

Abstract

This article describes the evolution of a Nazarov cyclization-based synthetic strategy targeting the anticancer, antiinflammatory, and insecticidal natural product (±)–rocaglamide. Initial pursuit of a polarized heteroaromatic Nazarov cyclization to construct the congested cyclopentane core revealed an unanticipated electronic bias in the pentadienyl cation. This reactivity was harnessed in a successful second-generation approach using an oxidation-initiated Nazarov cyclization of a heteroaryl alkoxyallene. Full details of these two approaches are given, as well as the characterization of undesired reaction pathways available to the Nazarov cyclization product. A sequence of experiments that led to an understanding of the unexpected reactivity of this key intermediate is described, which culminated in the successful total synthesis of (+)-rocaglamide.
Introduction
Several species of Aglaia, a genus of trees found primarily in the tropical forests of Southeast Asia and on the Pacific Islands, have long been used by native populations for housebuilding, firewood, tea fragrance, moth repellent, and as sources of medicinal substances. In the latter role, their extracts have been valued as heart stimulants and as treatments for fever, inflammation, diarrhea, and injuries.1 In 1982, King and coworkers2 isolated the compound rocaglamide (1a, Figure 1) from the chloroform-soluble fraction of the extract of dried roots and stems of Aglaia elliptifolia. Rocaglamide exhibited substantial antileukemic activity against P388 lymphocytic leukemia in CDF1 mice with optimal T/C values of ca. 156% at a non-toxic dose of 1.0 mg/kg. This natural product was the first of a large series of cyclopenta[b]benzofurans isolated from various Aglaia species over the subsequent years (e.g. 1b–1e; Figure 1), of which many were found to possess powerful bioactive characteristics. The compounds are potent insecticides1a,3 and NF-κB inhibitors,4 and display remarkable anticancer activity. Since King’s report describing the anticancer behavior of rocaglamide, testing of an expanding array of both naturally-occurring and synthetic rocaglamide derivatives in both in vitro and in vivo models has revealed anticancer activity across a broad array of human cancer cell lines.1 Studies on the cellular mechanism of rocaglamide and its derivatives have shown that cyclopenta[b]benzofurans function through both cytostatic and cytotoxic modes of action, which can depend on both the derivative and the cell line.
Figure 1.

Rocaglamide and Analogs.
Rocaglamide itself displays potent and selective cytotoxicity through consistent activation of the p38 MAP kinase pathway along with a long-term suppression of the survival MAP, an extracellular signal-regulated kinase.4d This affects pro-apoptotic bcl-2 proteins, triggering caspase-mediated apoptosis. This effect is highly selective for abnormal lymphocytes over healthy cells. Recently, rocaglamide was found to have a cytostatic effect in T cells infected with HTLV-1 associated T-cell leukemia/lymphoma. This is accomplished through inhibition of the MEK-ERK-MNK1 signaling pathway, thereby inhibiting phosphorylation of eIF4E. This prevents de novo synthesis of c-FLIP, a protein that causes resistance to receptor-mediated apoptosis. In the same cell, upregulation of CD95L was also shown. CD95L is a ligand which when bound to the CD95 receptor, causes apoptosis. Thus, rocaglamide sensitizes cancer cells to treatment with apoptosis-inducing ligands such as CD95L and TRAIL.5,6
The impressive biological profile of rocaglamide and the cyclopenta[b]benzofurans as a whole has stimulated considerable effort toward their synthesis over the past 20 years. The primary challenges in synthesizing them lay in the densely functionalized core. In particular, the fused cyclopentane ring is composed of five stereogenic carbons, of which the two fusion bond carbons are fully substituted. This compact architecture has lent itself to a variety of creative approaches toward its construction.
The first, executed by Trost7 and coworkers in 1990, used a 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)-mediated oxidative cyclization to form the rocaglamide skeleton as a single diastereomer, which after a stereochemical correction and functional group manipulations yielded (−)-rocaglamide. In addition to being the first synthesis of the natural product, it provided confirmation of its absolute stereochemical assignment. An approach pioneered by Taylor8 and Kraus9 in the late 1980’s and early 1990’s took advantage of a samarium (II) iodide-mediated intramolecular pinacol coupling to form the cyclopentane ring. Both quaternary fusion stereocenters were formed in the correct relative configuration. This method provided the basis for several later syntheses of rocaglamide and rocaglamide derivatives.10
More recently, Porco11 developed a [3+2] photocycloaddition to synthesize a cyclopenta[bc]pyran intermediate. This approach was first attempted by Staunton, who obtained incorrect regiochemistry of cycloaddition.12 This skeleton type is proposed1a,13 to be a key intermediate in the biosynthesis of cyclopenta[b]benzofuran and benzo[b]oxepine flavaglines. When subjected to base, this material underwent an α-ketol rearrangement to give a cyclopenta[b]benzofuran intermediate, which was concisely elaborated to rocaglamide. Treatment of the [3+2] product with lead tetraacetate formed the benzo[b]oxepine. This spectacularly concise and novel method of accessing all three flavagline core types through a biomimetic pathway was used in an enantioselective synthesis of rocaglamide,14 rocaglaol,14 silvestrol,15 and a variety of other natural and unnatural rocagamide derivatives.16 In 2009, our group published an annotated study on the total synthesis of rocaglamide which harnessed an oxidation-initiated Nazarov cyclization.17 In addition to the complete syntheses outlined above, a number of other approaches have been used to construct rocaglamide derivatives, including those published by Feldman,18 Umezawa,19 Schoop,20 Ragot,21 Magnus,22 and Moser.23
Results/Discussion
First Generation Synthetic Strategy
The initial strategy we envisioned for the synthesis of Rocaglamide capitalized on a Nazarov cyclization–oxyallyl cation trapping sequence,24,25,26 shown in Scheme 1.
Scheme 1.

First Generation Approach.
We planned to synthesize the requisite Nazarov substrate 3 from benzofuran precursor 2. Lewis acid activation of 3 would generate a pentadienyl cation of type I, which after electrocyclization generates oxyallyl cation of type II. Trapping of this intermediate with water26e would install the tertiary hydroxyl group while retaining the adjacent stereocenters formed via conrotatory Nazarov cyclization. Thus, planar benzofuran 3 would be converted to cyclopentanone 4 in one step, and intermediate 4 has been converted by Trost et al into (±)-rocaglamide.7
Nazarov substrate 3 was appealing as a starting point because of its polarized nature: it was expected to exhibit enhanced reactivity due to the low activation barrier for cyclization observed when one olefin is electron-poor and the other is electron-rich.25 Earlier work in our laboratory27 demonstrated that benzofuran is a competent “vinyl nucleophile” in polarized Nazarov cyclizations (Scheme 2), and we expected the additional electron-donating groups of 3 to further enhance its reactivity.
Scheme 2.

Catalytic Nazarov Cyclization of a Benzofuryl Vinyl Ketone.
To explore this strategy, benzofurans of type 2 (Scheme 1) were required. Vilsmeier formylation of 4,6-dimethoxybenzofurans reportedly occurs at the 7-position,28 so Friedel-Crafts acylation was not a viable strategy for their preparation. Instead, a cyclization/carbomethoxylation strategy was examined (eq 1). Ortho-alkynylphenol derivative 5, prepared via Sonagashira coupling29 of 2-iodo-5-methoxyphenol and phenylacetylene, could be efficiently converted into benzofuran methyl ester 6.30
 |
(1) |
Optimistic that this approach would allow preparation of an appropriately functionalized benzofuran, compounds of type 8 were prepared via the protocol shown in Scheme 2. 2-hydroxy-4,6-dimethoxybenzaldehyde31 was O-alkylated with (2-(chloromethoxy)ethyl)trimethylsilane (SEMCl) or ((chloromethoxy)methyl)benzene (BOMCl) in the presence of DMAP and N,N-diisopropylethylamine in dichloromethane to furnish the corresponding aryl ethers. These compounds were subjected to the Bestmann-Ohira modification32 of the Seyferth-Gilbert homologation33 using dimethyl (1-diazo-2-oxopropyl)phosphonate and K2CO3 in methanol which provided terminal alkynes 7a and 7b. Sonogashira coupling with 1-iodo-4-methoxybenzene furnished internal alkynes 8a and 8b.
Unfortunately, the transformation performed in eq 1 was not effective for electron-rich systems of type 8 after removal of the protecting group. Reports of a related cycloisomerization by Yamamoto34 and Fürstner35 (eq 2) were then explored. Again, electron-rich alkynes 8 did not cyclize, even though these reactions were highly efficient on systems with fewer electron-donating groups.
 |
(2) |
However, we found that modification of Yamamoto's conditions (equation 2, top) to include excess ((chloromethoxy)methyl) benzene induced cyclization of 8b to afford benzofuran 9 in 31% yield (77% conversion) using 10 mol % platinum (II) chloride (Table 1, entry 1). It was later discovered that gold (III) chloride efficiently promoted the reaction (entry 2), but high catalyst loading (25 mole %) was necessary for optimal results, and the reaction did not scale well (entry 3).
Table 1.
Cycloisomerization of BOM-protected Phenol 8b to Benzofuran 9.
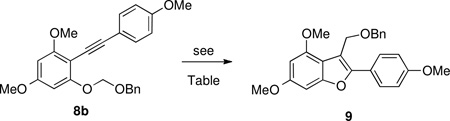 | ||||
|---|---|---|---|---|
| entry | Conditions | Conversion (%) | Yield | |
| 1 | PtCl2 (10 mol %), COD BOMCl, PhCH3 | 77% | 31% | |
| 2 | AuCl3 (25 mol %), COD BOMCl, PhCH3 | 100% | 60% | |
| 3a | AuCl3 (25 mol %), COD BOMCl, PhCH3 | 66% | 45% | |
BOM= benzyloxymethyl
Results upon scaling up by a factor of 50.
Ultimately, Nazarov substrate 3 could be prepared from 2-hydroxy-2-(4-methoxyphenyl)acetonitrile as shown in Scheme 4. Upon stirring with phloroglucinol in hydrogen chloride–saturated diethyl ether, the cyanohydrin undergoes a Hoesch reaction36 which after acidic hydrolysis provided a diphenolic benzofuranone. Dimethyl sulfate methylation of the free hydroxyl groups yielded dimethylated benzofuranone 10. Treatment of 10 with vinyl magnesium bromide in the presence of cerium chloride37 followed by elimination afforded 3-vinylbenzofuran 11. Wacker oxidation38 gave ketone 12. Claisen condensation with dimethylcarbonate gave the β-ketoester 13, and Knoevenagel condensation yielded Nazarov substrate 3 as a 1:3.3 mixture of E/Z isomers.
Scheme 4.

Synthesis of Nazarov Precursor 3.
Despite extensive experimentation, the Nazarov cyclization / trapping sequence could not be realized (Scheme 5). Various catalysts (scandium (III) trifluoromethanesulfonate, aluminum chloride, hydrochloric acid, boron trifluoride diethyl etherate) were examined in the presence of water or sodium acetate, which were intended to serve as nucleophiles capable of trapping the oxyallyl cation.26 No cyclization products were detected under any of the reaction conditions examined; the only transformations observed were efficient isomerization of the E/Z mixture to the Z alkylidene β-ketoester isomer, retro-Knoevenagel condensation, and even retro-Claisen condensation to deliver precursor ketone 12.
Scheme 5.

Subjection of 3 to Lewis Acidic Conditions.
Strategy Revision: Adjusting the Reactivity of the Pentadienyl Cation
Since none of the experiments on alkylidene β-ketoester 3 ever showed any evidence of cyclization chemistry, it was necessary to reevaluate our synthetic strategy. It was apparent that the Lewis acid was activating 3 toward nucleophilic attack by water, as evidenced by the isolation of the retro-Knoevenagel product (Scheme 5), but no cyclization was occurring. A report from the Magnus group in 2005 described conrotatory cyclization of a different system, via ionization of a mixed ketal (Scheme 6),22,39 which led us to view the reactivity of 3 differently.
Scheme 6.

Magnus: Nazarov Cyclization to Access the Rocaglate Core
This result suggests that the C1 terminus (see I; C4=EWG; Scheme 7) of the pentadienyl cation exhibits electrophilic character, which is competent to react with the nucleophilic α-position of the silyl enol ether (i.e., C5, the nucleophilic terminus of the pentadienyl cation). Extrapolating to our alkylidene β-ketoester substrate 3, the pentadienyl cation generated by Lewis acid activation may be more accurately visualized with positive charge localized at the C1 terminus. The carbocation at C1 would be stabilized by both oxygen and the p-methoxybenzyl group (Scheme 7). With an electron-withdrawing ester at C4, C5 is also electrophilic, and the system is not favorably polarized for cyclization. However, polarization of the system would be restored if the ester at C4 were replaced with an electron-donating substituent (see I; C4=EDG), improving both kinetic reactivity and stability of the oxyallyl cation intermediate (II; C4=EDG).
Scheme 7.

Reactivity Paradigm for Electron-Rich Benzofuryl Pentadienyl Cations.
To pursue these ideas, we chose to explore a strategy involving oxidation of a heteroaryl alkoxyallene.40,41,42 Treatment of an alkoxyallene of type 14 with an oxidant would produce the target pentadienyl cation I (C4=O−), either directly or via an intermediate allene oxide (Scheme 8).
Scheme 8.

New Synthetic Design: Generation of Pentadienyl Cation via Oxidation of Alkoxyallene
The reactivity of this pentadienyl cation was expected to be similar to the Magnus system (Scheme 6), leading to cyclization to a diosphenol of type 15. Unlike other methods for generating pentadienyl cations for Nazarov cyclization, Lewis acid would not play a role as promoter or catalyst in either activation of substrate 14 or cyclization of intermediate I. Epoxidation of simple vinyl allenes (Figure 2; X=H) as the first step in a Nazarov cyclization sequence has been described previously,43 and has been identified as part of a biosynthetic pathway in the formation of prostanoids in plants and corals.44 However, no examples of oxidation/cyclization of vinyl alkoxyallenes (Figure 1; X=OR) had been disclosed.45
Figure 2.

Substituted Vinylallenes
In many cases, the isomerization of propargylic ethers to alkoxyallenes is kinetically favored under basic conditions.46 To this end, several propargylic ethers were synthesized according to Scheme 9. Oxidative cleavage of 3-vinylbenzofuran 11 provided aldehyde 16, which was then treated with lithium phenylacetylide or ethynylmagnesium bromide to form the corresponding propargylic alcohols 17a and 17b. O-Alkylation then furnished target propargylic ethers 18a–d.
Scheme 9.

