked to many human cancers, including colorectal, breast, ovarian, hepatocellular carcinoma, melanoma and neuroectodermal tumours (Lindvall et al, 2007; Lustig & Behrens, 2003; Polakis, 2007; Saif & Chu, 2010). Its role has been best characterized in colorectal cancer, where mutations in the tumour suppressor APC lead to activation of Wnt signalling. Medulloblastoma arise often in patients with the Turcot syndrome, a subgroup of which is genetically characterized by germline mutations in the APC gene (Hamilton et al, 1995). Mutations in other Wnt pathway members, including β-catenin and Axin2, and aberrant production of Wnt ligands also have been associated with cellular transformation and tumour development (Behrens & Lustig, 2004; Klaus & Birchmeier, 2008; Reya & Clevers, 2005). Recent results identified a correlation between aberrant activation of Wnt/β-catenin signalling and progression in astrocytoma (Liang et al, 2009; Liu et al, 2010a; Sareddy et al, 2009). Despite the importance of Wnt signalling in tumourigenesis, there is still a lack of druggable targets in the Wnt pathway to modulate its activity and potentially inhibit tumour growth (Barker & Clevers, 2006).
Wnt proteins are highly conserved secreted, cysteine-rich glycoproteins. They initiate at least three intracellular signalling cascades: the canonical/β-catenin-dependent Wnt pathway, the planar cell polarity (PCP) and the Wnt/Ca2+ pathway (McDonald & Silver, 2009). Different Wnt signalling pathways share several components including receptor complexes and the adaptor protein Dishevelled (Axelrod et al, 1998; Boutros et al, 1998), but diverge further downstream and control distinct molecular and physiological outcomes (Cadigan & Peifer, 2009; Seifert & Mlodzik, 2007).
The molecular mechanisms governing the maturation and secretion of Wnt ligands in the producing cells are only beginning to emerge. We previously identified Evi/Wntless/Sprinter/GPR177, a highly conserved seven-pass transmembrane protein, as a component of the Wnt secretion machinery (Banziger et al, 2006; Bartscherer et al, 2006; Goodman et al, 2006). Evi acts as a Wnt cargo receptor, shuttling between the Golgi and the plasma membrane and is required for exocytosis of Wnt proteins (reviewed in Bartscherer & Boutros, 2008; Eaton, 2008). Evi is essential for Wnt secretion and its loss leads to accumulation of Wnts in the producing cell (Banziger et al, 2006; Bartscherer et al, 2006). Genetic inactivation of Evi in Drosophila leads to early embryonic patterning defects that phenocopy Wnt/Wg-depletion. Loss-of-function of Evi in mice causes embryonic lethality due to disruption of axial patterning (Fu et al, 2009).
Here, we demonstrate that Evi is overexpressed in human astrocytic gliomas relative to normal adult brain tissue. Our experiments show that Evi is required for glioma cell growth ex vivo and in vivo. Loss of Evi resulted in downregulation of cell cycle and survival genes. These experiments identify Evi as a potential molecular marker of human glioma and establish a functional role of Evi in the pathogenesis of human brain tumours.
RESULTS
Evi is overexpressed in astrocytomas
Evi is ubiquitously expressed during mouse embryonic development with particular prominent expression in the developing head structures. Expression persists in adult tissues with distinct expression pattern and levels in different organs (Jin et al, 2010; Yu et al, 2010; our unpublished data). Evi is essential for Wnt-dependent developmental processes (Fu et al, 2009). To assess Evi expression during brain tumourigenesis, we analysed an expression profiling database of 71 diffuse astrocytic tumours of different malignancy grades [WHO grade II: n = 8; WHO grade III: n = 11; WHO grade IV (primary glioblastoma: n = 42 and secondary glioblastoma: n = 10)] (Toedt et al, 2011). Strikingly, we found that Evi is strongly overexpressed in astrocytic gliomas of all malignancy grades as compared to control tissue (Fig 1A). This finding was confirmed in an independent data set from the Molecular Brain Neoplasia Database (REMBRANDT) (Madhavan et al, 2009), which also revealed a significant upregulation of Evi transcripts in gliomas. Moreover, higher levels of Evi expression were associated with shorter overall survival of glioma patients (p = 0.013) (Fig S1 of Supporting information). Evi expression levels showed no association with gender, age, TP53, IDH1 or IDH2 mutation status or with MGMT promoter methylation status (data not shown).
Figure 1. The Wnt secretion factor Evi is overexpressed in astrocytic gliomas.
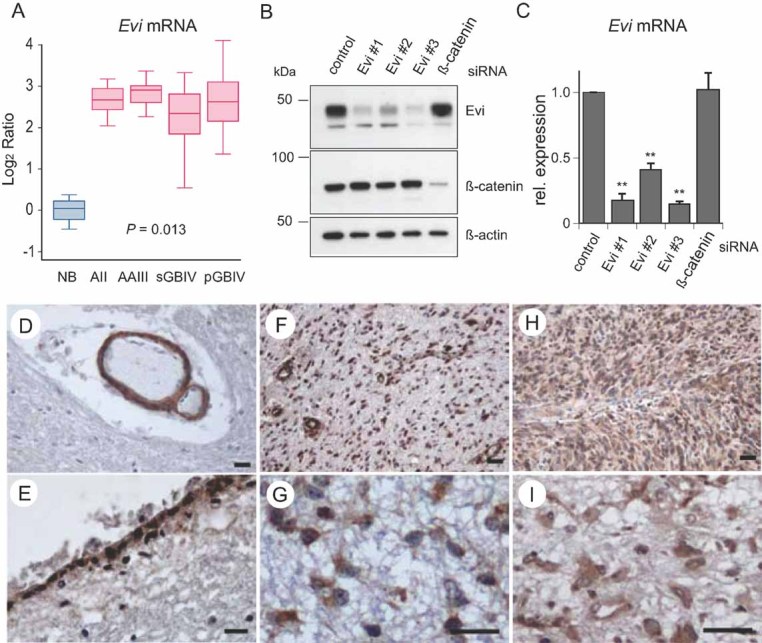
- A. Log2-gene expression ratios normalized to the mean expression in NB samples are shown for diffuse astrocytoma WHO grade II (AII), anaplastic astrocytoma WHO grade III (AAIII), secondary glioblastoma WHO grade IV (sGBIV) and primary glioblastoma WHO grade IV (pGBIV). Median RNA expression is indicated by horizontal bars; boxes show the 25th and 75th percentile range, whiskers mark the 5th and 95th percentiles; maximum and minimum values are depicted as horizontal bars.
- B, C. The specificity of the antibody against Evi was confirmed by siRNA silencing of the target protein. U87MG cells were transfected with three-independent siRNAs to silence Evi. Silencing of gene expression was validated by Western blot and quantified real-time RT-PCR confirming robust downregulation of Evi expression. β-Actin was detected as loading control. PCR-data are expressed as mean ± SD of three-independent experiments (**p < 0.01).
- D, E. Representative immunohistochemical stainings for Evi on tissue sections of NB and astrocytic gliomas of different WHO grades. (D) NB, Evi-positive vascular smooth muscle cells; (E) NB, Evi-positive ependymal cells.
- F, G. Evi-positive tumour cells in a diffuse astrocytoma WHO grade II.
- H, I. Evi-positive tumour cells in a primary glioblastoma WHO grade IV. Scale bar: 100 µm.
In order to examine the expression of Evi at the protein level, we raised an antibody against its N-terminus. The antibody recognized a protein with the expected size of ∼45 kDa, which was lost or reduced after depletion by siRNA targeting Evi (Fig 1B). Both Evi #1 and Evi #3 siRNA revealed a strong reduction of Evi protein and mRNA levels (Fig 1B and C). Immunofluorescence analysis of U87MG cells revealed strong perinuclear Evi staining (Fig S2A of Supporting information), consistent with previous reports from other cell types (Fu et al, 2009; Korkut et al, 2009). Next, we examined Evi protein levels in normal and astrocytic tumour tissue. In normal brain (NB) tissue, Evi was restricted to vascular smooth muscle cells, ependymal cells, few neurons and some astrocytes in NB tissue (Fig 1D and E). In contrast, and consistent with the mRNA expression data, Evi protein was highly expressed in tumour cells of both low-grade and high-grade gliomas (Fig 1F–I). Taken together, our data demonstrates that Evi is strongly overexpressed in diffuse astrocytic gliomas irrespective of the WHO grade.
Evi gain-of-function enhances Wnt-reporter-activity
Next, we asked whether Evi overexpression is sufficient to lead to an increase in Wnt signalling. To this end, we generated Evi overexpressing embryonic stem cells (ESCs) by targeted insertion of a C-terminal tagged Evi-YFP into the ROSA26-locus (Fig S3 of Supporting information). Immunofluorescence analysis of ROSA26::Evi-YFP ESC confirmed Evi-YFP fusion protein expression with perinuclear enrichment (Fig 2A). Quantitative RT-PCR revealed elevated Evi transcription in ROSA26::Evi-YFP ESCs and Western blot analysis with an antibody against Evi detected endogenous and Evi-YFP fusion protein (Fig 2B and C).
Figure 2. Evi overexpressing ESCs showed increased Wnt response.
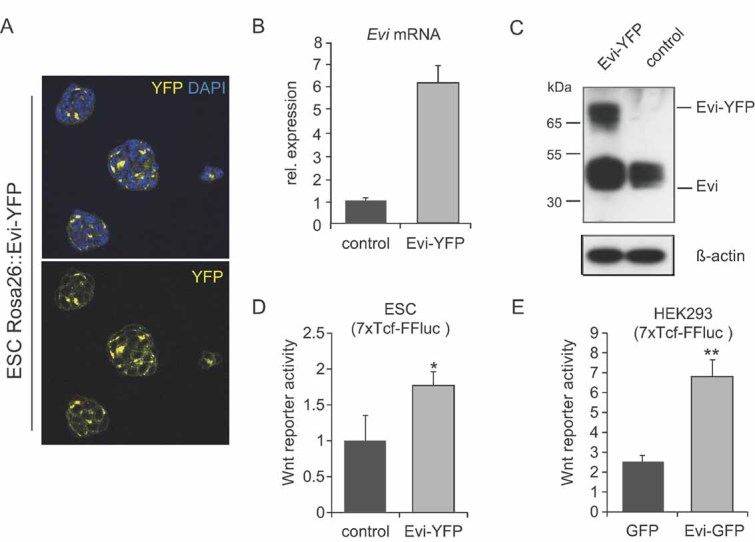
- Immunofluorescence of endogeneous expression of Evi-YFP in ESC colonies.
- Relative mRNA expression levels of Evi were analysed by quantitative RT-PCR. Evi-ESCs expressed increased Evi levels.
- Western blot of Evi overexpressing ESCs. Evi-ESCs expressed Evi-YFP fusion protein.
- Evi-ESCs stably transfected with 7TCF Firefly luciferase reporter showed increased Wnt reporter activity (*p < 0.05).
- Transfection of 7TCF Firefly luciferase reporter containing HEK293 cells with Evi-GFP plasmid led to increased Wnt reporter activity compared to transfection with GFP control vector (**p < 0.01).
We then tested whether overexpression of Evi is sufficient to induce Wnt signalling activity. As shown in Fig 2D and E, both ROSA26::Evi-YFP ESCs and Evi transfected HEK293 cells lead to a higher Wnt reporter activity, indicating that an increase in Evi levels can lead to overactivation of Wnt signalling pathways.
Evi is required for proliferation of glioma cells
In order to analyse the functional role of Evi in glioma cells, we silenced Evi in glioblastoma cell lines and glioblastoma-derived cancer stem-like cells. Glioblastoma cell lines express varying levels of Evi transcripts as determined by qPCR analysis. Among these, U87MG, A172 and T98G cells showed the highest level of Evi expression (Fig S2B of Supporting information).
Evi is an essential Wnt secretion factor for canonical and non-canonical Wnt ligands. To test the effect of Evi silencing on Wnt ligand secretion, we examined the ability of conditioned media from wild-type (WT) and Evi silenced U87MG cells to induce Wnt reporter activation in HEK293 cells. Depletion of Evi led to significantly reduced Wnt response in HEK293 7TCF firefly luciferase reporter cells (Fig S2C of Supporting information).
Next, we examined the consequences of Evi silencing on cell viability in glioblastoma cell lines. Depletion of Evi by RNAi resulted in significant inhibition of cell viability in U87MG, A172 and U251MG cells compared to transfections with control siRNAs, while no change in viability was observed in T98G cells (Fig 3A and Fig S4A and B of Supporting information). In contrast, β-catenin silencing only significantly affected the viability of U87MG and U251MG cells, mostly to a lesser extent. We identified further cell lines, including LN229 that showed a decrease in cell viability upon Evi silencing. Two out the tested six cell lines (T98G and LN18) were not dependent on Evi for growth (Fig 3A and Fig S5A and B of Supporting information), indicating that they might harbour additional growth promoting aberrations. All Evi-dependent glioblastoma cell lines (except LN18) are PTEN mutant, however, a synergism between PTEN and Wnt signalling would need further confirmation. To provide additional evidence that Evi silencing affects cell survival, we performed a colony formation assay with U251MG cells. Downregulation of Evi led to significantly fewer colonies compared to controls (Fig 3B and Fig S4 of Supporting information). Taken together, we showed that depleting Evi reduced the viability of four out of six glioblastoma cell lines.
Figure 3. Evi is required for proliferation and survival of glioblastoma cell lines.
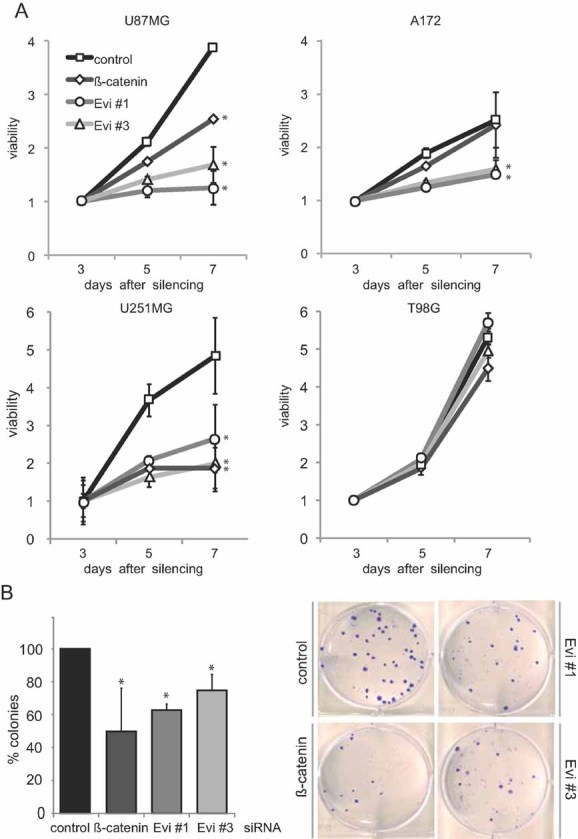
- Viability of RNAi transduced U87MG cells, A172, U251MG cells and T98G was determined by CellTiter-Glow assay and revealed reduced viability of Evi-RNAi transduced cells compared to control cells. β-catenin silencing had significant effect on viability of U787MG and U251MG cells. Evi and β-catenin silencing had no significant effect on proliferation of T98G cells (*p < 0.05).
- Evi and β-catenin silencing caused reduced colony formation in U251MG cells. Representative example of three-independent experiments is shown (right). Data are expressed as mean ± SD of three-independent experiments (*p < 0.05).
Long-term shRNA-based silencing of Evi expression through lentiviral transduction similarly led to reduced U87MG cell viability as observed in the siRNA-based experiments (Figs S4 and S5C of Supporting information). In addition, we analysed cycle profiles of U87MG cells after Evi depletion. Evi RNAi led to a G1 arrest, indicated by significantly increased number of cells in G1-phase in Evi-silenced cells compared to control shRNA-silenced cells (53.4% vs. 47.6%) (Fig S5D of Supporting information). Concomitantly, the number of cells in S-phase was reduced in Evi silenced cells (6.5% vs. 12.1%). In contrast, the subG1-fraction of the cells was not significantly changed. A further FACS-based analysis of Annexin V-positive cells after Evi silencing revealed a slight increase in apoptotic cells indicating that the observed decrease in cell viability was primarily due to decreased proliferation but also affects cell death (Fig S5E of Supporting information). Therefore, we conclude that Evi predominantly is required for cell proliferation and viability of glioblastoma cells.