Synthesis of Propargylic Ethers 18a–d.
Contrary to expectations, no isomerization occurred using base-catalyzed or -promoted allene isomerization conditions for 18a (Table 2, Entry 1). For this compound, there are two possible isomers of the allenylanion intermediate (see III and IV, Table 2), and the results indicate that protonation occurs at the propargylic position rather than the allenyl position. However, the lithium anion of 18a or 18b could be trapped with chlorotrimethylsilane or tributyltin chloride to afford the corresponding allenylsilane 19a or allenylstannanes 19b and 19c (Entries 2–4). Allenylsilane 19a was prone to hydrolysis and isolation led to partial decomposition. The selectivity may be attributed to the steric bulk of the electrophile, which reacts at the terminal rather than the benzofuranylic position of the allenyl anion (i.e. IV rather than III). Based on this finding, we examined isomerization of 18c, hoping the steric bulk of the ortho-methoxy group might discourage reprotonation at the propargylic position, but no isomerization was observed (Entry 5). Interestingly, we found that 18d, with no substitution at the alkyne terminus, readily isomerized to the terminal allene 20 efficiently and in high yield in the presence of Triton B® in dimethylsulfoxide47 (Entry 7).
Table 2.
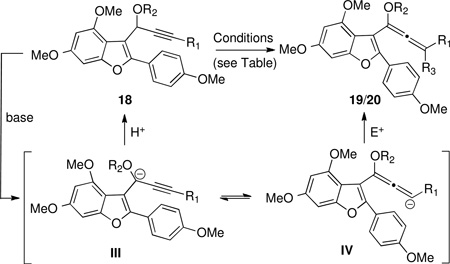 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | R1 | R2a | conditionsb | product (R3) | yield (%) |
| 1 | 18a | Ph | Et | A, B, C, D, E, F | c | – |
| 2 | 18a | Ph | Et | G | 19a (SiMe3) | 40 |
| 3 | 18a | Ph | Et | H | 19b (SnBu3) | 95 |
| 4 | 18b | Ph | PMB | H | 19c (SnBu3) | 94 |
| 5 | 18c | Ph | OMB | I, J, K, L, M | c | – |
| 6 | 18d | H | PMB | N | 20 (H) | ~90 |
PMB=p-methoxybenzyl; OMB=o-methoxybenzyl
Conditions: A) KOt-Bu, MeOH, THF. B) MeLi, THF then MeOH. C) MeLi, THF then ZnCl2 then NH4Cl. D) t-BuLi, THF then imidazole. E) t-BuLi, THF then ZnCl2 then NH4Cl. F) Triton B®, DMSO, 50°C. G) 1.3 equiv. t-BuLi then TMSCI, THF, −30°C. H) t-BuLi, Et2O, −40°C then Bu3SnCl. I) t-BuLi, THF then MeOH. J) t-BuLi, TMEDA, THF then MeOH. K) t-BuLi, TMEDA, THF then t-BuOH. L) t-BuLi, TMEDA, THF then 2,6-di-t-butylphenol. M) t-BuLi, TMEDA, THF then imidazole. N) Triton B® (40% w/w MeOH), DMSO, 50°C
Propargylic ether was isolated unchanged.
When allenylsilane 19a was subjected to either meta-chloroperoxybenzoic acid (m-CPBA) or dimethyldioxirane (DMDO), decomposition occurred. However, allenylstannane 19b underwent the desired Nazarov cyclization in the presence of meta-chloroperoxybenzoic acid with potassium carbonate in dichloromethane/hexanes in 34% yield to give 15a as a single diastereomer (Table 1, entry 1). However, ethyl ester 21 was identified as the major product of the reaction. Solvent polarity, oxidant and reaction acidity were found to impact product ratio (Table 3, entries 2–7). Optimal conditions were found to be m-CPBA (3.5 equiv) in degassed N,N-dimethylformamide at 0°C, providing the desired product 15a in 50% yield, with ester 21 as a minor byproduct (4.3:1 ratio; entry 5).
Table 3.
Optimization of Oxidative Nazarov Cyclization.a
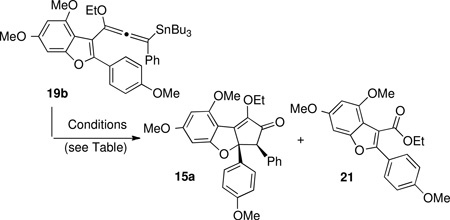 | ||||
|---|---|---|---|---|
| Entry | solvent | oxidant | temp | ratio (15a:21) |
| 1 | DCM / hexane (1:1) | m-CPBA (1 equiv.) | r.t. | 1: 1.2 |
| 2 | toluene | m-CPBA (1 equiv.) | r.t. | 1: 4 |
| 3 | DMF | m-CPBA (1 equiv.) | r.t. | 1.6: 1 |
| 4 | DMF | m-CPBA (1 equiv.) p-TSA (5 mol %) | −10 – 0 °C | 1: 2 |
| 5 | DMF | m-CPBA (3.5 equiv.) | 0 °C | 4.3: 1 |
| 6 | DMF | CF3CO3H | −10 – 0 °C | 1: 6 |
| 7 | sulfolane | m-CPBA (3.5 equiv.) | −10 – 0 °C | 1.2: 1 |
Interestingly, we found that DMDO promoted cyclization of terminal allene 20 even more efficiently, giving a 63% yield of diosphenol 22 over 2 steps from the propargylic ether precursor without any detectable ester byproduct (Scheme 10). Although intermediate 22 lacks the phenyl group of the natural product, it may be possible to synthesize rocaglamide derivatives from this material through α-arylation chemistry.48
Scheme 10.

Oxidation-Initiated Nazarov Cyclization of Terminal Alkoxyallene 20
Proposed Mechanism of Electrocyclization
Since treatment of alkoxyallene 19b with m-CPBA leads to the formation of two products, a mechanistic hypothesis that accounts for this behavior is required. Direct oxidation at the central carbon of the allene 19b or epoxidation to the corresponding allene oxide followed by ring opening would give pentadienyl cation 23 (Scheme 11). Protodestannylation, most likely from the α-stannylketone, would produce pentadienyl cation 24, relieving the steric crowding at the terminal carbons of the pentadienyl cation 23. Conrotatory electrocyclization of the enolate with phenyl situated in an exo position provides 15a, which is the only isomer observed in the product mixture.
Scheme 11.

Proposed Mechanism for Oxidation-Initiated Nazarov Cyclization of 19a
Ester 21 is thought to arise from oxidation at the benzofuranylic carbon, an electron-rich carbon center, to give intermediate hemiketal 25, followed by expulsion of phenylacetylene. Consistent with this proposal, it was also observed that if the stannyl allene were allowed to stand neat, prior to exposure with m-CPBA, it was slowly transformed to ester 21. Therefore, oxidation by adventitious oxygen may also account for the formation of 21, which explains why this reaction pathway is minimized in degassed solvent.
Completion of the Synthesis
Our next objective was to convert intermediate 15a into the natural product, which required installation of a hydroxyl group at the sp2 carbon at the ring junction, conversion of the enol ether into a hydroxy group, and addition of an appropriate carbon unit to the ketone (Scheme 12).
Scheme 12.

Remaining Manipulations to Synthesize Rocaglamide from Intermediate 15a.
Unfortunately, attempts to functionalize 15a triggered an unexpected cleavage of the ring C-O bond (Scheme 13). Thus, exposure of ketone 15a to basic conditions, acidic conditions, nucleophiles (sodium cyanide) or hydride reagents gave ring-opened products, of which 26 and 27 are just two examples.49 In one case, treatment of ketone 15a with 1,4-diazabicyclo[2.2.2]octane (DABCO) led to 26 (Scheme 13), while treatment with lithium triethylborohydride gave an initially unidentified compound 27. We found that 27 could be functionalized with methyl chloroformate to give a carbonate 28a, or with Comin's reagent50 to give a triflate 28b. From 1H and 13C NMR data, we deduced that compounds 27, 28a and 28b had the same carbon skeleton (see supporting information). Then, the mixed ketal structure of 28a was determined by X-ray crystallographic analysis, which led us to propose analogous mixed ketal structures for 27 and 28b, as shown in Scheme 13. It was also found that upon isolation and exposure to air, mixed ketal 27 was converted to pyrone 29.
Scheme 13.

Ring-Opening of Intermediate 15a.
A proposed mechanism for the formation of reduction product 27 is shown in Scheme 14. Stereoselective borohydride reduction of 15a, due to the overwhelming steric influence of the aryl groups,51 would lead to enol ether 30. Once the enol ether is no longer in conjugation with the ketone, it would be destabilized and may collapse to expel the phenolate oxygen, leading to intermediate 31. Intramolecular attack by the phenolate, under neutral, anhydrous conditions, would then form mixed ketal 27, as a single diastereomer. This process could also be viewed as a concerted [1,3]-shift of the oxygen, converting 30 to 27 directly, without opening the ring.
Scheme 14.

Proposed Mechanism for Formation of Hydride Reduction Product 27.
Upon standing overnight, or rapidly in the presence of acid, mixed ketal 27 is converted to pyrone 29 through a sequence involving a net oxidation. One possible mechanism involves hydrolysis of 27 to give ketone 32, followed by oxidation at the α-position of the enol form by adventitious oxygen of 32 (see 33, Scheme 15).52 Fragmentation of hydroperoxide 34 would produce ketene 35, which is set up to undergo 6π electrocyclization to give the observed pyrone 29.
Scheme 15.

Proposed Mechanism for Formation of Pyrone 29.
After finding that our preferred approaches for functionalization of ketone 15a led to cleavage of the ring C-O bond, we sought reaction conditions that would not generate an enolate at the ketone α-position or activate the tetrahydrofuran oxygen, yet would still deliver a useful synthetic intermediate. Thus, we explored oxidation protocols for installing the hydroxyl group at C8b (see Scheme 12). Oxidation of 15a did not occur upon treatment with DMDO, while oxidation with ceric ammonium nitrate or DDQ led to a complex mixture of products. The analogous para-methoxybenzyl ether 15b was prepared from 19c (Scheme 16). We hypothesized that while oxidation of 15a would produce reactive intermediate 36a, oxidation of 15b might afford neutral intermediate 36b (Figure 3) Indeed, treatment of 15b with DDQ effected both cleavage of the PMB group and oxidation at carbon 8b, and diosphenol 37 was isolated in 71% yield.
Scheme 16.

Synthesis and DDQ Oxidation of 15b.
Figure 3.

Putative Intermediates Generated upon Oxidation of 15a and 15b
Treatment of 37 with N-(2-pyridyl)bis(trifluoromethanesulfonimide) provided triflate 38, and palladium-catalyzed carbonylation in the presence of methanol installed the ester to give 39 (Scheme 17). This completed assembly of the carbon skeleton of the natural product.
Scheme 17.

Installation of the α-Carbomethoxy Group.
Compound 39 could be transformed directly to β-ketoester 40 via hydrogenation in the presence of PtO2. However, purification of 40 obtained from the hydrogenation was difficult and reaction results were erratic. More reliable outcomes were obtained if hydrogenation was conducted on the trimethylsilyl ether 41, followed by deprotection of β-ketoester 42 to give 40, consistent with the findings of Trost7 (Scheme 18).
Scheme 18.

Preparation of β-Ketoester 40 via Hydrogenation of Alkylidene β-Ketoester 39.
Templated reduction with sodium triacetoxyborohydride yielded aglafolin (1e) as a single diastereomer (Scheme 19). Saponification of aglafolin gave rocagloic acid (1d) in 82% yield, which after dicyclohexylcarbodiimide-mediated amide coupling yielded (±)–rocaglamide (1a) in 60% yield. All spectral data were identical to literature values for the natural product.10c
Scheme 19.

Preparation of Natural Products Aglafolin, Rocagloic Acid and Rocaglamide from β-ketoester 40.
In summary, the total synthesis of (±)–rocaglamide was achieved starting from benzofuranone 10. The key step was oxidation-triggered Nazarov cyclization of the highly functionalized alkoxyallene 19c to give 15b (Scheme 20). The adjacent stereocenters of the rocaglamide system were created via conrotatory electrocyclization. However, due to an unexpected, irreversible ring-opening, the Nazarov cyclization product could not be functionalized using either acidic or basic conditions. Ultimately, the requisite tertiary hydroxyl group was installed by oxidation of 15b under neutral conditions, which allowed completion of the synthesis via Trost's intermediate 39. Ongoing work in the laboratory is focused on developing an enantioselective strategy for the synthesis of rocaglamide and its congeners, and investigation of the antiproliferative activity of this natural product class.
Scheme 20.

Summary: Total Synthesis of Rocaglamide.
Experimental Section
General Methods
Reactions were carried out in oven- or flame-dried glassware under argon with magnetic stirring unless otherwise specified. Dry solvents were obtained from a solvent purification system. Reagents were used as received unless otherwise specified. n-Butyllithium was titrated periodically with diphenylacetic acid. Thin layer chromatography was visualized with p-anisaldehyde/heat, potassium permanganate/heat, vanillin/heat, or a UV lamp. 1H NMR and 13C NMR spectroscopy were performed at ambient temperature. Dimethyldioxirane (DMDO) was prepared according to the procedure of Adam,53 and the concentration assayed by preparing an NMR sample containing 5 µL thioanisole and 50 µL DMDO solution and comparing relative peak areas of the aromatic protons of thioanisole and its corresponding sulfoxide.
5-Methoxy-2-(2-phenylethynyl)phenol (5)
In dry diethylamine (15 mL), sparged with argon for 10 min., 2-iodo-5-methoxyphenol (0.50 g, 2.0 mmol) was suspended. To the suspension, trans-dichlorobis(triphenylphosphine)palladium (II) (7.0 mg, 0.0099 mmol) and copper (I) iodide (3.8 mg, 0.020 mmol) were added. Phenylacetylene (0.25 g, 2.40 mmol) was added via syringe and the reaction was allowed to stir for 12 h at room temperature. The reaction was then warmed to 45°C and stirred for an additional 1.5 h. The reaction was allowed to cool, whereupon the solvent was removed in vacuo. The residue was taken up in dichloromethane (15 mL), washed with water (15 mL), the organic layer was dried over Na2SO4, then filtered over an alumina plug and concentrated. If a solid was isolated, it was triturated with hexanes and vacuum filtered to afford 5-methoxy-2-(2-phenylethynyl)phenol 5 (260 mg, 59%) as a gray powder, or the pure material was obtained by flash chromatography using hexane/ethyl acetate (6:1). IR (neat) cm−1 3450, 2220, 1616, 1504, 1245, 1145, 1022. 1H NMR (400 MHz, CDCl3) δ7.61–7.57 (2H, m), 7.47–7.37 (4H, m), 6.55 (1H, d, J = 2.4 Hz), 6.50 (1H, dd, J = 8.6, 2.4 Hz), 5.87 (1H, s, broad), 3.86 (3H, s). 13C NMR (125 MHz, CDCl3) δ161.7, 158.0, 132.5, 131.5, 128.5, 122.8, 107.5, 102.1, 100.3, 95.3, 83.3, 55.5. HRMS m/z calc. for C15H12O2: [M+H+] 224.0832, found 224.0839.
Methyl 6-methoxy-2-phenylbenzofuran-3-carboxylate (6)
Alkynylphenol 5 (0.300 g, 1.34 mmol) was dissolved in a mixture of 7:3 methanol/tetrahydrofuran (85 mL). The solution was sparged with carbon monoxide for 20 min. To the solution palladium (II) iodide (48 mg, 0.13 mmol), thiourea (10 mg, 0.13 mmol), cesium carbonate (3.05 g, 9.37 mmol), and carbon tetrabromide (3.11 g, 9.37 mmol) were added. The flask was sealed and stirred under 1 atm of carbon monoxide at 50°C for 5 h. The reaction was allowed to cool and concentrated in vacuo. The residue was extracted with dichloromethane (2 × 20 mL), washed with brine, and the organic layer was dried over Na2SO4 and passed through a silica plug. The filtrate was concentrated and the residue subjected to flash column chromatography on silica gel using 2:1 dichloromethane/hexanes to afford methyl 6-methoxy-2-phenylbenzofuran-3-carboxylate 6 (290 mg, 77%) as a white solid. IR (neat) cm−1 3063, 2999, 2950, 2836, 1716, 1622, 1492, 1278, 1225, 1150, 1115, 1089, 767, 690. 1H NMR (400 MHz, CDCl3) δ8.02 (d, J = 8.0 Hz, 2H), 7.92 (d, J = 8.7 Hz, 1H), 7.50–7.46 (3H, m), 7.07 (d, J = 2.2 Hz, 1H), 6.99 (dd, J = 8.7, 2.2 Hz, 1H), 3.94 (3H, s), 3.90 (3H, s). 13C NMR (100 MHz, CDCl3) δ164.6, 159.9, 158.6, 154.8, 130.0, 129.3, 128.6, 128.2, 123.0, 120.3, 113.1, 108.7, 95.5, 55.8, 51.6. HRMS m/z calc. for C17H14O4Na [M+Na+] 305.0784, found 305.0784.
(2-((2-ethynyl-3,5-dimethoxyphenoxy)methoxy)ethyl)trimethylsilane (7a)
To a solution of 2-hydroxy-4,6-dimethoxybenzaldehyde31 (1.00 g, 5.50 mmol), dry N,N-diisopropylethylamine (1.15 mL, 6.60 mmol), and 4-(N,N-dimethylamino)pyridine (732 mg, 0.550 mmol) in dry dichloromethane (18.3 mL) was added (2-(chloromethoxy)ethyl)trimethylsilane (1.05 mL, 6.00 mmol) slowly. The reaction was stirred at 40°C for 3.5 h, then N,N-diisopropylethylamine (1.91 mL, 11.0 mmol) and (2-(chloromethoxy)ethyl)trimethylsilane (1.94 mL, 11.0 mmol) were added. After refluxing for 1.25 h, the reaction was diluted with dichloromethane (150 mL). The solution was washed with a 10% w/w aqueous solution of copper (II) sulfate (3 × 100 mL), brine (40 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo to afford a residue which was purified by flash chromatography on silica gel (90:10-50:50 hexanes/ethyl acetate) to afford 2,4-dimethoxy-6-((2-(trimethylsilyl)ethoxy)methoxy)benzaldehyde (1.09 g, 63%).
The aldehyde from the previous reaction (1.09 g, 3.50 mmol) was dissolved in methanol (12 mL) and potassium carbonate (1.20 g, 8.75 mmol) added. After cooling to 0 °C, a solution of dimethyl (1-diazo-2-oxopropyl)phosphonate (1.34 g, 6.98 mmol) in methanol (5.0 mL) was added slowly. The resulting slurry was stirred to room temperature for 18 h at which time it was filtered and concentrated in vacuo. The resulting oil was dissolved in ethyl acetate (130 mL) and washed with saturated aqueous ammonium chloride (3 × 70 mL), brine (50 mL), dried over anhydrous MgSO4 and concentrated in vacuo to provide an oil which was purified by flash chromatography on silica gel (99:1-85:15 hexanes/ethyl acetate) to afford (2-((2-ethynyl-3,5-dimethoxyphenoxy)methoxy)ethyl)trimethylsilane 7a (430 mg, 41%). 1H NMR (400 MHz, CDCl3) δ6.40 (d, J = 2.2 Hz, 1H), 6.16 (d, J = 2.2 Hz, 1H), 5.31 (s, 2H), 3.89 (s, 3H), 3.85–3.80 (m, 5H), 3.47 (s, 1H), 1.00–0.94 (m, 2H), 0.08–−0.03 (s, 10H).
(2-((3,5-dimethoxy-2-((4-methoxyphenyl)ethynyl)phenoxy)methoxy)ethyl)trimethylsilane (8a)
To an argon-sparged solution of alkyne 7a (430 mg, 1.43 mmol), 4-iodoanisole (257 mg, 1.10 mmol), dry triethylamine (0.46 mL, 3.30 mmol), and copper (I) iodide (54 mg, 0.29 mmol) in tetrahydrofuran (11 mL) was added tetrakis(triphenylphosphine) palladium (0) (165 mg, 0.14 mmol). The slurry was stirred at room temperature for 14 h, then concentrated in vacuo to remove most of the tetrahydrofuran. The residue was dissolved in ethyl acetate (100 mL), washed with a 10% w/w aqueous solution of copper (II) sulfate (3 × 75 mL), brine (60 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo to afford a residue which was purified by flash chromatography on silica gel (98:2-86:14 hexanes/ethyl acetate) to afford (2-((3,5-dimethoxy-2-((4-methoxyphenyl)ethynyl)phenoxy)methoxy)ethyl)trimethylsilane 8a (141 mg, 30%) along with starting terminal alkyne (191 mg, 44% recovery). IR (neat) cm−1 2935, 2829, 1608, 1513, 1245, 1161, 1122, 1074, 1008, 830. 1H NMR (400 MHz, CDCl3) δ7.49 (d, J = 8.9 Hz, 1H), 6.86 (d, J = 8.9 Hz, 1H), 6.39 (d, J = 2.2 Hz, 1H), 6.18 (d, J = 2.2 Hz, 1H), 5.31 (s, 2H), 3.89 (s, 3H), 3.84–3.70 (m, 2H), 3.65 (s, 6H), 1.03–0.94 (m, 2H), 0.00 (s, 9H). 13C NMR (100 MHz, CDCl3) δ161.8, 161.1, 160.17, 159.2, 133.0, 116.6, 113.8, 96.5, 96.0, 94.1, 93.7, 92.3, 80.5, 66.5, 56.1, 55.4, 55.3, 18.1, −1.4. HRMS m/z calc. for C23H3105Si [M+H+] 415.1941, found 415.1956.
1-((benzyloxy)methoxy)-2-ethynyl-3,5-dimethoxybenzene (7b)
To a solution of 2-hydroxy-4,6-dimethoxybenzaldehyde31 (6.76 g, 37.0 mmol), dry N,N-diisopropylethylamine (19.4 mL, 111 mmol), and 4-(N,N-dimethylamino)pyridine (451 mg, 3.70 mmol) in dry dichloromethane (221 mL) was added ((chloromethoxy)methyl)benzene (15.5 mL, 111 mmol) slowly. The reaction was stirred at room temperature for 12 h, then N,N-diisopropylethylamine (6.47 mL, 37.1 mmol) and ((chloromethoxy)methyl)benzene (5.15 mL, 37.1 mmol) were added. After refluxing for 1 h, the reaction was diluted with dichloromethane (250 mL). The solution was washed with a 10% w/w aqueous solution of copper (II) sulfate (3 × 200 mL), brine (70 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo to afford a residue which was purified by flash chromatography on silica gel (90:10-50:50 hexanes/ethyl acetate) to afford 2-((benzyloxy)methoxy)-4,6-dimethoxybenzaldehyde (7.3 g, 65%). 1H NMR (400 MHz, CDCl3) δ10.44 (s, 1H), 7.45–7.32 (m, 5H), 6.46 (d, J = 2.0 Hz, 1H), 6.20 (d, J = 2.0 Hz, 1H), 5.43 (s, 2H), 4.81 (s, 2H), 3.94 (s, 3H), 3.89 (s, 3H).
The aldehyde from the previous reaction (217 mg, 0.71 mmol) was dissolved in methanol (2.2 mL) and potassium carbonate (215 mg, 1.56 mmol) added. After cooling to 0°C, a solution of dimethyl (1-diazo-2-oxopropyl)phosphonate (230 mg, 1.28 mmol) in methanol (1.3 mL) was added slowly. The resulting slurry was stirred to room temperature over 14 h at which time it was filtered and concentrated in vacuo. The resulting oil was dissolved in ethyl acetate (70 mL) and washed with saturated aqueous ammonium chloride (3 × 70 mL), brine (50 mL), dried over anhydrous MgSO4 and concentrated in vacuo to provide an oil which was was purified by flash chromatography on silica gel (96:4-85:15 hexanes/ethyl acetate) to afford 1-((benzyloxy)methoxy)-2-ethynyl-3,5-dimethoxybenzene 7b (93 mg, 43%). 1H NMR (400 MHz, CDCl3) δ7.40–7.27 (m, 5H), 6.42 (d, J = 2.1 Hz, 1H), 6.15 (d, J = 2.1 Hz, 1H), 5.36 (s, 2H), 4.76 (s, 2H), 3.87 (s, 3H), 3.78 (s, 3H), 3.48 (s, 1H).
1-((benzyloxy)methoxy)-3,5-dimethoxy-2-((4-methoxyphenyl)ethynyl)benzene (8b)
To an argon-sparged solution of alkyne 7b (1.65 g, 5.50 mmol), 4-iodoanisole (983 mg, 4.20 mmol), dry triethylamine (1.76 mL, 12.6 mmol), and copper (I) iodide (160 mg, 0.84 mmol) in tetrahydrofuran (42 mL) was added tetrakis(triphenylphosphine) palladium (0) (485 mg, 0.42 mmol). The slurry was stirred at room temperature for 12 h, then concentrated in vacuo to remove most of the tetrahydrofuran. The residue was dissolved in ethyl acetate (200 mL), washed with a 10% w/w aqueous solution of copper (II) sulfate (3 × 150 mL), brine (90 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo to afford a residue which was purified by flash chromatography on silica gel (98:2-75:25 hexanes/ethyl acetate) to afford 1-((benzyloxy)methoxy)-3,5-dimethoxy-2-((4-methoxyphenyl)ethynyl)benzene 8b (1.5 g, 67%) along with starting alkyne 7 (172 mg, 10% recovery). IR (neat) cm−1 3000, 2950, 2840, 1608, 1575, 1512, 1454, 1335, 1245, 1160, 1065, 1121, 831. 1H NMR (400 MHz, CDCl3) δ7.49 (d, J = 8.7 Hz, 2H), 7.38–7.28 (m, 5H), 6.85 (d, J = 8.7 Hz, 2H), 6.41 (d, J = 2.1 Hz, 1H), 6.18 (d, J = 2.1 Hz, 1H), 5.36 (s, 2H), 4.80 (s, 2H), 3.88 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H). 13C NMR (100 MHz, CDCl3) δ161.8, 161.2, 159.9, 159.2, 137.1, 133.0, 128.5, 128.3, 127.9, 116.4, 113.8, 96.5, 96.2, 94.1, 92.9, 92.5, 80.5, 70.1, 56.1, 55.5, 55.3. HRMS m/z calc. for C25H2505 [M+H+] 405.1702, found 405.1712.
3-((benzyloxy)methyl)-4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran (9)
To a solution of alkyne 8b (1.5 g, 3.7 mmol), ((chloromethoxy)methyl)benzene (0.1 mL, 0.74 mmol), and 1,5-cyclooctadiene (0.36 mL, 3.0 mmol) in toluene (37 mL) was added gold (III) chloride (280 mg, 0.93 mmol). The reaction was stirred for 16 h then filtered through a short plug of silica gel eluting with dichloromethane (150 mL) and ethyl acetate (150 mL). The filtrate was concentrated in vacuo to provide a residue which was purified by flash chromatography on silica gel (98:2-82:18 hexanes/ethyl acetate) to afford 3-((benzyloxy)methyl)-4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran 9 (680 mg, 45%) along with starting alkyne 8b (523 mg, 34% recovery). IR (neat) cm−1 2928, 2914, 2840, 2178, 2161, 1597, 1458, 1249, 1072, 1015, 821. 1H NMR (400 MHz, CDCl3) δ7.32–7.26 (m, 2H), 7.17 (d, J = 8.8 Hz, 2H), 6.95–6.93 (m, 3H), 6.64 (d, J = 8.8 Hz, 2H), 6.43 (d, J = 2.1 Hz, 1H), 6.21 (d, J = 2.1 Hz, 1H), 5.24 (d, J = 7.1 Hz, 1H), 5.14 (d, J = 7.1 Hz, 1H), 4.43 (d, J = 11.8 Hz, 1H), 4.35 (d, J = 11.8 Hz, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 3.65 (s, 3H). 13C NMR (100 MHz, CDCl3) δ162.1, 158.7, 155.6, 137.1, 131.4, 128.9, 128.4, 128.3, 127.9, 127.7, 124.2, 113.5, 109.5, 93.1, 92.4, 91.7, 77.2, 69.5, 56.1, 55.4, 55.0. HRMS m/z calc. for C25H2505 [M+H+] 405.1702, found 405.1703.
4,6-Dimethoxy-2-(4-methoxyphenyl)benzofuran-3(2H)-one (10)
Potassium cyanide (57.4 g, 881 mmol) was added to a stirring mixture of distilled water (200 mL) and ethyl acetate (230 mL). Once dissolved, sodium bisulfite (NaHSO3, 91.7 g, 881 mmol) was added and stirring continued. Note: The order of addition is important, addition of KCN to a solution of NaHSO3 results in a violent reaction. To this mixture, freshly distilled p-anisaldehyde was added (17.9 mL, 147 mmol), the flask was then sealed with an outlet and stirred vigorously for 20 h. The reaction was poured into 300 mL distilled water, washed with distilled water (3 × 200 mL), then brine. The organic layer was dried over MgSO4 and concentrated. The residue was dissolved in 150 mL of toluene and ~30 mL hexanes were added with swirling. The solution was allowed to stand, whereupon crystallization occurred. The product was filtered, washed with hexane and dried in vacuo to give 2-hydroxy-2-(4-methoxyphenyl)acetonitrile (23.7 g, 99%) 1H NMR (400 MHz, CDCl3) δ7.46 (d, J = 9.0 Hz, 2H), 6.96 (d, J = 9.0 Hz, 2H), 4.48 (d, J = 7.0Hz, 1H), 3.84 (s, 3H), 2.74 (d, J = 7.0 Hz, 1H).