Evi silencing induces apoptosis in stem-like glioma cells
We next analysed whether Evi is required for proliferation and survival in primary stem-like glioma cells (SLGCs) obtained from patient-derived glioblastoma samples. Previous studies have shown that spheroid-forming cells isolated from human glioblastomas and cultured under serum-free conditions were enriched for glioma cancer stem cells (Wan et al, 2010). We tested two SLGC lines (NCH421k and NCH644) which are derived from a subpopulation of glioma tumour cells that are capable of expanding into an actively tumour after intracranial xenograph transplantation (Campos et al, 2010).
Light microscopic analysis of SLGC spheres transduced with Evi shRNA versus control shRNA revealed strong morphological differences: Evi silenced spheres were significantly smaller and lost their packed condensed morphology compared to control spheres (Fig 4A and Fig S4 of Supporting information). Moreover, the amount of viable cells was significantly reduced in Evi depleted spheroid cultures (Fig 4B). Further analysis of apoptotic cells after Evi shRNA transduction revealed an increase in apoptosis as demonstrated by a significant rise in the sub-G1 fraction with no significant changes in cell cycle distribution (Fig 4C and D). These experiments demonstrate that Evi is required for cell survival in primary patient-derived glioblastoma cells.
Figure 4. Depletion of Evi induces apoptosis in glioblastoma-derived cancer stem-like cells.

- Neurosphere shape and size was disturbed after Evi silencing. Scale bar: 100 µm.
- Reduction in cell number compared to control transfected spheres 7 days after infection (**p < 0.01; ***p < 0.001).
- Lentiviral shRNA silencing of Evi expression in NCH421k and NCH644 cells led to an increase in the sub-G1 fraction (*p < 0.05; **p < 0.01).
- Representative graphs of cell cycle distribution. Data are expressed as mean ± SD of three-independent experiments.
Effect of Evi depletion in U87MG on tumour cell migration and in vivo tumour growth
Glioblastoma are characterized by pronounced invasion of tumour cells into the surrounding healthy tissue (Tysnes & Mahesparan, 2001). To examine the consequences of Evi depletion in glioma cells on cell migration, we performed transwell migration experiments. As shown in Fig 5A and B, siRNA-based silencing of Evi expression caused a 32% inhibition of migration of glioma cells; a more robust lentiviral shRNA-induced downregulation of Evi led to an even stronger 63% decrease in migratory behaviour.
Figure 5. Wnt secretion is important for tumour cell migration and tumour formation in xenograft models.
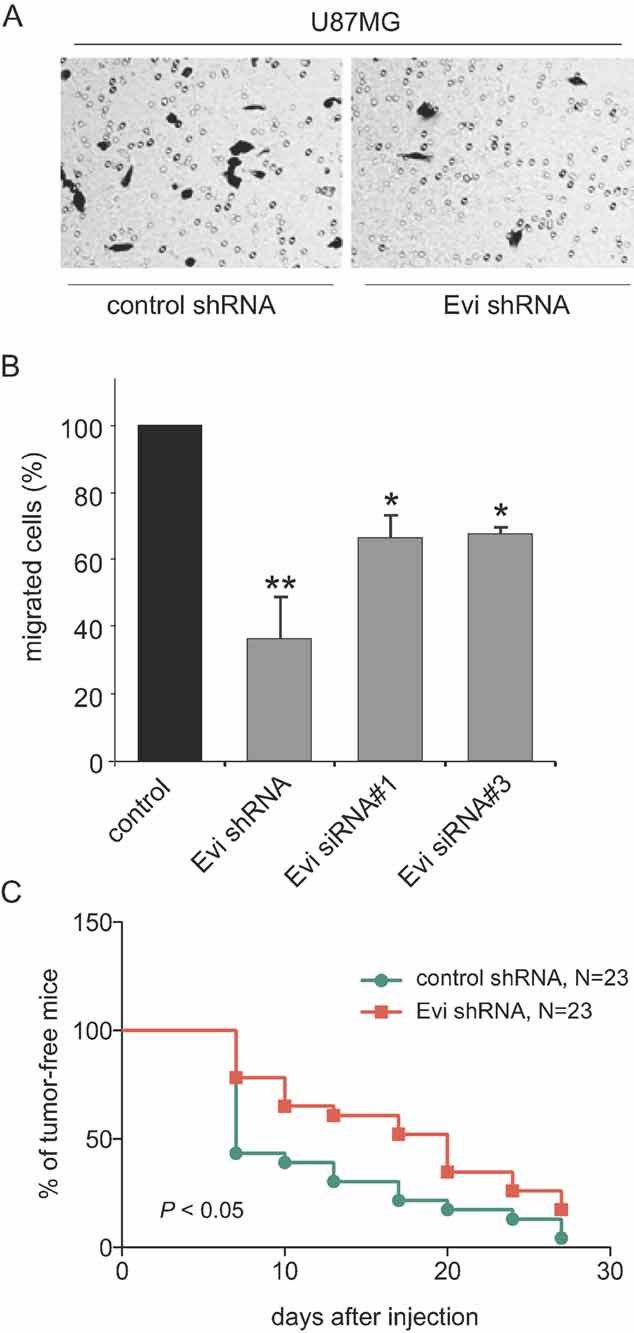
- Evi shRNA U87MG cells showed less transwell migration compared to control. Similar effect was achieved by siRNA transfection.
- Migration experiments were done as short-term assays to exclude anti-proliferative effects. Values represent mean ± SD from three-independent experiments (*p < 0.05; **p < 0.01).
- Reduced in vivo growth of glioma cells transfected with shRNA targeting Evi. The appearance of U87MG glioma xenografts formed by Evi shRNA or control transfected cells was reduced in the Evi downregulated glioma cells.
We then examined the effect of Evi silencing on glioma tumourigenesis in vivo by comparing the growth of subcutaneously grafted control and Evi shRNA transduced U87MG tumours. Correlating with the reduced proliferative and migratory capacity of Evi silenced U87MG cells in culture, shRNA-based downregulation of Evi caused a significant reduction of glioma tumourigenesis (Fig 5C). Silencing of Evi induced a delay in the onset of tumour growth indicating that depletion of Evi affected growth and survival of glioblastoma cells after xenotransplantation. Median tumour take time point of the control cells is 7 days in contrast to 20 days after injection of Evi silenced cells. Our experiments showed that Evi interferes with tumour-promoting characteristics like tumour cell migration and tumour initiation.
Silencing of Evi leads to downregulation of pro-proliferative genes and interleukins in glioma cells
To identify genes that are transcriptionally controlled by Evi, we compared expression profiles of glioma cells after transfection with two-independent Evi (siRNA Evi #1 and Evi #3) and control siRNAs (Fig 6A). In addition, we performed expression profiling after β-catenin silencing in order to subselect genes regulated through a canonical Wnt pathway (Fig 6A).
Figure 6. Evi controls cell cycle and interleukin expression.