To a 3-necked flask containing dry diethyl ether (440 mL) was added 2-hydroxy-2-(4-methoxyphenyl)acetonitrile (26.6 g, 163 mmol) and phloroglucinol (20.6 g, 163 mmol, dried in vacuo 1h @ 35 in. Hg, 100°C). The flask was cooled to 0°C with mechanical stirring. Dry HCl (g) was bubbled through slowly for 1 h. After ~30 min a precipitate developed which could range in appearance from orange and sticky to off-white and free flowing. Once a full hour of gas flow had elapsed, the gas was stopped, the precipitate was scraped from the walls of the reaction vessel and the flask refrigerated for 3 days. Once removed from the cold, the supernatant was vacuum suctioned from the flask, and replaced with fresh diethyl ether. The precipitate was triturated in this same manner several times with diethyl ether (3 × 300 mL). To remove the residual ether that could not be removed by vacuum suctioning, the flask was connected to a vacuum pump directly and warmed until the precipitate was freely flowing. At this point the trituration with ether was repeated followed by and additional evacuation on a pump. The resulting powder was then dissolved in 0.1 M HCl (aq.) (356 mL) and heated to 112°C with a reflux condenser in place. The solution was refluxed for ~3 h, at which time a precipitate formed. The reaction was cooled, the precipitate was filtered, washed with distilled water and dried in vacuo overnight to yield the di-phenolic benzofuranone (11.25 g, 25%). 1H NMR (500 MHz, CD3OD) δ7.23 (d, J = 9.0 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 6.03 (d, J = 2.0 Hz, 1H), 5.94 (d, J = 2.0 Hz, 1H), 5.45 (s, 1H), 3.79 (s, 3H).
Once dry, the product of the above reaction was dissolved in degassed acetone (300 mL, degassed by five vacuum/argon cycles under stirring). This solution was cannulated to a suspension of dimethyl sulfate (23.5 mL, 248 mmol) and potassium carbonate (34.3 g, 248 mmol) in 150 mL of degassed acetone and the mixture was refluxed under argon for 30 min. After this time, methanol (200 mL) was added and the mixture was filtered (to remove the remaining solids) and concentrated. The resulting solid was filtered, washed with 100 mL of methanol and dried in vacuo overnight to give 4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3(2H)-one 10 (8.60 g, 69%) as a clear brown powder. 1H NMR (500 MHz, CDCl3) δ7.31 (d, J = 9.0 Hz, 2H), 6.89 (d, J = 9.0 Hz, 2H), 6.22 (d, J = 2.0 Hz, 1H), 6.04 (d, J = 2.0 Hz, 1H), 5.43 (s, 1H), 3.90 (s, 3H), 3.89 (s, 3H), 3.79 (s, 3H); (lit.8b 1H NMR).
4,6-Dimethoxy-2-(4-methoxyphenyl)-3-vinylbenzofuran (11)
To a stirring solution of benzofuranone 10 (10.3 g, 34.4 mmol) in dry tetrahydrofuran (300 mL) at room temperature, vinyl magnesium bromide (100 mL 1M soln. in tetrahydrofuran, 100 mmol) was added rapidly via cannula. The reaction was stirred ~15 min., until no starting material remained as judged by TLC. The reaction was quenched with a saturated aqueous solution of ammonium chloride and made slightly acidic by the addition of 1N HCl (aq.). The mixture was poured into distilled water/ethyl acetate (100 mL/250 mL) and extracted. The aqueous phase was extracted with ethyl acetate (100 mL) and the combined organic extracts were washed successively with distilled water and brine. The organic layer was dried over MgSO4, filtered and concentrated. The crude material was re-dissolved in toluene (200 mL) and para-toluenesulfonic acid (5 mg) was added. The reaction was stirred briefly (no more than 7 min.), until most material was converted to the least polar product by TLC (Rf ~0.8, 80:20 hexane/ethyl acetate, blue under UV). The reaction was filtered over a silica plug washing with ethyl acetate and concentrated. The resulting residue was submitted to flash column chromatography (75:25 hexane/ethyl acetate) to give the desired 4,6-dimethoxy-2-(4-methoxyphenyl)-3-vinylbenzofuran 11 (7.11 g, 67%) as a white solid. Note: Pre-stirring the ketone at room temperature for 2 h with 1.5 equivalents of anhydrous cerium (III) chloride made it possible to decrease the amount of Grignard reagent to 1.5 equivalents, but did not increase the yield of the reaction.
IR (neat) cm−1 2997, 2953, 2938, 2905, 2835, 1618, 1591, 1501, 1250, 1148, 1113, 1103. 1H NMR (500 MHz, CDCl3) δ7.70 (d, J = 8.5 Hz, 2H), 6.99–6.95 (m, 3H), 6.65 (d, J = 2.0 Hz, 1H), 6.35 (d, J = 2.0Hz, 1H), 5.99 (dd, J = 17.5 Hz, 2.0 Hz, 1H), 5.40 (dd, J = 11.5 Hz, 2.0 Hz, 1H), 3.91 (s, 3H), 3.86 (s, 6H). 13C NMR (125 MHz, CDCl3) δ159.5, 158.9, 156.1, 154.6, 150.7, 129.2, 127.7, 123.9, 118.3, 114.3, 114.0, 111.6, 94.5, 88.0, 55.7, 55.4, 55.3. HRMS m/z calc. for C19H18O4 [M+] 310.1200, found 310.1196.
1-(4,6-Dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)methylketone (12)
In a round bottom flask, vinyl benzofuran 11 (0.139 g, 0.447 mmol) was dissolved in 10 mL of a 7:1 mixture of N,N-dimethylformamide and distilled water. To this solution, palladium (II) chloride (7.9 mg, 0.045 mmol) and copper (I) chloride (49 mg, 0.049 mmol) were added. The reaction was placed under an oxygen atmosphere and warmed to 65°C. After 4 h at this temperature the reaction was cooled, filtered over celite (ethyl acetate), and poured into distilled water. The aqueous layer was extracted with ethyl acetate (3 × 25 mL) and the combined organic extracts were washed with distilled water, brine and then dried over MgSO4. After filtration, the filtrate was concentrated and the resulting residue was submitted to flash column chromatography (80:20 hexanes/ethyl acetate) to give 1-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)methylketone 12 (135 mg, 93%). IR (neat) cm−1 3012, 2961, 2947, 2838, 1689, 1608, 1501, 1463, 1453, 1414, 1251, 1217, 1148, 1110. 1H NMR (400 MHz, CDCl3) δ7.72 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 6.60 (d, J = 1.6 Hz, 1H), 6.30 (d, J = 1.6 Hz, 1H), 3.81 (s, 3H), 3.77 (s, 3H), 3.76 (s, 3H), 2.57 (s, 3H). 13C NMR (100 MHz, CDCl3) δ199.8, 160.3, 159.6, 155.2, 153.3, 151.6, 128.7, 122.4, 117.1, 113.9, 110.6, 94.8, 88.0, 55.6, 55.5, 55.2, 32.3. HRMS m/z calc. for C19H19O5 [M+H+] 327.1227, found 327.1230.
Methyl 3-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)-3-oxopropanoate (13)
In a 2-neck round bottom flask outfitted with a Liebig condenser, sodium hydride (60% in mineral oil, 195 mg, 4.88 mmol) and potassium hydride (30% in mineral oil, 653 mg, 4.88 mmol) were washed first with hexanes (3 mL), then with dry tetrahydrofuran (3 mL) under argon. The hydride reagents were suspended in dry tetrahydrofuran (50 mL), dimethyl carbonate (1.12 mL, 13.3 mmol) was added via syringe and the flask was warmed to 65°C. Once at that temperature, ketone 12 (1.45 g, 4.44 mmol), dissolved in dry tetrahydrofuran (15 mL), was added slowly. The reaction was refluxed until the starting ketone was consumed, cooled to room temperature and quenched with a saturated aqueous solution of ammonium chloride. The reaction was made acidic by the addition of 1N HCl (aq.) and poured into distilled water (100 mL). The aqueous phase was extracted with ethyl acetate (2 × 30 mL), the combined organic extracts were washed with distilled water, brine and then dried over MgSO4. Filtration of the organic phase, followed by concentration and chromatography (silica gel, 85:15 hexanes/ethyl acetate) gave methyl 3-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)-3-oxopropanoate 13 (1.13 g, 66%). IR (neat) cm−1 3010, 2955, 2838, 1738, 1667, 1608, 1501. 1H NMR (400 MHz, CDCl3) δ7.84 (d, J = 8.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 6.68 (d, J = 2.0 Hz, 1H), 6.38 (d, J = 2.0 Hz, 1H), 4.04 (s, 2H), 3.91 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 3.66 (s, 3H). 13C NMR (100 MHz, CDCl3) δ193.1, 168.0, 160.6, 159.7, 155.2, 153.8, 153.1, 129.2, 122.0, 115.8, 113.9, 110.0, 95.0, 88.1, 55.7, 55.3, 52.1, 50.2.
Methyl 2-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-carbonyl)-3-phenylacrylate (3)
β-Ketoester 13 (1.13 g, 2.93 mmol) was dissolved in benzene (20 mL). To the solution, glacial acetic acid (0.08 mL, 1.3 mmol), piperidine (0.03 mL, 0.3 mmol), and benzaldehyde (0.31 mL, 3.1 mmol) were added. The flask was fitted with a Dean-Stark trap and Liebig condenser and the reaction was heated to 110°C. Once the reaction finished (as determined by TLC) the flask was cooled, the reaction was poured into distilled water (50 mL), diluted with ethyl acetate (50 ml) and extracted (2 × 25 mL). The organic extract was washed with 1N HCl (150 mL), a saturated aqueous solution of sodium bicarbonate (200 mL), and brine. The organic layer was dried over Na2SO4, filtered and concentrated. The resulting residue was subjected to flash chromatography (80:20 hexane/ethyl acetate) to yield methyl 2-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-carbonyl)-3-phenylacrylate 3 (1.24 g, 89%) as a 1 : 3.3 mixture of E/Z isomers. IR (neat) cm−1 3006, 2949, 2837, 1729, 1651, 1615, 1502. 1H NMR (400 MHz, CDCl3) δ7.90 (d, J = 8.8 Hz, 2H, E), 7.79 (s, 1H, E), 7.74 (d, J = 9.2 Hz, 2H, Z), 7.37 (s, 1H, Z), 7.37–7.31 (m, 4H, Z + 3H, E), 7.15–7.09 (m, 1H, E), 6.98–6.92 (m, 3H, Z + 2H, E), 6.70 (d, J = 2.0 Hz, 1H, Z), 6.50 (d, J = 2.0 Hz, 1H, E), 6.34–6.28 (m, 1H, E + Z), 3.90 (s, 3H, Z), 3.89 (s, 3H, E), 3.87 (s, 3H, Z), 3.86 (s, 3H, E), 3.82 (s, 3H, Z), 3.80 (s, 3H, E + Z), 3.78 (s, 3H, E). 13C NMR (100 MHz, CDCl3) major isomer δ191.1, 167.7, 160.2, 159.7, 155.1, 153.6, 152.3, 144.4, 136.1, 133.1, 131.2, 130.7, 129.4, 128.8, 128.1, 122.1, 114.2, 112.4, 94.9, 87.9, 55.8, 55.7, 55.3, 52.6. HRMS m/z calc. for C28H25O7 [M+H+] 473.1595, found 473.1596.
4,6-Dimethoxy-2-(4-methoxyphenyl)benzofuran-3-carboxaldehyde (16)
To a stirring solution of vinyl benzofuran 11 (5.00 g, 16.1 mmol) in acetone (240 mL), t-butanol (35 mL) and distilled water (12 mL) were added and the reaction vessel was flushed with argon. To this solution N-methylmorpholine-N-oxide (3.60 g, 20.5 mmol) was added followed by osmium tetroxide (2.4 mL of a 4% solution in H2O, 0.4 mmol). The reaction was stirred at room temperature for ~3 h, until the starting material was consumed. The reaction was concentrated to an oil via rotary evaporation and then diluted with tetrahydrofuran (410 mL) and distilled water (150 mL). To this solution, sodium periodate (7.50 g, 35.0 mmol) was added. The reaction was stirred until the intermediary diol was converted to aldehyde 16. Upon completion the reaction was poured into a saturated aqueous sodium thiosulfate solution and extracted with ethyl acetate (2 × 200 mL). The organic extracts were combined and washed several times with additional sodium thiosulfate until the aqueous layer stopped developing a pink coloration. The organic extract was washed with distilled water, brine and then dried over MgSO4. The organic phase was filtered and concentrated to yield 4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-carboxaldehyde 16 (5.05 g, quantitative), which was used without further purification. IR (neat) cm−1 3003, 2966, 2941, 2910, 2887, 2835, 1674, 1622, 1597, 1539, 1497, 1350, 1313, 1250, 1150, 1107. 1H NMR (400 MHz, CDCl3) δ10.59 (s, 1H), 8.22 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.8 Hz, 2H), 6.69 (d, J = 2.0 Hz, 1H), 6.43 (d, J = 2.0 Hz, 1H), 3.95 (s, 3H), 3.89 (s, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3) δ187.5, 161.5, 159.6, 158.4, 155.3, 154.6, 130.3, 121.9, 116.2, 113.8, 110.6, 95.5, 88.0, 55.8, 55.7, 55.4. HRMS m/z calc. for C18H17O5 [M+H+] 313.1071, found 313.1078; mp = 145°C–149°C.
1-[4,6-Dimethoxy-2-(4-methoxy-phenyl)-benzofuran-3-yl]-3-phenyl-prop-2-yn-1-ol (17a)
Phenylacetylene (2.70 mL, 24.6 mmol) was dissolved in 80 mL dry tetrahydrofuran and cooled to −78°C. To this solution, n-butyllithium (11.0 mL of a 1.9M solution in hexane, 20.9 mmol) was added dropwise under argon. The solution was allowed to stir for 10 min. after which a solution of aldehyde 16 (5.05 g, 16.1 mmol) in dry tetrahydrofuran (120 mL) was added via cannula. The reaction was allowed to warm to room temperature and quenched with a saturated aqueous solution of ammonium chloride. The mixture was poured into distilled water and the aqueous phase was made neutral by the addition of 1N HCl (aq.). The mixture was extracted with ethyl acetate (2 × 200 mL) and the combined organic extracts were washed with brine. The organic layer was dried over Na2SO4 and concentrated. Precipitation in methyl-t-butyl ether afforded 1-[4,6-dimethoxy-2-(4-methoxy-phenyl)-benzofuran-3-yl]-3-phenyl-prop-2-yn-1-ol 17a. (5.20 g, 78%). IR (neat) cm−1 3474, 3003, 2935, 2836, 1621, 1597, 1501, 1255, 1146, 1111. 1H NMR (400 MHz, CDCl3) δ7.63 (d, J = 6.8 Hz, 2H), 7.40 (m, 2H), 7.28 (m, 3H), 7.03 (d, J = 6.8 Hz, 2H), 6.73 (d, J = 1.6 Hz, 1H), 6.45 (d, J = 1.6 Hz, 1H), 5.85 (d, J = 9.2 Hz, 1H), 4.63 (d, J = 9.2 Hz, 1H), 4.06 (s, 3H), 3.87 (s, 6H). 13C NMR (125 MHz, CDCl3) δ160.0, 159.3, 156.2, 152.3, 149.7, 131.7, 129.3, 128.3, 128.2, 122.9, 122.8, 115.6, 114.3, 111.2, 95.0, 89.4, 89.1, 84.5, 57.6, 56.2, 55.9, 55.4. HRMS m/z calc. for C28H26O5 [M+] 414.1462, found 414.1471; mp = 119°C–121°C.
3-(1-ethoxy-3-phenylprop-2-ynyl)-4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran (18a)
Propargylic alcohol 17a (3.70 g, 9.00 mmol) was dissolved in 60 mL of dry tetrahydrofuran and potassium hydride (0.740 g, 18.5 mmol, from the 30% suspension in mineral oil which was washed 3 times with hexanes) was added. The reaction was stirred for ~5 min. until steady gas evolution was observed. At this point, ethyl iodide (2.22 mL, 27.8 mmol) is added and the reaction stirred until the intermediary propargyl alcohol is consumed. The excess potassium hydride is quenched by the addition of a saturated aqueous solution of ammonium chloride and the mixture is poured into distilled water and made neutral by the addition of 1N HCl (aq.). The mixture was extracted with ethyl acetate (2 × 100 mL) and the combined organic extracts were washed successively with distilled water and brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was subjected to flash column chromatography (80:20 hexane/ethyl acetate) to afford 3-(1-ethoxy-3-phenylprop-2-ynyl)-4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran 18a (2.57 g, 64%). IR (neat) cm−1 2969, 2936, 2907, 2878, 2836, 1622, 1611, 1592, 1501, 1490, 1296, 1252, 1215, 1149, 1110, 1060, 1029. 1H NMR (500 MHz, CDCl3) δ8.03 (d, J = 9.0 Hz, 2H), 7.23–7.17 (m, 3H), 7.13–7.07 (m, 2H), 7.00 (d, J = 7.2 Hz, 2H), 6.67 (d, J = 1.5 Hz, 1H), 6.36 (d, J = 1.5 Hz, 1H), 6.29 (s, 1H), 3.94 (s, 3H), 3.94–3.88 (m, 1H), 3.86 (s, 6H), 3.73–3.67 (m, 1H), 1.31 (t, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ159.8, 158.9, 155.3, 154.4, 152.7, 131.7, 130.0, 128.1, 128.0, 123.5, 122.9, 113.5, 112.6, 112.2, 94.6, 88.1, 87.5, 84.8, 64.0, 63.2, 55.8, 55.5, 55.4, 15.3. HRMS m/z calc. for C28H26O5Na [M+Na+] 465.1672, found 465.1666; mp = 98°C–100°C.
3-(1-(4-methoxybenzyloxy)-3-phenylprop-2-ynyl)-4,6-dimethoxy-2-(4-methoxyphenyl) benzofuran (18b)
To a solution of propargylic alcohol 17a (350 mg, 0.84 mmol) in N,N-dimethylformamide (7.1 mL) at 0°C was added sodium hydride (60% dispersion in mineral oil, 40 mg, 1.01 mmol) in one portion. The resulting suspension was stirred for 1 h at 0°C over which time it turned red. p-methoxybenzyl chloride (0.14 mL, 1.01 mmol) was added and the suspension stirred to room temperature overnight. The reaction mixture was diluted with saturated aqueous ammonium chloride (100 mL) and a 5% w/w aqueous solution of LiCl (80 mL). The resulting aqueous mixture was extracted with ethyl acetate (3 × 80 mL) and the combined organics washed with a 5% w/w aqueous solution of lithium chloride (3 × 90 mL), brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The resulting oil was purified by flash chromatography on silica gel (95:5-60:40 hexanes/ethyl acetate to provide 3-(1-(4-methoxybenzyloxy)-3-phenylprop-2-ynyl)-4,6-dimethoxy-2-(4-methoxyphenyl) benzofuran 18b (6.19 g, 96%) as a thick yellow oil. IR (neat) cm−1 3002, 2956, 2936, 2906, 2834, 1611, 1507, 1464, 1249, 1149, 1113. 1H NMR (400 MHz, CDCl3) δ8.01 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.8 Hz, 2H), 7.27–7.17 (m, 5H), 6.99 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 2.0 Hz, 1H), 6.35 (d, J = 2.0 Hz, 1H), 6.28 (s, 1H), 4.83 (d, J = 11.6 Hz, 1H), 4.75 (d, J = 11.6 Hz, 1H), 3.91 (s, 3H), 3.90 (s, 3H), 3.86 (s, 6H). 13C NMR (100 MHz, CDCl3) δ159.8, 159.2, 158.9, 155.4, 154.4, 152.8, 131.7, 130.1, 130.0, 128.7, 128.2, 128.1, 123.4, 122.9, 114.0, 113.7, 113.5, 112.4, 112.0, 94.6, 88.0, 87.2, 69.9, 62.3, 55.8, 55.4, 55.35, 55.3. HRMS m/z calc. for C34H30O6 [M+] 534.2037, found 534.2051.
4,6-dimethoxy-3-(1-((2-methoxybenzyl)oxy)-3-phenylprop-2-yn-1-yl)-2-(4-methoxyphenyl)benzo-furan (18c)
To a solution of propargylic alcohol 17a (350 mg, 0.84 mmol) in dry N,N-dimethylformamide (7 mL) at 0°C was added sodium hydride (60% dispersion in mineral oil, 41 mg, 1.0 mmol) in one portion. The deep orange suspension was stirred for 30 min, after which o-methoxybenzyl chloride54 (160 mg, 1.0 mmol) was added slowly. The mixture was stirred for 2.5 h, then diluted with saturated aqueous ammonium chloride (150 mL) and a 5% w/w aqueous solution of LiCl (80 mL). The resulting aqueous mixture was extracted with dichloromethane (3 × 100 mL) and the combined organics washed with a 5% w/w aqueous solution of lithium chloride (3 × 90 mL), brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The resulting oil was purified by flash chromatography on silica gel (95:5-60:40 hexanes/ethyl acetate to provide 4,6-dimethoxy-3-(1-((2-methoxybenzyl)oxy)-3-phenylprop-2-yn-1-yl)-2-(4-methoxyphenyl)benzofuran 18c (382 mg, 85%). IR (neat) cm−1 2930, 2840, 1600, 1500, 1250, 1120, 1035, 753. 1H NMR (400 MHz, CDCl3) δ8.02 (d, J = 8.9 Hz, 2H), 7.45 (d, J = 7.4 Hz, 1H), 7.34–7.04 (m, 6H), 7.01–6.89 (m, 3H), 6.86 (d, J = 8.2 Hz, 1H), 6.65 (d, J = 1.9 Hz, 1H), 6.35 (s, 2H), 4.88 (dd, J = 58.3, 12.3 Hz, 1H), 3.85 (s, 3H), 3.84 (s, 3H), 3.81 (s, 3H), 3.78 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 159.8, 158.8, 157.4, 155.3, 154.5, 152.9, 131.7, 130.0, 129.5, 128.6, 128.1, 128.0, 126.6, 123.6, 120.4, 113.5, 112.6, 112.2, 110.1, 94.6, 88.0, 87.5, 85.2, 65.3, 63.0, 55.8, 55.4, 55.4, 55.3.
1-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)prop-2-yn-1-ol (17b)
To ethynylmagnesium bromide (0.5M in tetrahydrofuran, 1.8 mL, 0.88 mmol) at −78°C was added a solution of aldehyde 16 (250 mg, 0.8 mmol) in tetrahydrofuran (8 mL). After addition, the cooling bath was removed and the reaction solution was stirred at room temperature for 16 h. The solution was quenched with saturated aqueous ammonium chloride (50 mL) and partially concentrated in vacuo to remove tetrahydrofuran. Distilled water (150 mL) was added, then the mixture extracted with dichloromethane (3 × 150 mL). The combined organic layers were washed with brine (100 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The resulting orange residue was purified by flash chromatography on silica gel (95:5-6:4 hexanes/ethyl acetate) to provide 1-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)prop-2-yn-1-ol 17b (85%). 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.8 Hz, 2H), 6.70 (d, J = 1.9 Hz, 1H), 6.43 (d, J = 1.9 Hz, 1H), 5.62 (dd, J = 11.6, 2.3 Hz, 1H), 4.63 (d, J = 11.6 Hz, 1H), 4.04 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 2.56 (d, J = 2.3 Hz, 1H).
4,6-dimethoxy-3-(1-((4-methoxybenzyl)oxy)prop-2-yn-1-yl)-2-(4-methoxyphenyl)benzofuran (18d)
To a solution of propargylic alcohol 17b (218 mg, 0.64 mmol) in dry N,N-dimethylformamide (5.4 mL) at 0°C was added sodium hydride (60% dispersion in mineral oil, 31 mg, 0.77 mmol) in one portion. The deep red suspension was stirred for 45 min, after which para-methoxybenzyl chloride (0.10 mL, 0.77 mmol) was added slowly. The mixture was stirred to room temperature over 16 h, then diluted with saturated aqueous ammonium chloride (70 mL) and a 5% w/w aqueous solution of lithium chloride (80 mL). The resulting aqueous mixture was extracted with ethyl acetate (3 × 150 mL) and the combined organics washed with a 5% w/w aqueous solution of lithium chloride (4 × 50 mL), brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The resulting yellow residue was purified by flash chromatography on silica gel (95:5-7:3 hexanes/ethyl acetate) to provide 4,6-dimethoxy-3-(1-((4-methoxybenzyl)oxy)prop-2-yn-1-yl)-2-(4-methoxyphenyl)benzofuran 18d (125 mg, 42%) as a pale yellow tar. IR (neat) cm−1 3280, 3000, 2920, 2820, 1612, 1508, 1251, 1150. 1H NMR (400 MHz, CDCl3) δ7.89 (d, J = 8.9 Hz, 2H), 7.26 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 6.63 (d, J = 1.9 Hz, 1H), 6.30 (d, J = 1.9 Hz, 1H), 6.02 (d, J = 2.4 Hz, 1H), 4.69 (d, J = 11.5 Hz, 1H), 4.60 (d, J = 11.5 Hz, 1H), 3.85 (s, 3H), 3.83 (s, 3H), 3.79 (s, 3H), 3.77 (s, 3H), 2.37 (d, J = 2.4 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 159.8, 159.2, 158.9, 155.4, 154.3, 152.5, 130.1, 129.7, 128.3, 123.1, 113.6, 113.5, 111.9, 111.7, 94.6, 87.9, 81.5, 73.6, 69.8, 61.6, 55.8, 55.4, 55.3. HRMS m/z calc. for C28H26O6Na [M+Na+] 481.1627, found 481.1631.
(3-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)-3-((4-methoxybenzyl)oxy)-1-phenylpropa-1,2-dien-1-yl)trimethylsilane (19a)
Alkyne 18a (30 mg, 0.068 mmol) was dissolved in 2 mL of dry tetrahydrofuran and cooled to −40°C. This solution was treated dropwise with t-butyllithium (0.052 mL of a 1.7M solution in pentane, 0.088 mmol) and allowed to stir for 20 min. At this time, chlorotrimethylsilane (12 µL, 0.0947 mmol) was added and after 3 min. the reaction was quenched with a saturated aqueous solution of ammonium chloride (40 mL). The reaction was poured into distilled water (30 mL, extracted with ethyl acetate (3 × 25 mL), and the combined organic extracts washed with brine (30 mL). The organic layer was dried over MgSO4, filtered and concentrated. The resulting residue was submitted to flash column chromatography (6:1 hexanes/ethyl acetate) to yield (3-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)-3-((4-methoxybenzyl)oxy)-1-phenylpropa-1,2-dien-1-yl)trimethylsilane 19a (14 mg, 40%) 1H NMR (400 MHz, CDCl3) δ7.73 (d, J = 9.2 Hz, 2H), 7.27–7.21 (m, 3H), 7.14–7.08 (m, 2H), 6.66 (d, J = 9.2 Hz, 2H), 6.65 (d, J = 2.0 Hz, 1H), 6.32 (d, J = 2.0 Hz, 1H), 3.86 (m, 5H), 3.82 (s, 3H), 3.81 (s, 3H), 1.46 (t, J = 8.4 Hz, 3H), 0.17 (s, 9H). 13C NMR (100 MHz, CDCl3) δ203.0, 159.5, 158.0, 155.7, 154.0, 151.1, 138.9, 128.8, 127.6, 126.2, 124.6, 123.3, 117.5, 113.7, 112.9, 108.4, 94.3, 87.8, 64.0, 55.7, 55.2, 55.0, 15.1, −0.5.
(3S,3aR)-1-ethoxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-3-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one (15a)
Propargylic ether 18a (1.00 g, 2.26 mmol) was dissolved in 20 mL of dry diethyl ether and cooled to −40°C under argon. To this solution t-butyllithium (1.46 mL, 1.7M solution in hexanes, 2.49 mmol) was added dropwise. With the addition of the base the reaction color progressed from light yellow to a deep brown. Upon completion of addition the reaction was allowed to stir for 5 min and then tributyltin chloride (0.74 mL, 2.7 mmol) was added and the reaction was allowed to warm to room temperature. Upon the addition of the stannyl chloride the color of the reaction bleached back to yellow and eventually a white precipitate developed. The magnetic stir bar was removed and the reaction was concentrated and the residue was taken up in hexanes and immediately subjected to flash column chromatography (90:10 hexanes/ethyl acetate on silica deactivated with triethylamine) to afford tributyl(3-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)-3-ethoxy-1-phenylpropa-1,2-dien-1-yl)stannane 19b (1.57 g, 95%). This material was used without delay in subsequent reactions.
Stannyl allene 19b (1.55 g, 1.95 mmol) was dissolved in 20 mL of degassed N,N-dimethylformamide and m-chloroperoxybenzoic acid (1.66 g of 77% m-CPBA, 7.41 mmol w/r/t m-CPBA) were added all at once. The flask was flushed with argon and stirred. After ~3 h an additional 1 equiv. of m-CPBA was added and the reaction was stirred until the starting allene was consumed (an additional 3 h). At this time the reaction was poured into a solution of sodium thiosulfate in water, and extracted with ethyl acetate (2 × 50 mL). The organic extracts were washed with a saturated aqueous solution of sodium bicarbonate twice, distilled water and finally brine. The organic phase was dried over MgSO4, filtered and concentrated to give an orange residue. This was subjected to flash column chromatography (85:15 hexane/ethyl acetate). Note: fractions containing the desired product appear orange/blue under UV light. This material was concentrated, redissolved in the minimum amount of diethyl ether and allowed to stand. After a few moments a precipitate developed and the diethyl ether suspension is briefly refrigerated. After refrigeration the precipitate is filtered to yield pure (3S,3R)-1-ethoxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-3-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one 15a as a yellow powder (450 mg, 50%). 1H NMR (400 MHz, CDCl3) δ7.14–7.08 (m, 5H), 6.91–6.85 (m, 2H), 6.59 (d, J = 7.2 Hz, 2H), 6.18 (d, J = 1.2 Hz, 1H), 6.04 (d, J = 1.2 Hz, 1H), 5.57 (s, 1H), 4.56–4.50 (m, 1H), 4.15–4.09 (m, 1H), 3.80 (s, 3H), 3.67 (s, 3H), 3.09 (s, 3H), 1.45 (t, J = 5.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ198.4, 166.1, 165.6, 159.1, 157.8, 149.9, 144.9, 134.3, 132.0, 130.0, 127.8, 127.4, 127.0, 113.0, 104.7, 98.2, 93.0, 89.7, 68.4, 66.5, 55.9, 55.8, 55.1, 15.8. HRMS m/z calc. for C28H26O6Na [M+Na+] 481.1627, found 481.1631; mp = 131°C–135°C. Ethyl ester 21 was isolated as a byproduct in varying ratios relative to ketone 15a, depending on the reaction conditions employed. The structure was deduced based on 1H NMR: the chemical shifts were nearly identical to those measured for methyl ester 7.
(3S,3aR)-6,8-dimethoxy-1-((4-methoxybenzyl)oxy)-3a-(4-methoxyphenyl)-3-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one (15b)