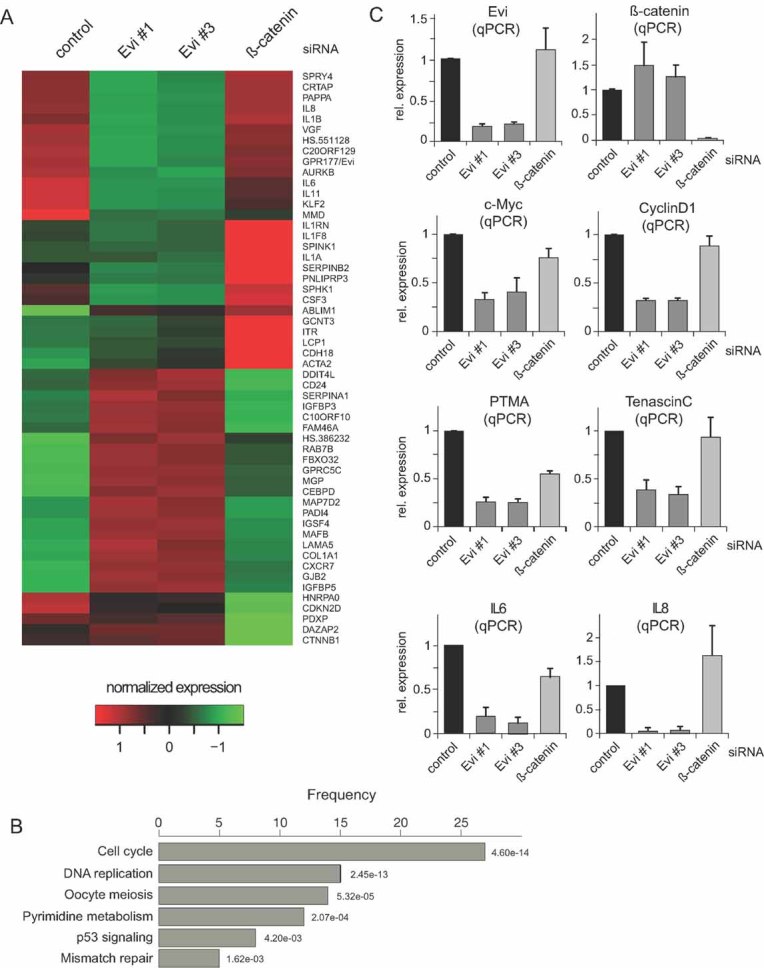
- Heatmap based on normalized U87MG gene expression values of RNAi Evi#1, Evi#3, β-catenin and control samples. Rows represent genes, which are differentially expressed (p < 0.01) and have an absolute log2-fold change >1.5 compared to control, in Evi#1, Evi#3 or β-catenin silencing experiments.
- Over-represented KEGG categories in set of differentially expressed genes (Fisher's exact test, p < 0.01). The length of the bars represents the number of genes within the set of differentially expressed genes that are annotated as part of the corresponding KEGG category. The numbers is red indicate the significance of the over-representation. The KEGG categories are not mutually exclusive. ‘Cell cycle’ is the most significant over-represented category.
- Cells were transduced with indicated siRNAs. The relative mRNA expression levels of Evi, β-catenin, cyclin D1, c-Myc, PTMA, tenascin-C, and IL8 after Evi silencing analysed by quantified RT-PCR. Data are expressed as mean ± SD of three-independent experiments.
The global expression profiles after Evi RNAi were highly similar for the two Evi siRNAs (R2 = 0.99) (Fig S6A of Supporting information), without obvious off-target effects. However, β-catenin silencing revealed little overlap in expression patterns compared to Evi depletion, suggesting that Evi silencing affects primarily the non-canonical/β-catenin-independent Wnt signalling branch in glioma cells. In total, we identified 139 differentially expressed genes between Evi RNAi and control treatments with a log odds ratio >10. Thirty genes displayed a log2-fold change ≥1.5 (Fig S6B of Supporting information). An analysis of the differential expression data at the level of KEGG categories revealed that Evi depletion strongly affected the expression of genes involved in cell cycle regulation, DNA replication, mismatch repair and nucleotide excision repair, among others (Fig 6B). Quantitative RT-PCR confirmed the regulation of c-Myc, cyclin D1, PTMA and tenascin-C by Evi (Fig 6C). Since loss-of Evi can affect the production of both canonical and non-canonical Wnt pathways, we also tested whether these genes were downregulated after silencing of β-catenin. As shown in Fig 6, β-catenin silencing reduced PTMA expression.
Members of the interleukin family including IL8, IL6, IL1B and IL11 were strongly downregulated after Evi depletion (Fig 6A and C and Fig S6 of Supporting information). Experiments with conditioned medium of parental U87MG cells rescued the downregulation of IL6 and IL8 after Evi silencing (Fig S7A of Supporting information). Similarly the viability effect of Evi depletion was abolished in the presence of conditioned medium (Fig S7B of Supporting information). High levels of IL6 and IL8 have been linked to tumour generation and poor prognosis in many cancer types, including glioblastoma (Hodge et al, 2005; Putoczki & Ernst, 2010; Samaras et al, 2009), however, little is known on how their expression is regulated. Recently, it has been shown that IL8 is a target gene of STAT3 in human glioblastoma cells (de la Iglesia et al, 2008a). Phosphorylated STAT3 directly binds to the IL8 promotor and inhibits IL8 transcription (de la Iglesia et al, 2008b). Therefore, we analysed the STAT3 status after Evi silencing compared to control transfected U87MG cells. Our data showed that Evi depletion increased phosphorylated STAT3 indicating that STAT3 activation is downstream of Evi and Wnt secretion (Fig 7A). Taken together, these results suggest that Wnt proteins regulate IL8 expression via the inhibition of STAT3 phophorylation (Fig 7B).
Figure 7. Downregulation of Evi repressed IL transcription by activation of STAT3.
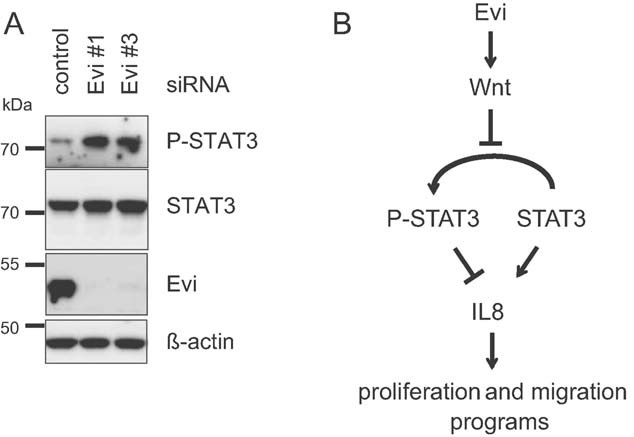
- Western blot of U87MG cell lysates after RNAi transfection against Evi (Evi #1 and Evi #3) showed increased levels of phosphorylated STAT3 compared to control transfection. β-Actin was detected as loading control. Representative example of three-independent experiments is shown.
- Model of the Evi-Wnt-STAT3-IL8 signalling link in human glioblastoma cells. Evi mediated Wnt secretion controls phosphorylation of STAT3. Downregulation of Evi leads to activation of STAT3 by phosphorylation. Phosphorylated STAT3 binds to IL8 promotor and represses IL8 transcription. Downregulation of IL8 reduces glioblastoma cell proliferation and invasiveness.
DISCUSSION
Despite recent advances in surgery and adjuvant therapy, the overall prognosis for patients with malignant brain tumours remains poor, emphasizing the need for an in-depth understanding of the molecular pathogenesis and the development of new concepts for cancer therapy. Aberrant activation of Wnt signalling is important in a variety of human cancers. In this study, we show that the Wnt-specific secretion factor Evi is highly overexpressed in brain tumours, indicating that the aberrant release of canonical and non-canonical Wnt is a potential driver of glioma tumourigenesis.
Wnt signalling and its contributions to tumourigenesis have been characterized in many tissues (Klaus & Birchmeier, 2008; Polakis, 2007; Reya and Clevers, 2005). For example, mutations in APC in colon cancer lead to the stabilization of β-catenin and subsequent increased expression of transcriptional target genes. Aberrant expression of Wnt proteins has also been implicated in tumour formation, including breast cancer (Bafico et al, 2004; Curtin and Lorenzi, 2010). Several antagonists have been identified that target different components in the Wnt pathway, including blocking protein–protein interaction of Fz and Dsh at the membrane, β-catenin and TCF in the nucleus, small molecule inhibitors of Tankyrases leading to an increase in Axin levels and antibodies against Dkk1 and LRP6, however, antagonists (and agonists) have not yet entered clinical development, making the Wnt pathway one of the few major signalling routes which are not yet addressable by targeted therapeutics (reviewed in Barker & Clevers, 2006; Takahashi-Yanaga & Kahn, 2010; Liu et al, 2009).