Propargylic ether 18b (2.6 g, 4.9 mmol) was dissolved in 70 ml of dry diethyl ether and cooled to −40°C under argon. To this solution t-butyllithium (3.0 mL, 1.7M solution in hexanes, 5.1 mmol) was added dropwise. With the addition of the base the reaction color progressed from light yellow to a deep brown. The reaction was allowed to stir for 5 min and then tributyltin chloride (1.5 mL, 5.6 mmol) was added. Upon the addition of the stannyl chloride the color of the reaction bleached back to yellow and eventually a white precipitate developed. The reaction was allowed to warm up to room temperature, the magnetic stir bar was removed and the reaction was concentrated and the residue was taken up in hexanes and immediately subjected to flash column chromatography (75:25 hexanes/ethyl acetate) to afford (3-(4-methoxybenzyloxy)-3-(4,6-dimethoxy-2-(4-methoxyphenyl)benzofuran-3-yl)-1-phenylpropa-1,2-dienyl)tributylstannane 19c (3.8 g, 94%). This material was used without delay in subsequent reactions. IR (neat) cm−1 3002, 2952, 2921, 2836, 1917, 1612, 1506, 1464, 1247, 1146, 1109. 1H NMR (500 MHz, CDCl3) δ7.77 (d, J = 9.0 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 7.25–7.18 (m, 3H), 6.88 (d, J = 8.5 Hz, 2H), 7.10–7.08 (m, 2H), 6.79 (d, J = 9.0 Hz, 2H), 6.67 (d, J = 2.0 Hz, 1H), 6.36 (d, J = 2.0 Hz, 1H), 4.89 (d, J = 11.0 Hz, 1H), 4.83 (d, J = 11.0 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 6H), 3.83 (s, 3H), 1.45–1.35 (m, 6H), 1.25–1.15 (m, 12H), 0.80 (t, J = 7.0 Hz, 9H). 13C NMR (125 MHz, CDCl3) δ200.1, 159.4, 159.1, 158.9, 155.8, 154.2, 150.8, 140.4, 130.3, 130.1, 129.5, 128.9, 128.2, 128.1, 126.3, 123.5, 123.2, 116.5, 113.7, 113.0, 109.1, 94.3, 87.8, 70.2, 55.7, 55.3, 55.2, 46.2, 29.0, 27.2, 11.6. HRMS m/z calc. for C34H30O6 [M+] 534.2037, found 534.2051.
Stannyl allene 19c (3.8 g, 4.6 mmol) was dissolved in 150 mL of degassed N,N-dimethylformamide and m-chloroperoxybenzoic acid (4.50 g of 77% m-CPBA, 18.4 mmol w/r/t m-CPBA) were added all at once. The flask was flushed with argon and stirred. After ~3 h and additional 1 equiv. of m-CPBA was added and the reaction was stirred until the starting allene was consumed (an additional 3 h). At this time the reaction was poured in distilled water, and extracted with ethyl acetate (2 × 200 mL). The organic extracts were washed with distilled water twice, a 10 % solution of sodium thiosulfate and finally brine. The organic phase was dried over MgSO4, filtered and concentrated to give an orange residue. This was subjected to flash column chromatography (80:20 hexanes/ethyl acetate). Note: fractions containing the desired product appear bright yellow under UV light. This material was concentrated, redissolved in the minimum amount of diethyl ether and allowed to stand. After a few moments a precipitate developed and the diethyl ether suspension was briefly refrigerated. After refrigeration the precipitate was filtered to yield pure (3S,3aR)-6,8-dimethoxy-1-((4-methoxybenzyl)oxy)-3a-(4-methoxyphenyl)-3-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one 15b as a yellow powder (960 mg, 38%). 1H NMR (400 MHz, CDCl3) δ7.51 (d, J = 8.4 Hz, 2H), 7.13–7.07 (m, 2H), 6.95 (d, J = 8.8 Hz, 2H), 6.82 (m, 2H), 6.78 (d, J = 8.4 Hz, 2H), 6.48 (d, J = 8.8 Hz, 2H), 6.15 (d, J = 2.0 Hz, 1H), 6.04 (d, J = 2.0 Hz, 1H), 5.64 (d, J = 11.7 Hz, 1H), 5.38 (d, J = 11.7 Hz, 1H), 4.52 (s, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.77 (s, 3H), 3.64 (s, 3H). 13C NMR (100 MHz, CDCl3) δ198.6, 166.3, 165.6, 159.6, 159.0, 157.8, 151.3, 143.9, 134.4, 131.7, 130.3, 130.0, 129.7, 128.6, 127.8, 127.4, 127.0, 113.8, 112.8, 98.3, 93.0, 89.6, 72.6, 66.6, 55.8, 55.8, 55.27, 55.0. HRMS m/z calc. for C34H31O7 [M+H+] 551.2064, found 551.2064; mp = 135°C–139°C.
4,6-dimethoxy-3-(1-((4-methoxybenzyl)oxy)propa-1,2-dien-1-yl)-2-(4-methoxyphenyl)benzofuran (20)
To a solution of propargylic ether 18d (177 mg, 0.386 mmol) in dimethylsulfoxide (1.0 mL) was added benzyltrimethylammonium hydroxide (40% wt/wt in MeOH, 0.1 mL, 0.20 mmol). The red solution was warmed to 40°C and stirred for 1 h, then poured into water (70 mL). The aqueous solution was extracted with ethyl acetate (3 × 80 mL) and the combined organics washed with brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo to provide 4,6-dimethoxy-3-(1-((4-methoxybenzyl)oxy)propa-1,2-dien-1-yl)-2-(4-methoxyphenyl)benzofuran 20 as a red-orange oil which was brought to the Nazarov cyclization step without delay. IR (neat) cm−1 2995, 2922, 2839, 2367, 2339, 1952, 1616, 1508, 1458, 1250, 1219, 1175, 1148, 1111, 1030. 1H NMR (400 MHz, CDCl3) δ7.78 (d, J = 8.9 Hz, 2H), 7.35 (d, J = 8.6 Hz, 2H), 6.88 (dd, J = 12.6, 8.8 Hz, 4H), 6.62 (d, J = 1.8 Hz, 1H), 6.31 (d, J = 1.8 Hz, 1H), 5.41 (s, 2H), 4.79 (s, 2H), 3.85 (s, 3H), 3.84 (s, 3H), 3.83 (s, 3H), 3.79 (s, 3H). 13C NMR (125 MHz, CDCl3) δ200.8, 159.4, 159.2, 159.0, 155.7, 154.2, 150.5, 129.9, 129.7, 127.9, 125.7, 123.4, 113.8, 113.7, 112.6, 107.9, 94.8, 88.8, 87.9, 77.2, 70.5, 55.8, 55.3, 55.3.
6,8-dimethoxy-1-((4-methoxybenzyl)oxy)-3a-(4-methoxyphenyl)-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one (22)
To allene 20 (180 mg, 0.39 mmol) in acetone (HPLC grade, 3 mL) was added a solution of dimethyldioxirane in acetone (0.084M, 7.0 mL, 0.58 mmol) slowly. The yellow solution was stirred for 10 min., then diluted with water (60 mL) and partially concentrated in vacuo to remove acetone. The resulting mixture was extracted with ethyl acetate (3 × 80 mL). The organic layers were combined and washed with brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The resulting orange residue was purified by flash chromatography on silica gel (95:5-75:25 hexanes/ethyl acetate) to provide 6,8-dimethoxy-1-((4-methoxybenzyl)oxy)-3a-(4-methoxyphenyl)-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one 22 (116 mg, 63% over 2 steps from propargylic ether 18d) as a pale yellow tar. IR (neat) cm−1 2880, 2834, 1703, 1606, 1509, 1246, 1146, 1083, 1028, 812. 1H NMR (400 MHz, CDCl3) δ7.38 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.9 Hz, 2H), 6.16 (d, J = 1.9 Hz, 1H), 6.01 (d, J = 1.9 Hz, 1H), 5.45 (d, J = 11.5 Hz, 1H), 5.27 (d, J = 11.5 Hz, 1H), 3.82 (s, 3H), 3.80 (s, 6H), 3.73 (s, 3H), 3.17 (d, J = 15.8 Hz, 1H), 2.86 (d, J = 15.8 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ199.5, 166.3, 165.8, 159.5, 159.1, 157.8, 154.6, 143.9, 134.3, 130.0, 129.6, 125.8, 113.8, 113.7, 104.5, 93.0, 92.9, 89.4, 72.8, 55.8, 55.7, 55.2, 55.2, 51.6. HRMS m/z calc. for C28H27O7 [M+H+] 475.1757, found 475.1753.
2-ethoxy-3-(2-hydroxy-4,6-dimethoxyphenyl)-4-(4-methoxyp6,8-dimethoxy-1-((4-methoxybenzyl)oxy)-3a-(4-methoxyphenyl)-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one phenyl)-5-phenylcyclopenta-2,4-dienone (26)
Nazarov product 15a (59 mg, 0.00013 mmol) was dissolved in 3 mL of dichloromethane. To this solution 1,4-diazabicyclo[2.2.2]octane (3 drops) was added. Upon addition of base, the reaction immediately developed a dark coloration. The solvent was removed via rotary evaporation and the crude product was subjected to flash column chromatography eluting with 4:1 hexane/ethyl acetate to give 2-ethoxy-3-(2-hydroxy-4,6-dimethoxyphenyl)-4-(4-methoxyp6,8-dimethoxy-1-((4-methoxybenzyl)oxy)-3a-(4-methoxyphenyl)-3,3a-dihydro-2H-cyclopenta[b]benzofuran-2-one henyl)-5-phenylcyclopenta-2,4-dienone 26 as a dark brown oil (53 mg, 90%). 1H NMR (400 MHz, CDCl3) δ7.23–7.17 (m, 5H), 6.83 (d, J = 8.0 Hz, 2H), 6.63 (d, J = 8.0 Hz, 2H), 6.25 (d, J = 4.0 Hz, 1H), 5.83 (d, J = 4.0 Hz, 1H), 4.35–4.29 (m, 1H), 4.26–4.20 (m, 1H), 3.80 (s, 3H), 3.74 (s, 3H), 3.06 (s, 3H), 1.29 (t, J = 5.0 Hz, 8H). 13C NMR (100 MHz, CDCl3) δ195.0(q), 162.6(q), 159.8(q), 158.5(q), 155.1(q), 142.9(q), 131.2(q), 130.1(CH), 15.6(CH3), 129.5(CH), 128.1(CH), 127.0(CH), 126.7(q), 125.4(q), 118.8(q), 113.1(CH), 101.5(q), 94.2(CH), 92.1(CH), 67.9(CH2), 55.5(CH3), 55.3(CH3), 55.2(CH3).
(2S,3S,3aS)-3a-ethoxy-6,8-dimethoxy-1-(4-methoxyphenyl)-2-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-3-ol (27)
Lithium triethylborohydride (0.42 mL of a 1M solution in tetrahydrofuran, 0.42 mmol) was diluted with dry tetrahydrofuran (6 mL) and cooled to −30°C. To this solution Nazarov product 15a (60 mg, 0.013 mmol) in 2 mL dry tetrahydrofuran was added dropwise. The reaction was allowed to stir for ~2 min. and was then poured into a saturated aqueous solution of sodium bicarbonate. The aqueous phase was extracted with ethyl acetate and the organic phase was washed with brine, dried over MgSO4, filtered and concentrated to give (2S,3S,3aS)-3a-ethoxy-6,8-dimethoxy-1-(4-methoxyphenyl)-2-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-3-ol 27 which was used crude for subsequent reactions (40 mg, 67%). Column chromatography could be performed with triethylamine-deactivated silica, eluting with 3:1 hexanes/ethyl acetate. 1H NMR (500 MHz, CDCl3) δ7.43–7.37 (m, 5H), 7.25 (d, J = 8.5 Hz, 2H), 6.80 (d, J = 8.5 Hz, 2H), 6.27 (d, J = 2.0 Hz), 6.18 (d, J = 2.0 Hz, 1H), 4.63 (dd, J = 12.0 Hz, 8Hz, 1H), 4.49 (d, J = 8.0 Hz, 1H), 3.88 (s, 3H), 3.83 (s, 3H), 3.77 (s, 3H), 3.57–3.51 (m, 1H), 3.48–3.52 (m, 1H), 2.42 (d, J = 12.5 Hz, 1H), 1.00 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ166.1(q), 163.42(q), 159.4(q), 155.3(q), 138.5(q), 134.9(q), 132.5(q), 130.5(CH), 130.2(CH), 129.6(q), 127.9(CH), 127.2 (CH), 117.0(q), 112.5(CH), 104.3(q), 92.8(CH), 89.1(CH), 77.2(CH), 62.2(CH), 59.2(CH2), 55.8(CH3), 55.4(CH3), 55.1(CH3), 15.07(CH3).
(2S,3S,3aS)-3a-ethoxy-6,8-dimethoxy-1-(4-methoxyphenyl)-2-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-3-yl methyl carbonate (28a)
Mixed ketal 27 (35 mg, 0.075 mmol) was dissolved in 2 mL of dry tetrahydrofuran and cooled to −78°C. n-butyllithium (0.06 mL of a 1.98M solution in pentane, 0.113 mmol) was added slowly and the reaction was allowed to stir for 2 min. After this time, freshly distilled methyl chloroformate (0.012 mL, 0.150 mmol) was added. The reaction was allowed to stir for ~3 min. and was then poured into a saturated aqueous solution of sodium bicarbonate. The aqueous phase was extracted with ethyl acetate and the organic phase was washed with brine, dried over MgSO4, filtered and concentrated. The residue was subjected to flash column chromatography (3:1 hexanes/ethyl acetate) to give (2S,3S,3aS)-3a-ethoxy-6,8-dimethoxy-1-(4-methoxyphenyl)-2-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-3-yl methyl carbonate 28a (40 mg, 90%). 1H NMR (500 MHz, CDCl3) δ7.36–7.30 (m, 5H), 7.19 (d, J = 9.0 Hz, 2H), 6.75 (d, J = 9.0 Hz, 2H), 6.24 (s, 1H), 6.12 (s, 1H), 5.42 (d, J = 8.0 Hz, 1H), 4.72 (d, J = 8.0 Hz, 1H), 3.83 (s, 3H), 3.78 (s, 3H), 3.75 (s, 3H), 3.69 (s, 3H), 3.60–3.54 (m, 1H), 3.51–3.45 (m, 1H), 1.03 (t, J = 8.8 Hz, 3H).
(2S,3S,3aS)-3a-ethoxy-6,8-dimethoxy-1-(4-methoxyphenyl)-2-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-3-yl trifluoromethanesulfonate (28b)
Mixed ketal 27 (69 mg, 0.15 mmol) was dissolved in 4 mL of dry tetrahydrofuran and cooled to −78°C. n-butyllithium (0.09 mL of a 2.5M solution in pentane, 0.223 mmol) was added slowly and the reaction was allowed to stir for 2 min. After this time, 2-[N,N-bis(trifluoromethanesulfonyl)amino]-5-chloropyridine (117 mg, 0.297 mmol) in 0.5 mL dry tetrahydrofuran was added. The reaction was allowed to stir for ~3 min without cooling after which silica gel was added. The reaction was concentrated and the resulting residue was subjected to flash column chromatography (4:1 hexanes/ethyl acetate) to give (2S,3S,3aS)-3a-ethoxy-6,8-dimethoxy-1-(4-methoxyphenyl)-2-phenyl-3,3a-dihydro-2H-cyclopenta[b]benzofuran-3-yl trifluoromethanesulfonate 28b (84 mg, 95%) 1H NMR (400 MHz, CDCl3) δ7.42–7.36 (m, 5H), 7.16 (d, J = 8.4 Hz, 2H), 6.76 (d, J = 8.4 Hz, 2H), 6.27 (d, J = 2.0 Hz, 1H), 6.14 (d, J = 2.0 Hz, 1H), 5.50 (d, J = 8.0 Hz, 1H), 4.67 (d, J = 8.0 Hz, 1H), 3.84 (s, 3H), 3.79 (s, 3H), 3.70 (s, 3H), 3.58–3.52 (m, 1H), 3.49–3.43 (m, 1H), 1.01 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ166.0, 163.7, 159.6, 155.5, 136.0, 132.8, 132.5, 130.3, 130.1, 128.2, 128.0, 127.9, 115.7, 112.5, 103.4, 93.0, 89.2, 85.0, 60.5, 59.8, 55.7, 55.2, 55.0, 15.0.
3-(2-hydroxy-4,6-dimethoxyphenyl)-4-(4-methoxyphenyl)-6-phenyl-2H-pyran-2-one (29)
The mixed ketal 27 was purified via flash column chromatography on silica gel. The combined product containing fractions were evaporated in a round bottom flask until a thin film remained. The product containing flask was exposed to air for 12 h upon which time the residue was resubjected to flash column chromatography (3:1 hexanes/ethyl acetate) to yield pyrone 29 (84%). 1H NMR (400 MHz, CDCl3) δ7.62 (s, 1H), 7.28–7.22 (m, 3H), 7.04–6.98 (m, 2H), 6.77 (d, J = 8.8 Hz, 2H), 6.56 (d, J = 8.8 Hz, 2H), 6.12 (d, J = 2.0 Hz, 1H), 6.08 (s, 1H), 5.88 (d, J = 2.0 Hz, 1H), 3.72 (s, 3H), 3.68 (s, 3H), 3.43 (s, 3H).
(3aS,8bR)-2,8b-dihydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-3-phenyl-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-1-one (37)
Nazarov product 15b (264 mg, 0.480 mmol) was dissolved in 5 mL of dichloromethane. To this solution 1 mL of distilled water was added followed by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (489 mg, 2.16 mmol). The reaction was stirred at room temperature for 12 h. Upon completion the reaction was poured into distilled water and extracted with ethyl acetate (2 × 20 mL). The organic layer was washed exhaustively with distilled water to remove the excess DDQ. The organic layer was then washed with brine, dried over MgSO4, filtered and concentrated. The resulting residue was subjected to flash column chromatography (60:40 ethyl acetate/hexanes) to yield (3aS,8bR)-2,8b-dihydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-3-phenyl-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-1-one 37 (158 mg, 71%) as a white solid. IR (neat) cm−1 3432, 3315, 1710, 1598, 1512, 1500, 1393. 1H NMR (400 MHz, CDCl3) δ7.99 (d, J = 5.6 Hz, 2H), 7.37–7.29 (m, 5H), 6.88 (d, J = 6.8 Hz, 2H), 6.16 (d, J = 1.2 Hz, 1H), 6.06 (d, J = 1.2 Hz, 1H), 3.82 (s, 3H), 3.78 (s, 3H), 3.77 (s, 3H), 2.88 (broad s, 1H). 13C NMR (100 MHz, CDCl3) δ195.6, 164.4, 161.7, 159.7, 158.7, 148.3, 132.9, 131.2, 130.7, 129.3, 128.3, 127.9, 113.9, 105.0, 98.0, 92.7, 88.8, 83.9, 55.7, 55.6, 55.2. HRMS m/z calc. for C26H22O7Na [M+Na+] 469.1258, found 469.1265; mp = 95°C–100°C.
(3aS,8bR)-8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-2-yl trifluoromethanesulfonate (38)
Diosphenol 37 (105 mg, 0.235 mmol) was dissolved in 4 mL of dry tetrahydrofuran and cooled to 0°C under argon. To this solution freshly prepared potassium bis(trimethylsilyl)amide (0.59 mL of a 1 M solution in tetrahydrofuran, 0.59 mmol) was added dropwise. The solution was stirred for 3 min whereupon 2-[N,N-bis(trifluoromethanesulfonyl)amino]-5-chloropyridine (168 mg, 0.47 mmol) was added slowly in 1 mL of dry tetrahydrofuran. The reaction was stirred at 0°C for 30 min and then poured into distilled water (20 mL). The aqueous layer was extracted with ethyl acetate (3 × 25 mL) and the combined organic extracts were washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The resulting residue was subjected to flash column chromatography (60:40 hexanes/ethyl acetate) to yield (3aS,8bR)-8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-2-yl trifluoromethanesulfonate 38 (112 mg, 83%) as a yellow solid. IR (neat) cm−1 3445, 2940, 2843, 1740, 1624, 1597, 1512, 1427, 1215. 1H NMR (400 MHz, CDCl3) δ7.58 (d, J = 8.8 Hz, 2H), 7.43–7.33 (m, 5H), 6.95 (d, J = 8.8 Hz, 2H), 6.13 (d, J = 2.0 Hz, 1H), 6.08 (d, J = 2.0Hz, 1H), 3.82 (s, 3H), 3.76 (s, 3H), 3.03 (s, 1H). 13C NMR (100 MHz, CDCl3) δ191.2, 164.7, 161.1, 160.1, 158.7, 152.7, 142.5, 131.8, 130.4, 128.8, 128.2, 127.6, 126.0, 114.3, 105.2, 97.1, 93.1, 88.9, 84.6, 55.8, 55.7, 55.3. HRMS m/z calc. for C27H21O9F3SNa [M+Na+] 601.0751, found 601.0734; mp = 179°C–183°C.
(3aS,8bR)-methyl 8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-2-carboxylate (39)
The enol triflate 38 (112 mg, 0.194 mmol) was dissolved in 4 mL of dry tetrahydrofuran. The solution was saturated with carbon monoxide for 5 min whereupon tetrakistriphenylphosphine palladium (0) (56 mg, 0.048 mmol), methanol (0.62 mL, 16 mmol), and N,N-diisopropylethylamine (0.135 mL, 0.776 mmol) were added. The reaction was warmed to 65°C for 6 h. Upon consumption of the starting material the reaction was poured into distilled water and extracted with ethyl acetate (3 × 20 mL). The organic extract was washed with distilled water and then brine. The organic layer was dried over MgSO4, filtered and concentrated. The resulting residue was subjected to flash column chromatography (50:50 hexanes/ethyl acetate) to yield (3aS,8bR)-methyl 8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-2-carboxylate 39 (69 mg, 79%) as a white solid. IR (neat) cm−1 3445, 2955, 2843, 1740, 1613, 1219, 1150. 1H NMR (400 MHz, CDCl3) δ7.38–7.29 (m, 7H), 6.93 (d, J = 8.8 Hz, 2H), 6.13 (d, J = 2.0 Hz, 1H), 6.08 (d, J = 2.0 Hz, 1H), 3.82 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 3.77 (s, 3H), 3.24 (s, 1H). 13C NMR (100 MHz, CDCl3) δ196.2, 166.1, 164.2, 164.1, 160.9, 159.8, 158.5, 133.0, 131.6, 130.9, 129.4, 128.3, 127.5, 125.8, 114.1, 105.8, 98.5, 92.8, 88.6, 86.9, 55.6, 55.6, 52.5, 55.2. HRMS m/z calc. for C28H24O8 [M+] 488.1466, found 488.1482; mp = 189°C–191°C.
(2R,3S,3aR,8bR)-methyl 8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-2,3,3a,8b-tetrahydro-1H-cyclopenta[b]benzofuran-2-carboxylate (40)
α, β-unsaturated ester 39 (52 mg, 0.11 mmol) was dissolved in 10 mL of absolute ethanol, platinum oxide (10 mg) was added and the solution was saturated with hydrogen gas for 10 min and stirred at room temperature for 3 h. Upon consumption of the starting material the reaction was filtered over celite and the filtrate concentrated. The resulting residue was subjected to flash column chromatography (50:50 hexanes/ethyl acetate) to yield (2R,3S,3aR,8bR)-methyl 8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-2,3,3a,8b-tetrahydro-1H-cyclopenta[b]benzofuran-2-carboxylate 40 (33 mg, 65%). Note: purification of 40 obtained under these reaction conditions was difficult and reaction results were erratic.
(3aS,8bR)-methyl-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-8b-((trimethylsilyl)oxy)-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-2-carboxylate (41)
Alkylidene β-ketoester 39 (69 mg, 0.15 mmol) was dissolved in 3 mL of benzene. To this solution, N,N-diisopropylethylamine (0.080 mL, 0.046 mmol) and 4-dimethylaminopyridine (2 mg, 0.015 mmol) were added. Chlorotrimethyl silane (0.039 mL, 0.306 mmol) was added and the reaction was stirred at room temperature for 30 min. Upon consumption of the starting material the reaction was filtered over a plug of silica (ethyl acetate) and the filtrate concentrated to yield 3aS,8bR)-methyl-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-8b-((trimethylsilyl)oxy)-3a,8b-dihydro-1H-cyclopenta[b]benzofuran-2-carboxylate 41 (83 mg, 96%). IR (neat) cm−1 2959, 2932, 2917, 2847, 1740, 1597, 1427, 1219. 1H NMR (400 MHz, CDCl3) δ7.34–7.32 (m, 3H), 7.27–7.23 (m, 4H), 6.91 (d, J = 9.2 Hz, 2H), 6.11 (d, J = 2.0Hz, 1H), 6.06 (d, J = 2.0Hz, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.75 (s, 3H), –0.03 (s, 9H). 13C NMR (100 MHz, CDCl3) δ194.6, 164.5, 164.3, 159.7, 161.0, 158.7, 133.5, 131.7, 130.3, 129.1, 128.3, 128.2, 126.9, 113.6, 106.6, 99.1, 92.5, 89.2, 88.5, 55.5, 55.3, 55.1, 52.3, 1.0. HRMS m/z calc. for C31H32O8Si [M+] 560.1861, found 560.1874.
(2R,3S,3aR,8bR)-methyl-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-8b-((trimethylsilyl)oxy)-2,3,3a,8b-tetrahydro-1H-cyclopenta[b]benzofuran-2-carboxylate (42)
Silyl ether 41 (76 mg, 0.11 mmol) was dissolved in 15 mL of absolute ethanol, platinum oxide (10 mg) was added and the solution was saturated with hydrogen gas for 10 min and stirred at room temperature for 6 h. Upon consumption of the starting material the reaction was filtered over celite and the filtrate concentrated. The resulting residue was subjected to flash column chromatography (80:20 Hexane/Ethyl acetate) to yield (2R,3S,3aR,8bR)-methyl-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-8b-((trimethylsilyl)oxy)-2,3,3a,8b-tetrahydro-1H-cyclopenta[b]benzofuran-2-carboxylate 42 (42 mg, 55%). 1H NMR (500 MHz, CDCl3) δ7.11–7.05 (m, 3H), 6.92 (d, J = 8.5 Hz, 2H), 6.80 (m, 2H), 6.64 (d, J = 8.5 Hz, 2H), 6.32 (d, J = 2.0 Hz, 1H), 6.08 (d, J = 2.0 Hz, 1H), 4.08 (d, J = 13.5 Hz, 1H), 3.94 (d, J = 13.5 Hz, 1H), 3.87 (s, 3H), 3.79 (s, 3H), 3.72 (s, 3H), 3.62 (s, 3H), −0.38 (s, 9H). 13C NMR (125 MHz, CDCl3) δ201.2, 167.7, 165.1, 161.4, 159.4, 158.7, 135.8, 128.5, 128.0, 127.8, 127.0, 126.4, 112.6, 105.7, 99.7, 92.6, 89.6, 58.4, 56.4, 55.6, 55.3, 55.1, 52.7, 51.3, 0.6. HRMS m/z calc. for C31H34O8SiNa [M+Na+] 585.1915, found 585.1941; mp = 155°C–158°C
(2R,3S,3aR,8bR)-methyl 8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-2,3,3a,8b-tetrahydro-1H-cyclopenta[b]benzofuran-2-carboxylate (40)
Silyl ether 42 (50 mg, 0.09 mmol) was dissolved in 3 mL of dry methanol. To this solution potassium fluoride (100 mg, 1.7 mmol) was added. The reaction was stirred at room temperature for 6 h after which the starting material was consumed. The methanol solution was poured into saturated ammonium chloride solution and extracted twice with ethyl acetate (2 × 25 mL). The combined organic extracts were washed with distilled water and brine and then dried over MgSO4. The organic phase was filtered and concentrated to yield (2R,3S,3aR,8bR)-methyl 8b-hydroxy-6,8-dimethoxy-3a-(4-methoxyphenyl)-1-oxo-3-phenyl-2,3,3a,8b-tetrahydro-1H-cyclopenta[b]benzofuran-2-carboxylate 40 (45 mg, quantitative) which was used without further purification. The compound was obtained as a 1:1 to 3:1 mixture of the keto- and the enolic forms. 1H NMR (400 MHz, CDCl3) δ10.60 (s, 1H, enol), 7.10–7.06 (m, 3H), 7.01 (d, J = 9.2 Hz, 2H, enol), 6.95–6.91 (m, 3H), 6.68 (d, J = 9.2 Hz, 2H, ketone), 6.53 (d, J = 9.2 Hz, 2H, enol), 6.35 (d, J = 2.0 Hz, 1H, ketone), 6.19 (d, J = 2.0 Hz, 1H, enol), 6.11 (d, J = 2.0 Hz, 1H, ketone), 6.05 (d, J = 2.0 Hz, 1H, enol), 4.48 (s, 1H, enol), 4.24 (d, J = 13.2 Hz, 1H, ketone), 3.88 (d, J = 13.2 Hz, 1H, ketone), 3.86 (s, 3H, ketone), 3.83 (s, 3H, enol), 3.81 (s, 3H, ketone), 3.79 (s, 3H, enol), 3.71 (s, 3H, ketone), 3.66 (s, 3H, ketone), 3.65 (s, 3H, enol), 3.59 (s, 3H, enol). 13C NMR (100 MHz, CDCl3) δ203.4, 172.0, 170.1, 167.3, 165.0, 163.6, 161.0, 160.8, 158.9, 158.6, 158.5, 158.1, 138.9, 135.4, 129.1, 129.0, 128.0, 127.9, 127.7, 127.2, 127.0, 126.6, 113.2, 112.0, 106.1, 105.8, 102.3, 100.7, 99.3, 92.9, 92.4, 89.9, 89.1, 88.9, 88.5, 56.4, 56.9, 55.7, 55.6, 55.1, 54.9, 52.9, 51.9, 51.5. HRMS m/z calc. for C28H26O8Na [M+Na+] 513.1520, found 513.1524.
Aglafolin (1e)
Sodium borohydride (80 mg, 2.1 mmol) was slowly and carefully added in 3 mL of glacial acetic acid. After 30 min of stirring the resulting solution was added to a solution of β-ketoester 20 (25 mg, 0.050 mmol) in 1 mL of dry acetonitrile. The reaction was stirred at room temperature for 15 h, poured into water and extracted with ethyl acetate (3×10 mL). The combined organic layers were washed with distilled water (10 mL) and brine and then dried over MgSO4. The organic phase was filtered and concentrated and the resulting residue was subjected to flash column chromatography (80:20 ethyl acetate/hexanes) to yield aglafolin 1e (14 mg, 56%) as a single stereoisomer. IR (neat) cm−1 3483, 3005, 2951, 2839, 1744, 1601, 1501, 1439, 1250. 1H NMR (400 MHz, CDCl3) δ7.09 (d, J = 9.2 Hz, 2H), 7.07–7.01 (m, 3H), 6.88–6.81 (m, 2H), 6.65 (d, J = 9.2 Hz, 2H), 6.26 (d, J = 1.6 Hz, 1H), 6.10 (d, J = 1.6 Hz, 1H), 5.01 (dd, J = 6.8, 1.2 Hz, 1H), 4.28 (d, J = 14.4 Hz, 1H), 3.88 (dd, J = 14.4, 6.4 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.75 (s, 3H), 3.63 (s, 3H), 3.62 (s, 1H). 13C NMR (100 MHz, CDCl3) δ170.5, 164.1, 160.9, 158.7, 157.0, 136.9, 128.9, 128.3, 127.8, 127.7, 126.5, 112.7, 107.7, 101.9, 93.7, 92.6, 89.5, 79.5, 55.7, 55.6, 55.1, 55.0, 51.9, 50.4. HRMS m/z calc. for C28H28O8Na [M+Na+] 515.1676, found 515.1701; mp = 89°C–91°C.
Rocagloic acid (1d)
Aglafolin 1e (14 mg, 0.028 mmol) was dissolved in 6 mL of a 5:1 mixture of tetrahydrofuran and distilled water. Lithium hydroxide (10 mg, 0.42 mmol) was added and the reaction was stirred at room temperature for 12 h. The mixture was diluted with 20 mL of dichloromethane and washed with 10 mL of 1N HCl, the organic layer was extracted with dichloromethane (2 × 5 mL), dried over MgSO4, filtered and concentrated to yield rocagloic acid 1d (11 mg, 82%) which was used without further purification. IR (neat) cm−1 3600-3200, 2923, 2853, 1715, 1610, 1513, 1453. 1H NMR (400 MHz, CDCl3) δ7.10 (d, J = 8.8 Hz, 2H), 7.14–7.00 (m, 3H), 6.93–6.85 (m, 2H), 6.68 (d, J = 8.8 Hz, 2H), 6.29 (d, J = 2.0 Hz, 1H), 6.12 (d, J = 2.0 Hz, 1H), 5.06 (d, J = 6.4, Hz, 1H), 4.28 (d, J = 15.2 Hz, 1H), 4.07 (dd, J = 14.0, 6.4 Hz, 1H), 3.89 (s, 3H), 3.88 (s, 3H), 3.72 (s, 3H). 13C NMR (100 MHz, CDCl3) δ174.0, 164.1, 160.8, 158.6, 156.8, 136.8, 128.9, 127.8, 127.7, 126.6, 126.2, 112.7, 107.5, 101.8, 93.6, 92.6, 89.4, 79.3, 55.8, 55.7, 55.0, 54.9, 50.1. (lit.14).
Rocaglamide (1a)
Rocagloic acid 1d (11 mg, 0.023 mmol) was dissolved in 4 mL of dry dichloromethane. Dicyclohexylcarbodiimide (12 mg, 0.058 mmol), 4-dimethylaminopyridine (19 mg, 0.15 mmol) and dimethylamine hydrochloride (6 mg, 0.07 mmol) were added and the reaction was stirred at room temperature for 15 h. The reaction was quenched with 1N HCl and extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with distilled water, brine and then dried over MgSO4. The organic phase was filtered and concentrated and the resulting residue was subjected to flash column chromatography (ethyl acetate) to yield rocaglamide 1a as a white solid (7 mg, 60%). Eventually rocaglamide can be recrystallized from dichloromethane/hexanes to give translucent crystals. IR (neat) cm−1 3500-3250, 2935, 2838, 1620, 1512, 1496. 1H NMR (400 MHz, CDCl3) δ7.12 (dd, J = 6.8, 2.0 Hz, 2H), 7.08–7.00 (m, 3H), 6.90–6.86 (m, 2H), 6.69 (dd, J = 6.8, 2.4 Hz, 2H), 6.29 (d, J = 2.0 Hz, 1H), 6.12 (d, J = 2.0 Hz, 1H), 4.94 (dd, J = 6.4, 2.0 Hz, 1H), 4.57 (d, J = 13.4Hz, 1H), 4.07 (dd, J = 13.4, 6.4 Hz, 1H), 4.03 (bs, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.72 (s, 3H), 3.33 (s, 3H), 2.95 (s, 3H), 1.81 (s, 1H). 13C NMR (100 MHz, CDCl3) δ169.5, 163.9, 161.0, 158.6, 157.2, 137.6, 128.8, 127.7, 127.6, 127.1, 126.2, 112.7, 107.6, 101.7, 94.0, 92.5, 89.2, 78.5, 56.0, 55.6, 55.0, 47.7, 37.0, 35.7. HRMS m/z calc. for C29H31O7NNa [M+Na+] 528.1993, found 528.1981; mp = 119°C–121°C. (lit.10c 1H NMR, 13C NMR, HRMS, mp).
Supplementary Material
Scheme 3.