Evi as a potential ‘druggable’ target is one of the most ‘upstream’ core components of the Wnt signalling pathway and is required for the export of Wnts ligands. Originally, the GPCR-like transmembrane protein Evi was identified in a genetic screen in Drosophila as an essential and specific component for Wg export. In vertebrates, it has been shown that Evi binds to and is required for the release of Wnt1, Wnt3 and Wnt5a (Fu et al, 2009). Since only a single gene exists in vertebrate as well as in invertebrate genomes (in contrast to most other Wnt pathway components), Evi is believed to be involved in the secretion of all Wnt proteins, affecting both canonical and non-canonical Wnt ligands (Gordon & Nusse, 2006; van Amerongen & Nusse, 2009). These properties make Evi an interesting target for modulating aberrant Wnt signalling at the source of production.
We found that Evi expression is upregulated in human astrocytic glioma tissues of different WHO grades when compared to NB tissue. In diffuse astrocytomas WHO grade II, Evi expression was strongly increased and remained at high levels in anaplastic astrocytomas WHO grade III and glioblastomas WHO grade IV, indicating that high-levels of Evi may be required for early neoplastic transformation. This also points towards the secretion of Wnt ligands as the limiting step of the signalling cascade in cancerous brain tissues, rendering increased expression of the Wnt cargo-receptor Evi essential for both tumour initiation and tumour growth. Indeed, previous studies showed upregulation in brain tumours of both canonical and non-canonical Wnt signalling components including Wnt1, Wnt2 and Wnt5a (Liu et al, 2010c; Pu et al, 2009; Yu et al, 2007). In glioma cell lines, such as U87MG, non-canonical Wnt5a is the most abundant Wnt ligand (Fig S8 of Supporting information, Kamino et al, 2011). Wnt5a has oncogenic and anti-oncogenic properties, depending on the tumour type. In colorectal cancer, for example, Wnt5a has tumour suppressive function (Dejmek et al, 2005). For brain tumours it has been reported that Wnt5a stimulates cell motility and infiltrative activity of tumour cells. Moreover, Wnt5a expression correlates with brain malignancy (Kamino et al, 2011). In addition it has been shown that Wnt-driven PCP signalling suppresses endothelial cell proliferation and migration supporting tumour promoting function of non-canonical Wnt signalling cascades (Ju et al, 2010). Further studies will be required to identify the Wnt ligands that are influenced by the upregulation of Evi. Since glioblastoma cell lines and tumour specimens express both canonical and non-canonical Wnt, it is tempting to speculate the modulation of Evi might disturb a finely tuned balance between canonical and non-canonical signalling required for homoestasis. While Evi has been shown to have specific phenotypes in Wnt signalling, at this stage it cannot be excluded that it influences additional processes in glioma.
Ex vivo experiments revealed that loss-of Evi in different glioblastoma cell lines and glioblastoma derived cancer stem-like cultures affected cell proliferation, migration and apoptotic cell death. The requirement of Evi for glioblastoma cell survival was further confirmed by the observed reduced tumourigenesis in xenograft models. Furthermore, the inhibition of cell proliferation after Evi silencing in glioblastoma cell lines was accompanied by cell cycle arrest in G1. However, not all glioblastoma cell lines are dependent on Evi. This finding is currently mechanistically not understood.
Evi depletion interferes with the cell cycle progression. C-myc and cyclin D1 genes are associated with tumour proliferation and previous studies identified that cyclin D1 regulates G1-to-S phase transition (Liu et al, 2010c). The nuclear oncogenic protein PTMA is involved in cell proliferation but also in apoptosis (Letsas & Frangou-Lazaridis, 2006). PTMA expression is also positively regulated by the transcription factor c-myc, indicating that downregulation of c-myc by Evi depletion may contribute to reduced PTMA expression. Like other oncoproteins, tenascin-C overexpression correlates with a variety of cancer types and tumour cell lines. Tenascin-C is an extracellular matrix molecule, which modulates adhesion and is highly expressed in the microenvironment of most solid tumours. High tenascin-C expression correlates with malignancy in astrocytic tumours and has been associated with less favourable prognosis (Orend & Chiquet-Ehrismann, 2006). Previous studies revealed that tenascin-C expression is associated with proliferation and invasiveness of tumour cells. Reduced tenascin-C expression after Evi silencing might contribute to less mobility and invasion of Evi targeted tumour cells.
Expression profiling analysis of U87MG cells after Evi silencing revealed a strong reduction in transcription of several interleukin genes. Signalling functions of IL6 and IL8 have been implicated in glioma stimulating both cell survival and tumour growth. High expression of IL6 and IL8 was found to be associated with glioma malignancy through promotion of proliferation, survival and invasiveness (Hodge et al, 2005; Liu et al, 2010b; Putoczki & Ernst, 2010; Samaras et al, 2009). Evi overexpression leads to an increase in Wnt response likely due to an increased Wnt secretion, suggesting that enhanced Wnt signalling stimulates tumour cell growth possibly by upregulation of interleukins and other pro-oncogenic factors. We found that β-catenin silencing has no significant effect on interleukin expression, suggesting that β-catenin-independent Wnt signalling is important for transcriptional regulation of interleukins. IL8 transcription is repressed by activated STAT3 in PTEN-mutated glioblastoma cell lines (de la Iglesia et al, 2009). The identification of Evi and Wnt secretion as a novel regulator of STAT3 activation in glioma cells might define a new connection between Wnt signalling and IL8 production affecting tumour cell growth (Fig 7B). Recent results showed that the chemokine receptor CXCR7 is highly expressed in glioma and is probably required for directionality of cell migration (Hattermann et al, 2010). Our expression analysis revealed that silencing of Evi significantly increased CXCR7 expression. Previous studies suggested that signalling events downstream of CXCR7 include an activation of Jak2/STAT3 as well as MAPK pathways (Gao et al, 2007). Therefore, we hypothesize that Evi might control STAT3 phosphorylation via CXCR7 expression.
Together, our data demonstrate that Evi-mediated Wnt signalling regulates proliferation, survival and migration of glioma cells, which are critical aspects of the pathogenesis of human brain tumours. Our study also shows for the first time that upregulation of a growth-factor specific secretory proteins can contribute to tumourigenesis, indicating that the Wnt cargo-receptor might be a limiting factor in situations when Wnt proteins are overexpressed. Targeting Evi may represent a potential strategy for therapeutic intervention as its inhibition affects multiple Wnts and would limit overactivation of both canonical and non-canonical Wnt signalling.
MATERIALS AND METHODS
Construct of the Evi targeting vector
For expression of Evi as a C-terminal tagged Evi-GFP fusion protein, human and mouse Evi cDNA was cloned into pEGFP-N1 expression vector. The coding sequence was cloned into EcoRI and BamHI sites. The gene-targeting ROSA26-β-geo construct used to target WT ESCs by homologous recombination was generated based on constructs published by Soriano, 1999. The conditional Evi-construct was integrated into the first XbaI-site after exon 1 of the ROSA26 gene. The flanking genomic sequence (1.1 kb 5′-prime arm and 4.3 kb 3′-prime arm) between two EcoRV-sites was cloned in the targeting vector and used for homologous recombination. We generated the ROSA26::Evi-YFP ESC line by loxP gene targeting and Cre-mediated deletion of the β-geo-cassette. Genotyping was done with the following primer pairs: WT: gtcgctctgagttgttatcag, ccagatgactacctatcctcc; Evi floxed: gtcgctctgagttgttatcag, gacgacagtatcggcctcaggaag.
Cell lines and tumour models
Human glioblastoma cell lines U87MG, A172, T98G, U251MG, LN18 and LN229 cell lines were kindly provided by Dr. W. Roth and Dr. P. Angel (DKFZ). Cells were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) with 10% foetal bovine serum (Invitrogen) at 37°C and 5% CO2 in a humidified atmosphere. The investigated glioma stem-like cell line NCH421k and NCH644 were previously established from a primary glioblastoma patient undergoing surgical resection as approved by the Institutional Review Board at the Medical Faculty, University of Heidelberg. This cell line was genotyped and phenotypically characterized (Campos et al, 2010). NCH421k and NCH644 were cultivated at 37°C in a humidified incubator with 5% CO2 as floating aggregates (neurospheres) on uncoated tissue culture dishes. Glioma stem-like cell medium consisted of Dulbecco's modified Eagle medium/F-12 medium containing 20% BIT serum-free supplement, basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) at a concentration of 20 ng/ml each (all Provitro, Berlin, Germany).
Mouse ESCs were cultured on mouse embryonic fibroblasts or gelatine-coated dishes in DMEM containing 15% FCS, 2 mM l-glutamin, MEM non-essential amino acids, 1 mM MEM sodium pyruvate, 100 µM β-mercaptoethanol and 1000 U/ml LIF (Millipore). Cells were routinely splitted every second day.
Mission RNAi clones targeting Evi were obtained from Sigma–Aldrich (TRCN0000133858 [Evi shRNA#1], TRCN0000138901 [Evi shRNA#2], TRCN0000138525 [Evi shRNA#3], Mission Non-Target shRNA Control Vector). pCF826:pLenti 7xTCF Firefly luciferase//SV40-PuroR (TCF luciferase reporter) construct was kindly provided by Roel Nusse (Fuerer & Nusse, 2010). Lentiviral particles were produced according to the manufacturer's instruction (Sigma–Aldrich). Produced lentiviruses were concentrated by ultra centrifugation using SW41 rotor (Beckman Coulter, Fullerton, CA, USA). Titer was measured by detecting GFP positive HEK293T cells using flow cytometry. Before transduction, neurospheres were dissociated by trypsinization. Transductions were performed at five of multiplicity of infection (MOI ∼ 5), conferring ∼90% transduction efficiency without significant cytotoxicity in negative control samples. Stable infected glioblastoma cell lines were selected in a medium containing 1 µg/ml puromycin (Invitrogen). Stable cell lines were maintained as polyclonal cell populations. Transient siRNA transfections of all cell lines were conducted using Dharmafect Reagent (ThermoFisher). siRNAs against human Evi, β-catenin and control siRNAs had the targeting sequences:
Pool of GCUGAAACAUGCAGUUGUA, GAUAAAGGCUACUGUUGGA, CCACUAAUGUCCAGCGUUU, ACAAGUAGCUGAUAUUGAU (β-catenin) and P002070-01-20 (control).
Cells were transfected in 384-, 24- or 6-well plates using 20 nM siRNA. siRNA transfected cells were cultured for 3 days prior to use for immunocytochemistry, Western blot and qPCR experiments.
Cell proliferation, colony forming assay and transwell migration assay
To assess cell viability, 500 cells were plated in quadruplicates in 384-well plates and viability was measured at different time points using CellTiter-Glo (Promega) according to the manufacturer's protocol. For rescue experiments, the medium of siRNA-transduced cells was daily changed against conditioned medium obtained from parental U87MG cells. Evi-YFP ESC and HEK293 T7 TCF firefly luciferase reporter cells were used for the Wnt-reporter assays. Luciferase assay was performed 2 days after seeding of the cells or adding conditioned medium to the Wnt reporter cells. Reporter activity was normalized to cell viability (CTG assay).
Colony forming assay was performed with U251MG cell. Cells were transfected with siRNA. After 2 days 1000 cells per 6-well were plated in triplicates and incubated for 2 weeks before they were stained with 0.1% crystal violet. For migration assays, invasion chambers (Corning) were used according to the manufacturer's instructions. Briefly, uncoated transwell membrane filter inserts (6.5 mm in diameter, 8-µm pore size and 10-mm-thick polycarbonate membrane) were placed in a 24-well tissue culture plates. Cells (1 × 105) suspended in DMEM containing 10% serum were pipetted in duplicate into the top chambers and DMEM containing 10% FBS was added to each bottom chamber. After 16 h incubation at 37°C, non-migrating cells were removed from the upper face of the filter using cotton swabs and cells on the lower filter surface were fixed and stained with haematoxylin (Sigma). The number of cells per microscopic field was counted light microscopically. The average number of migrating cells within seven random fields was calculated.
Western blotting
Cell pellets were dissolved in lysis buffer containing 8 M urea, 0.1 M NaH2PO4, 10 mM Tris–HCl. Cell pellets used for detection of phosphorylated STAT3 were lysed in same buffer with additional phosphatase inhibitors (Roche). Lysates were incubated on ice for 10 min and the centrifuged at maximum speed for 20 min. The supernatants were collected and protein concentrations were determined by BCA method. Protein (10–30 µg) was separated on 4–12% NuPage gradient gels and transferred to PVDF-membranes. Membranes were blocked and incubated overnight at 4°C or 1 h at RT with one of the following antibodies: anti-Evi (1:500, rabbit) and anti-β-catenin (1:1000, mouse, BD Transduction Laboratories #610154). Blots were then incubated with corresponding horseradish peroxidase-conjugated secondary antibodies (1:10000; Sigma). To confirm equal loading of the proteins, the blots were also immunoprobed with a mouse monoclonal antibody against β-actin (1:1000000; Sigma). The antiserum against Evi was generated against the peptide FTSPKTPEHEGRYYNC of the first extracellular loop.
Immunohistochemistry and cytochemistry
Non-neoplastic tissue samples from autopsy brains and human tumour tissues from neurosurgical biopsy samples were obtained from the Department for Neuropathology, Institute for Pathology, University of Heidelberg. All samples were analysed in an anonymized manner as approved by the local institutional ethics boards. In total, immunohistochemistry was carried out on 16 tumours (4 diffuse astrocytomas WHO grade II, 4 anaplastic astrocytomas WHO grade III and 8 glioblastomas WHO grade IV) and 3 NB tissue samples. Immunohistochemical studies were performed on formalin-fixed and paraffin-embedded specimen. Briefly, sections were deparaffinized in xylene and passed through graded alcohols and further rehydrated in phosphate buffered saline (PBS). Antigen unmasking was carried out by microwaving the sections for 10 min in 10 mM citrate buffer (pH 6.0). Sections were then treated with 1%H2O2 for 30 min to block endogenous peroxidase followed by incubation with Avidin/Biotin blocking solution (Vector) for 1 h at RT in a humid chamber. The sections were then incubated overnight at 4°C with primary antibodies against Evi (1:200). Peroxidase-conjugated secondary antibody (1:200, Dako) was used for 1 h incubation time at RT followed by 30 min incubation with AB-complex. Diaminobenzidine (DAB) in buffer was used until sections developed colour. Sections were then counterstained using haematoxylin. Negative control experiments included omission of the primary antibody.
For immunocytochemistry, siRNA transfected U87MG cells were fixed in 4% PFA/PBS for 10 min at RT. After washing steps, blocking was done with 1% BSA–PBS for 30 min. The primary antibody (1:200) against Evi was incubated overnight at 4°C. Fluorescein isothiocyanate (FITC)-labelled secondary antibody (1:800) was added for 1 h at RT. ESCs were seeded on glass slides without feeder cells. After fixation with 4%PFA/PBS nuclei were counterstained with Hoechst dye. Images were taken with a ZEISS LSM 510 META Confocal Microscope at 63× magnification.
Flow cytometry
Lentivirally shRNA transduced U87MG, NCH421k and NCH466 cells were cultured for 16 h (U87MG, 20% confluence) or 7 days (NCH421k and NCH644) prior to analysis. Cells were harvested and stained with 200 µg/ml propidium iodide, 0.1% NaAzide and 0.1% Triton X-100, 10 µg/ml RNAses for 3 h. A total of 20,000 nuclei were examined by FACS Array (BD Bioscience). AnnexinV staining was performed 6 days after siRNA transfection with Annexin-V-Alexa 568 staining kit (Roche) according to the protocol.
Real-time transcription (RT)-PCR analysis
Total RNA was extracted using RNeasy extraction kit (Qiagen) according to the manufacturer's instructions. Reverse transcription and quantitative PCR was performed with 25 ng cDNA and LightCycler 480 Probes Master as described (Roche). Relative mRNA expression was calculated as a fold-change versus control. For calculation of Evi copy number, a purified Evi cDNA fragment was titrated and analysed by quantitative PCR in parallel with cellular cDNA samples.
Primer sequences:
Evi/Gpr177: F: TCATGGTATTTCAGGTGTTTCG, R: GCATGAGGAACTTGAACCTAAAA (probe #38, Roche).
β-Catenin: F: AGCTGACCAGCTCTCTCTTCA, R: CCAATATCAAGTCCAAGATCAGC (probe#21, Roche).
Cyclin D1: F: GAAGATCGTCGCCACCTG, R: GACCTCCTCCTCGCACTTCT (probe #67, Roche).
C-myc: F: CACCAGCAGCGACTCTGA, R: GATCCAGACTCTGACCTTTTGC (probe #34, Roche).
PTMA: F: CCTGCTAACGGGAATGCTAA, R: CTTCCTCTTCTTCGTCTACCTCA (probe #75, Roche).
IL-8: F: ATGGTTCCTTCCGGTGGT, R: AGACAGCAGAGCACACAAGC (probe #72, Roche).
Expression profiling of human glioma tissue samples
RNA expression of Evi in NB and tumour samples relative to human reference RNA (Stratagene, La Jolla, USA) was determined using microarray analysis as described (Toedt et al, 2011).
Expression profiling of Evi silencing experiments
RNA was extracted from two biological replicates of cells transfected with either Evi siRNA#1, Evi siRNA#3, β-catenin or control. The poly(A)+ fraction was isolated from each of the eight samples and used to probe an IlluminaHumanHT-12 v4 beadchip. These arrays have on average 15 beads per probe and cover more than 47,000 transcripts and known splice variants. The complete data set contained six samples (R2 of all normalized replicates >0.98). Using BeadStudio software (v3.2+), summary intensities for each bead type on the array were produced, and quantile normalization between samples was performed. The limma package (v 3.2.1), part of the Bioconductor package suite, was employed to test for differential expression (Wettenhall & Smyth, 2004). This test assumes a linear model for gene expression levels. The differential expression test between both Evi and control samples is based on the null hypothesis that the expression values of a gene in the samples come from the same distribution, and results in p-values for each gene and sample pair. Specifically, a simple design matrix was formed to fit a linear model to each gene expression value, where the coefficients corresponded to the RNA sources of interest (i.e. siRNA Evi #1, Evi #3 and control). Contrast of interest extracted from the fit where: (1) genes which respond to knockdown using siRNA Evi #1; (2) genes which respond to knockdown using Evi #3; (3) genes which respond similarly in both the knockdowns using siRNA Evi #1 and Evi #3. Data from the latter contrast is presented in this study. An empirical Bayes method was used to moderate standard errors and estimate log fold-change from the data, and a moderated t-statistic was used to assess differential expression. Genes which had an adjusted (Benjamini–Hochberg) p-value < 0.01 with respect to the third contrast listed above were regarded as differentially expressed, and used for further KEGG pathway analysis. Bioconductor GOStats (v2.14), Category (v2.12) and KEGG.db (v2.14) packages were used to perform a Fisher's exact test KEGG categories for over-representation in this gene set. Expression profiling data is available through ArrayExpress (Acc No: E-MTAB-776).
The paper explained
PROBLEM
Malignant gliomas are the most common and most malignant primary brain tumours in adults and associated with poor prognosis. Recent evidence supports the involvement of canonical and non-canonical Wnt signalling in glioma development and malignant progression. However, insights into the mechanism behind Wnt signalling in glioma and the identification of druggable targets that can be addressed to inhibit both signalling branches have been lacking.
RESULTS
We here describe strong, WHO grade-independent overexpression of Evi/Wls/GPR177 in human astrocytic glioma suggesting an involvement of Evi in the earliest stages of glioma tumourigenesis. Evi/GPR177 is an essential Wnt ligand secretion factors. Depletion of Evi expression by RNAi in glioma cells and primary glioblastoma-derived cancer stem-like cells led to reduced proliferation, cell cycle arrest and increased apoptosis. Correspondingly, transcriptome profiling identified a strong transcriptional downregulation of interleukins and genes associated with cell cycle regulation after Evi silencing. Migration experiments revealed a reduced capacity for migration of glioma cells upon Evi silencing and Evi depletion also reduced tumour growth of human glioma cells after xenotransplantation in mice.
IMPACT
Our data established a functional role of Evi/GPR177 in the molecular pathogenesis of human astrocytic gliomas. Evi regulates both canonical and non-canonical Wnt signalling in glioma cells with both branches likely contributing to malignancy. With its GPCR-like structural features Evi may serve an attractive novel target for therapeutic interventions.
Statistical analysis
Unless otherwise indicated, data are expressed as mean ± SD. Statistical significance was calculated by two-tailed Student's t-test with unequal variance. A p-value of less than 0.05 was considered statistically significant and marked by asterisks. Two asterisks represent p-values of less than 0.01. Three asterisks represent p-values of less than 0.001.
Acknowledgments
We thank Thomas Sandmann and Julia Gross for critical comments on the manuscript. We are grateful to Oksana Voloshanenko, Gerrit Erdmann and Christina Falschlehner for helpful advice and Wilfried Roth and Peter Angel for reagents. We thank the NCT tumour bank for tissue samples and the DKFZ Genomics and Proteomics Core Facility for expression profiling experiments. Research in the laboratory of MB was supported by a Marie-Curie Excellence Grant, the DFG Research Group 1036 and SFB873.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
IA and MB devised the concept and planned the experiments; IA, AB, GV and VG performed the experiments; CH, AD and GR provided and processed primary tumour samples and performed histopathological assessments of the tumour tissues; CHM established the SLGC cells and provided samples; BR and GK performed microarray data analysis; IA and MB wrote and edited the manuscript with the contributions from all authors.
For more information
ArrayExpress:
http://www.ebi.ac.uk/arrayexpress/
Rembrandt:
https://caintegrator.nci.nih.gov/rembrandt/
Pubmed:
http://www.ncbi.nlm.nih.gov/pubmed/
GenomeRNAi:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Axelrod JD, Miller JR, Shulman JM, Moon RT, Perrimon N. Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Dev. 1998;12:2610–2622. doi: 10.1101/gad.12.16.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafico A, Liu G, Goldin L, Harris V, Aaronson SA. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell. 2004;6:497–506. doi: 10.1016/j.ccr.2004.09.032. [DOI] [PubMed] [Google Scholar]
- Banziger C, Soldini D, Schutt C, Zipperlen P, Hausmann G, Basler K. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell. 2006;125:509–522. doi: 10.1016/j.cell.2006.02.049. [DOI] [PubMed] [Google Scholar]
- Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- Bartscherer K, Boutros M. Regulation of Wnt protein secretion and its role in gradient formation. EMBO Rep. 2008;9:977–982. doi: 10.1038/embor.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartscherer K, Pelte N, Ingelfinger D, Boutros M. Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell. 2006;125:523–533. doi: 10.1016/j.cell.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Behrens J, Lustig B. The Wnt connection to tumorigenesis. Int J Dev Biol. 2004;48:477–487. doi: 10.1387/ijdb.041815jb. [DOI] [PubMed] [Google Scholar]
- Boutros M, Paricio N, Strutt DI, Mlodzik M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell. 1998;94:109–118. doi: 10.1016/s0092-8674(00)81226-x. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Peifer M. Wnt signaling from development to disease: insights from model systems. Cold Spring Harb Perspect Biol. 2009;1:a002881. doi: 10.1101/cshperspect.a002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos B, Wan F, Farhadi M, Ernst A, Zeppernick F, Tagscherer KE, Ahmadi R, Lohr J, Dictus C, Gdynia G, et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin Cancer Res. 2010;16:2715–2728. doi: 10.1158/1078-0432.CCR-09-1800. [DOI] [PubMed] [Google Scholar]
- Curtin JC, Lorenzi MV. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget. 2010;1:563–577. doi: 10.18632/oncotarget.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Iglesia N, Konopka G, Lim KL, Nutt CL, Bromberg JF, Frank DA, Mischel PS, Louis DN, Bonni A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci. 2008a;28:5870–5878. doi: 10.1523/JNEUROSCI.5385-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008b;22:449–462. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Iglesia N, Puram SV, Bonni A. STAT3 regulation of glioblastoma pathogenesis. Curr Mol Med. 2009;9:580–590. doi: 10.2174/156652409788488739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejmek J, Dejmek A, Safholm A, Sjolander A, Andersson T. Wnt-5a protein expression in primary dukes B colon cancers identifies a subgroup of patients with good prognosis. Cancer Res. 2005;65:9142–9146. doi: 10.1158/0008-5472.CAN-05-1710. [DOI] [PubMed] [Google Scholar]
- Eaton S. Retromer retrieves Wntless. Dev Cell. 2008;14:4–6. doi: 10.1016/j.devcel.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Fu J, Jiang M, Mirando AJ, Yu HM, Hsu W. Reciprocal regulation of Wnt and Gpr177/mouse Wntless is required for embryonic axis formation. Proc Natl Acad Sci USA. 2009;106:18598–18603. doi: 10.1073/pnas.0904894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerer C, Nusse R. Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PLoS ONE. 2010;5:e9370. doi: 10.1371/journal.pone.0009370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells. 2007;27:857–865. doi: 10.1002/stem.23. [DOI] [PubMed] [Google Scholar]
- Goodman RM, Thombre S, Firtina Z, Gray D, Betts D, Roebuck J, Spana EP, Selva EM. Sprinter: a novel transmembrane protein required for Wg secretion and signaling. Development. 2006;133:4901–4911. doi: 10.1242/dev.02674. [DOI] [PubMed] [Google Scholar]
- Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, et al. The molecular basis of Turcot's syndrome. N Engl J Med. 1995;332:839–847. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- Hattermann K, Held-Feindt J, Lucius R, Müerköster SS, Penfold ME, Schall TJ, Mentlein R. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010;70:3299–3308. doi: 10.1158/0008-5472.CAN-09-3642. [DOI] [PubMed] [Google Scholar]
- Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120–129. doi: 10.1038/35052535. [DOI] [PubMed] [Google Scholar]
- Jin J, Morse M, Frey C, Petko J, Levenson R. Expression of GPR177 (Wntless/Evi/Sprinter), a highly conserved Wnt-transport protein, in rat tissues, zebrafish embryos, and cultured human cells. Dev Dyn. 2010;239:2426–2434. doi: 10.1002/dvdy.22369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju R, Cirone P, Lin S, Griesbach H, Slusarski DC, Crews CM. Activation of the planar cell polarity formin DAAM1 leads to inhibition of endothelial cell proliferation, migration, and angiogenesis. Proc Natl Acad Sci USA. 2010;107:6906–6911. doi: 10.1073/pnas.1001075107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamino M, Kishida M, Kibe T, Ikoma K, Iijima M, Hirano H, Tokudome M, Chen L, Koriyama C, Yamada K, et al. Wnt-5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP-2. Cancer Sci. 2011;102:540–548. doi: 10.1111/j.1349-7006.2010.01815.x. [DOI] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- Korkut C, Ataman B, Ramachandran P, Ashley J, Barria R, Gherbesi N, Budnik V. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell. 2009;139:393–404. doi: 10.1016/j.cell.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsas KP, Frangou-Lazaridis M. Surfing on prothymosin alpha proliferation and anti-apoptotic properties. Neoplasma. 2006;53:92–96. [PubMed] [Google Scholar]
- Liang J, Ge F, Guo C, Luo G, Wang X, Han G, Zhang D, Wang J, Li K, Pan Y, et al. Inhibition of PI3K/Akt partially leads to the inhibition of PrP(C)-induced drug resistance in gastric cancer cells. FEBS J. 2009;276:685–694. doi: 10.1111/j.1742-4658.2008.06816.x. [DOI] [PubMed] [Google Scholar]
- Lindvall C, Bu W, Williams BO, Li Y. Wnt signaling, stem cells, and the cellular origin of breast cancer. Stem Cell Rev. 2007;3:157–168. doi: 10.1007/s12015-007-0025-3. [DOI] [PubMed] [Google Scholar]
- Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Liu C, Tu Y, Sun X, Jiang J, Jin X, Bo X, Li Z, Bian A, Wang X, Liu D, et al. Wnt/beta-Catenin pathway in human glioma: expression pattern and clinical/prognostic correlations. Clin Exp Med. 2010a;11:105–112. doi: 10.1007/s10238-010-0110-9. [DOI] [PubMed] [Google Scholar]
- Liu Q, Li G, Li R, Shen J, He Q, Deng L, Zhang C, Zhang J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J Neurooncol. 2010b;100:165–176. doi: 10.1007/s11060-010-0158-0. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang L, Zhao S, Ji X, Luo Y, Ling F. Beta-catenin overexpression in malignant glioma and its role in proliferation and apoptosis in glioblastma cells. Med Oncol. 2010c;28:608–614. doi: 10.1007/s12032-010-9476-5. [DOI] [PubMed] [Google Scholar]
- Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003;129:199–221. doi: 10.1007/s00432-003-0431-0. [DOI] [PubMed] [Google Scholar]
- Madhavan S, Zenklusen JC, Sahni H, Fine HA, Buetow K. Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol Cancer Res. 2009;7:157–167. doi: 10.1158/1541-7786.MCR-08-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SL, Silver A. The opposing roles of Wnt-5a in cancer. Br J Cancer. 2009;101:209–214. doi: 10.1038/sj.bjc.6605174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orend G, Chiquet-Ehrismann R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006;244:143–163. doi: 10.1016/j.canlet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Pu P, Zhang Z, Kang C, Jiang R, Jia Z, Wang G, Jiang H. Downregulation of Wnt2 and beta-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009;16:351–361. doi: 10.1038/cgt.2008.78. [DOI] [PubMed] [Google Scholar]
- Putoczki T, Ernst M. More than a sidekick: the IL-6 family cytokine IL-11 links inflammation to cancer. J Leukoc Biol. 2010;88:1109–1117. doi: 10.1189/jlb.0410226. [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Riemenschneider MJ, Reifenberger G. Astrocytic tumors. Recent Results Cancer Res. 2009;171:3–24. doi: 10.1007/978-3-540-31206-2_1. [DOI] [PubMed] [Google Scholar]
- Saif MW, Chu E. Biology of colorectal cancer. Cancer J. 2010;16:196–201. doi: 10.1097/PPO.0b013e3181e076af. [DOI] [PubMed] [Google Scholar]
- Samaras V, Piperi C, Levidou G, Zisakis A, Kavantzas N, Themistocleous MS, Boviatsis EI, Barbatis C, Lea RW, Kalofoutis A, et al. Analysis of interleukin (IL)-8 expression in human astrocytomas: associations with IL-6, cyclooxygenase-2, vascular endothelial growth factor, and microvessel morphometry. Hum Immunol. 2009;70:391–397. doi: 10.1016/j.humimm.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Sareddy GR, Panigrahi M, Challa S, Mahadevan A, Babu PP. Activation of Wnt/beta-catenin/Tcf signaling pathway in human astrocytomas. Neurochem Int. 2009;55:307–317. doi: 10.1016/j.neuint.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Seifert JR, Mlodzik M. Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat Rev Genet. 2007;8:126–138. doi: 10.1038/nrg2042. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells. Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toedt G, Barbus S, Wolter M, Felsberg J, Tews B, Blond F, Sabel MC, Hofmann S, Becker N, Hartmann C, et al. Molecular signatures classify astrocytic gliomas by IDH1 mutation status. Int J Cancer. 2011;128:1095–1103. doi: 10.1002/ijc.25448. [DOI] [PubMed] [Google Scholar]
- Tysnes BB, Mahesparan R. Biological mechanisms of glioma invasion and potential therapeutic targets. J Neurooncol. 2001;53:129–147. doi: 10.1023/a:1012249216117. [DOI] [PubMed] [Google Scholar]
- van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- Wan F, Zhang S, Xie R, Gao B, Campos B, Herold-Mende C, Lei T. The utility and limitations of neurosphere assay, CD133 immunophenotyping and side population assay in glioma stem cell research. Brain Pathol. 2010;20:877–889. doi: 10.1111/j.1750-3639.2010.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004;20:3705–3706. doi: 10.1093/bioinformatics/bth449. [DOI] [PubMed] [Google Scholar]
- Yu JM, Jun ES, Jung JS, Suh SY, Han JY, Kim JY, Kim KW. Role of Wnt5a in the proliferation of human glioblastoma cells. Cancer Lett. 2007;257:172–181. doi: 10.1016/j.canlet.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Yu HM, Jin Y, Fu J, Hsu W. Expression of Gpr177, a Wnt trafficking regulator, in mouse embryogenesis. Dev Dyn. 2010;239:2102–2109. doi: 10.1002/dvdy.22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.