Preparation of Alkynes 8a and 8b.
Acknowledgments
We thank the National Institutes of Health (NIGMS R01 GM079364) and the ARRA (3R01 GM079364-03S2) for funding this work. We are also grateful to Dr. William Brennessel (University of Rochester) for solving structures by X-ray crystallography, Dr. Sandip Sur (University of Rochester) for assistance with NMR spectroscopy, and Dr. Alice Bergmann (University of Buffalo) and Dr. Furong Sun (University of Illinois at Urbana-Champaign) for carrying out high-resolution mass spectroscopy. A.J.F thanks Prof. Philip Magnus for helpful discussions throughout this synthetic journey.
Footnotes
Supporting Information Available: 1H and 13C spectra for all new compounds, X-ray crystal structure coordinates and files for compounds 15a, 28a, 29 and 38 in CIF format. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
John A. Malona, Email: jmalona@avilatx.com.
Kevin Cariou, Email: kcariou@yahoo.com.
William T. Spencer, III, Email: wspence2@z.rochester.edu.
Alison J. Frontier, Email: frontier@chem.rochester.edu.
References
- 1.(a) Proksch P, Edrada R, Ebel R, Bohnenstengel FI, Nugroho BW. Current Organic Chemistry. 2001;5:923–938. [Google Scholar]; (b) Kim S, Salim AA, Swanson SM, Kinghorn AD. Anti-Cancer Agents Med. Chem. 2006;6:319–345. doi: 10.2174/187152006777698123. [DOI] [PubMed] [Google Scholar]; (c) Ebada SS, Lajkiewicz N, Porco JA, Jr, Li-Weber M, Proksch P. Prog. Chem. Org. Nat. Prod. 2011;94:1–58. doi: 10.1007/978-3-7091-0748-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King ML, Chiang CC, Ling HC, Fujita E, Ochiai M, McPhail AT. J. Chem. Soc. Chem. Commun. 1982:1150–1151. [Google Scholar]
- 3.(a) Dreyer M, Nugroho BW, Bohnenstengel FI, Ebel R, Wray V, Witte L, Bringmann G, Muhlbacher J, Herold M, Hung PD, Kiet LC, Proksch P. J. Nat. Prod. 2001;64:415–420. doi: 10.1021/np000123x. [DOI] [PubMed] [Google Scholar]; (b) Koul O, Kaur H, Goomber S, Wahab S. J. Appl. Entomol. 2004;128:177–181. [Google Scholar]
- 4.(a) Baumann B, Bohnenstengel F, Siegmund D, Wajant H, Weber C, Herr I, Debatin KM, Proksch P, Wirth T. J. Biol. Chem. 2002;277:44791–44800. doi: 10.1074/jbc.M208003200. [DOI] [PubMed] [Google Scholar]; (b) Proksch P, Giaisi M, Treiber MK, Palfi K, Merling A, Spring H, Krammer PH, Min LW. J. Immunol. 2005;174:7075–7084. doi: 10.4049/jimmunol.174.11.7075. [DOI] [PubMed] [Google Scholar]; (c) Fahrig T, Gerlach I, Horvath E. Mol. Pharmacol. 2005;67:1544–1555. doi: 10.1124/mol.104.008177. [DOI] [PubMed] [Google Scholar]; (d) Cencic R, Carrier M, Galicia-Vazquez G, Bordeleau ME, Sukarieh R, Bourdeau A, Brem B, Teodoro JG, Greger H, Tremblay ML, Porco JA, Pelletier J. PLoS One. 2009;4:e5223. doi: 10.1371/journal.pone.0005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu JY, Giaisi M, Kohler R, Muller WW, Muhleisen A, Proksch P, Krammer PH, Li-Weber M. Cell Death Differ. 2009;16:1289–1299. doi: 10.1038/cdd.2009.42. [DOI] [PubMed] [Google Scholar]
- 6.Bleumink M, Kohler R, Giaisi M, Proksch P, Krammer PH, Li-Weber M. Cell Death Differ. 2011;18:362–370. doi: 10.1038/cdd.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trost BM, Greenspan PD, Yang BV, Saulnier MG. J. Am. Chem. Soc. 1990;112:9022–9024. [Google Scholar]
- 8.(a) Davey AE, Schaeffer MJ, Taylor RJK. J. Chem. Soc. Chem. Commun. 1991:1137–1139. [Google Scholar]; (b) Davey AE, Schaeffer MJ, Taylor RJK. J. Chem. Soc.-Perkin Trans. 1. 1992:2657–2666. [Google Scholar]
- 9.Kraus GA, Sy JO. J. Org. Chem. 1989;54:77–83. [Google Scholar]
- 10.(a) Dobler MR, Bruce I, Cederbaum F, Cooke NG, Diorazio LJ, Hall RG, Irving E. Tetrahedron Lett. 2001;42:8281–8284. [Google Scholar]; (b) Diedrichs N, Ragot JP, Thede K. Eur. J. Org. Chem. 2005:1731–1735. [Google Scholar]; (c) Hongsen L, Fu B, Wang MA, Li N, Liu WJ, Xie ZQ, Ma YQ, Qin Z. Eur. J. Org. Chem. 2008:1753–1758. [Google Scholar]
- 11.Gerard B, Jones G, Porco JA. J. Am. Chem. Soc. 2004;126:13620–13621. doi: 10.1021/ja044798o. [DOI] [PubMed] [Google Scholar]
- 12.Hailes HC, Raphael RA, Staunton J. Tetrahedron Lett. 1993;34:5313–5316. [Google Scholar]
- 13.(a) Dumontet V, Thoison O, Omobuwajo OR, Martin MT, Perromat G, Chiaroni A, Riche C, Pais M, Sevenet T. Tetrahedron. 1996;52:6931–6942. [Google Scholar]; (b) Bacher M, Hofer O, Brader G, Vajrodaya S, Greger H. Phytochemistry. 1999;52:253–263. [Google Scholar]
- 14.Gerard B, Sangji S, O'Leary DJ, Porco JA. J. Am. Chem. Soc. 2006;128:7754–7755. doi: 10.1021/ja062621j. [DOI] [PubMed] [Google Scholar]
- 15.Gerard B, Cencic R, Pelletier J, Porco JA. Angew. Chem. Int. Ed. 2007;46:7831–7834. doi: 10.1002/anie.200702707. [DOI] [PubMed] [Google Scholar]
- 16.(a) Adams TE, El Sous M, Hawkins BC, Hirner S, Holloway G, Khoo ML, Owen DJ, Savage GP, Scammells PJ, Rizzacasa MA. J. Am. Chem. Soc. 2009;131:1607–1616. doi: 10.1021/ja808402e. [DOI] [PubMed] [Google Scholar]; (b) Roche SP, Cencic R, Pelletier J, Porco JA. Angew. Chem. Int. Ed. 2010;49:6533–6538. doi: 10.1002/anie.201003212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Thuaud F, Ribeiro N, Gaiddon C, Cresteil T, Desaubry L. J. Med. Chem. 2011;54:411–415. doi: 10.1021/jm101318b. [DOI] [PubMed] [Google Scholar]
- 17.Malona JA, Cariou KC, Frontier AJ. J. Am. Chem. Soc. 2009;131:7560–7561. doi: 10.1021/ja9029736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldman KS, Burns CJ. J. Org. Chem. 1991;56:4601–4602. [Google Scholar]
- 19.Watanabe T, Shiraga Y, Takeuchi T, Otsuka M, Umezawa K. Heterocycles. 2000;53:1051–1064. [Google Scholar]
- 20.Schoop A, Greiving H, Gohrt A. Tetrahedron Lett. 2000;41:1913–1916. [Google Scholar]
- 21.Thede K, Diedrichs N, Ragot JP. Org. Lett. 2004;6:4595–4597. doi: 10.1021/ol0479904. [DOI] [PubMed] [Google Scholar]
- 22.Magnus P, Stent MAH. Org. Lett. 2005;7:3853–3855. doi: 10.1021/ol0513793. [DOI] [PubMed] [Google Scholar]
- 23.Giese MW, Moser WH. Org. Lett. 2008;10:4215–4218. doi: 10.1021/ol801435j. [DOI] [PubMed] [Google Scholar]
- 24.Nazarov cyclization lead references and reviews: Nazarov IN, Zaretskaya II. Izv. Akad. Nauk. SSSR, Ser. Khim. 1941:211. Santelli-Rouvier C, Santelli M. Synthesis. 1983:429. Habermas KL, Denmark SE, Jones TK. In: Organic Reactions. Paquette LA, editor. Vol. 45. New York: John Wiley & Sons Inc.; 1994. pp. 1–158. Frontier AJ, Collison C. Tetrahedron. 2005;61:7577. Pellissier H. Tetrahedron. 2005;61:6479. Tius MA. Eur. J. Org. Chem. 2005;11:2193. Nakanishi W, West FG. Curr. Opin. Drug Discov. 2009;12:732. Vaidya T, Eisenberg R, Frontier AJ. ChemCatChem. 2011;3:1531–1548.
- 25.Polarized Nazarov Cyclization: He W, Sun XF, Frontier AJ. J. Am. Chem. Soc. 2003;125:14278–14279. doi: 10.1021/ja037910b. He W, Herrick IR, Atesin TA, Caruana PA, Kellenberger CA, Frontier AJ. J. Am. Chem. Soc. 2008;130:1003–1011. doi: 10.1021/ja077162g.
- 26.Recent examples of “Interrupted” Nazarov cyclization: (a) Review on domino reactions of oxyallyl intermediates: Grant TN, Rieder CJ, West FG. Chem. Commun. 2009:5676. doi: 10.1039/b908515g. (b) Interception of oxyallyl cation with halogen nucleophiles: Marx VM, Cameron TS, Burnell DJ. Tetrahedron Lett. 2009;50:7213–7216. (c) Interception of oxyallyl cation with carbon nucleophiles: Marx VM, Burnell DJ. J. Am. Chem. Soc. 2010;132:1685–1689. doi: 10.1021/ja909073r. (d) Oxyallyl cation-azide cycloaddition: Scadang O, Ferguson MJ, West FG. Org. Lett. 2011;13:114–117. doi: 10.1021/ol102651k. (e) Wagner-Meerwein rearrangement of oxyallyl cation: Huang J, Leboeuf D, Frontier AJ. J. Am. Chem. Soc. 2011;133:6307–6317. doi: 10.1021/ja111504w. (f) Interception of oxyallyl cation with nitrogen heterocycles: Marx VM, LeFort FM, Burnell DJ. Adv. Synth. Catal. 2011;353:64–68.
- 27.Malona JA, Colbourne JM, Frontier AJ. Org. Lett. 2006;8:5661–5664. doi: 10.1021/ol062403v. [DOI] [PubMed] [Google Scholar]
- 28.(a) Demerseman P, Lechartier JP, Pene C, Cheutin A, Royer R. Bull. Soc. Chim. Fr. 1965:1473–1486. [Google Scholar]; (b) Royer R, Demerseman P, Laval-Jeantet AM, Rossignol JF, Cheutin A, Desvoye ML. Bull. Soc. Chim. Fr. 1968:1026–1035. [Google Scholar]
- 29.(a) Cassar L. J. Organomet. Chem. 1975;93:253–257. [Google Scholar]; (b) Dieck HA, Heck FR. J. Organomet. Chem. 1975;93:259–263. [Google Scholar]; (c) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467–4470. [Google Scholar]
- 30. Nan Y, Miao H, Yang Z. Org. Lett. 2000;2:297–299. doi: 10.1021/ol991327b. See supporting information for experimental details.
- 31.Prepared according to: Roelens F, Huvaere K, Dhooge W, Van Cleemput M, Comhaire F, De Keukeleire D. Eur. J. Med. Chem. 2005;40:1042–1051. doi: 10.1016/j.ejmech.2005.04.010.
- 32.(a) Ohira S. Synth. Commun. 1989;19:561–564. [Google Scholar]; (b) Müller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521. [Google Scholar]
- 33.(a) Seyferth D, Hilbert P, Marmor RS. J. Am. Chem. Soc. 1967;89:4811–4812. [Google Scholar]; (b) Seyferth D, Marmor RS. Tetrahedron Lett. 1970:2493–2496. [Google Scholar]; (c) Gilbert JC, Weerasooriya U. J. Org. Chem. 1979;44:4997. [Google Scholar]; (d) Gilbert JC, Weerasooriya U. J. Org. Chem. 1982;47:1837–1845. [Google Scholar]
- 34.Nakamura I, Mizushima Y, Yamamoto Y. J. Am. Chem. Soc. 2005;127:15022–15023. doi: 10.1021/ja055202f. [DOI] [PubMed] [Google Scholar]
- 35.Fürstner A, Davies PW. J. Am. Chem. Soc. 2005;127:15024–15025. doi: 10.1021/ja055659p. [DOI] [PubMed] [Google Scholar]
- 36.(a) Katamna C. Bull. Soc. Chim. Fr. 1970:2309–2322. [Google Scholar]; (b) Davey AE, Taylor RJK. J. Chem. Soc. Chem. Commun. 1987:25–27. [Google Scholar]
- 37.(a) Imamoto T, Kusumoto T, Tawarayama Y, Sugiura Y, Mita T, Hatanaka Y, Yokoyama M. J. Org. Chem. 1984;49:3904–3912. [Google Scholar]; (b) Molander GA. Chem. Rev. 1992;92:29–68. [Google Scholar]; (c) Dimitrov V, Kostova K, Genov M. Tetrahedron Lett. 1996;37:6787–6790. [Google Scholar]
- 38.For recent reviews, see Takacs JM, Jiang X-T. Curr. Org. Chem. 2003;7:369–396. Cornell CN, Sigman MS. Inorg. Chem. 2007;46:1903–1909. doi: 10.1021/ic061858d. Keith JA, Henry PM. Angew. Chem. Int. Ed. 2009;48:9039–9049. doi: 10.1002/anie.200902194.
- 39.This can also be viewed as an intramolecular Mukaiyama-Aldol reaction, with orbital overlap that requires classification as an electrocyclization.
- 40. Chan TH, Ong BS. Tetrahedron. 1980;36:2269. Smadja W. Chem. Rev. 1983;83:263–320. Shipman M, Thorpe HR, Clemens IR. Tetrahedron Lett. 1997;38:897–900. For nucleophilic cyclizations of allene oxides, see: Bertrand M, Dulcere JP, Gil G, Grimaldi J, Sylvestre-Panthet P. Tetrahedron Lett. 1976;17:3305. Bertrand JM, Dulcere JP, Gil G, Grimaldi J, Sylvestre-Panthet P. Tetrahedron Lett. 1976;17:3305–3308. Macomber RS. J. Org. Chem. 1978;43:1832–1833. Crandall JK, Batal DJ. Tetrahedron Lett. 1988;29:4791–4794. Crandall JK, Rambo E. Tetrahedron Lett. 1994;35:1489–1492. Crandall JK, Rambo E. Tetrahedron. 2002;58:7027–7036. Cordier P, Aubert C, Malacria M, Gandon V, Lacôte E. Chem. Eur. J. 2010;16:9973–9976. doi: 10.1002/chem.201000914.
- 41.For chemistry and applications of allene diepoxides, see: Ghosh P, Lotesta SD, Williams LJ. J. Am. Chem. Soc. 2007;129:2438–2439. doi: 10.1021/ja068813w. Lotesta SD, Kiren S, Sauers RR, Williams LJ. Angew. Chem. Int. Ed. 2007;46:7108. doi: 10.1002/anie.200701401. Ghosh P, Zhang Y, Emge TJ, Williams LJ. Org. Lett. 2009;11:4402–4405. doi: 10.1021/ol901755a. Zhang Y, Cusick JR, Ghosh P, Shangguan N, Katukojvala S, Inghrim J, Emge TJ, Williams LJ. J. Org. Chem. 2009;74:7707. doi: 10.1021/jo901320f. Ghosh P, Cusick JR, Inghrim J, Williams LJ. Org. Lett. 2009;11:4672. doi: 10.1021/ol901948d.
- 42.For chemistry of heteroatom-substituted allene oxides: Xiong H, Hsung RP, Berry CR, Rameshkumar C. J. Am. Chem. Soc. 2001;123:7174–7175. doi: 10.1021/ja0108638. Rameshkumar C, Xiong H, Tracey MR, Berry CR, Yao LJ, Hsung RP. J. Org. Chem. 2002;67:1339–1345. doi: 10.1021/jo011048d. Huang J, Hsung RP. J. Am. Chem. Soc. 2005;127:50–51. doi: 10.1021/ja044760b.
- 43.(a) Grimaldi J, Bertrand M. Tetrahedron Lett. 1969;38:3269. [Google Scholar]; (b) Grimaldi J, Bertrand M. Bull. Soc. Chim. Fr. 1971;3:957–962. [Google Scholar]; (c) Bertrand M, Dulcere J-P, Grimaldi J. C. R. Seances Acad. Sci. Ser. C. 1974;279:805. [Google Scholar]; (d) Ong BS, Chan TH. Tetrahedron Lett. 1976:3257–3260. [Google Scholar]; (e) Roumestant ML, Malacria M, Gore J, Grimaldi J, Bertrand M. Synthesis. 1976:755–757. [Google Scholar]; (f) Bertrand M, Dulcere JP, Gil G. Tetrahedron Lett. 1977:4403–4406. [Google Scholar]; (g) Doutheau A, Gore J, Malacria M. Tetrahedron. 1977;33:2393–2398. [Google Scholar]; (h) Malacria M, Roumestant ML. Tetrahedron. 1977;33:2813–2817. [Google Scholar]; (i) Malacria M, Gore J. J. Org. Chem. 1979;44:885–886. [Google Scholar]; (j) Bertrand M, Dulcere J-P, Gil G, Roumestant ML. Tetrahedron Lett. 1979;21:1845. [Google Scholar]; (k) Dulcere J-P, Grimaldi J, Santelli M. Tetrahedron Lett. 1981;22:3179. [Google Scholar]; (l) Doutheau A, Sartoretti J, Gore J. Tetrahedron. 1983;39:3059–3065. [Google Scholar]; (m) Doutheau A, Gore J, Diab J. Tetrahedron. 1985;41:329–338. [Google Scholar]; (n) Tolstikov GA, Romanova TY, Kuchin AV. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1987;36:557–561. [Google Scholar]; (o) Kim SJ, Cha JK. Tetrahedron Lett. 1988;29:5613. [Google Scholar]
- 44.Recent reviews on the lipoxygenase pathway for jasmonate formation: Schaller A, Stintzi A. Phytochemistry. 2009;70:1532. doi: 10.1016/j.phytochem.2009.07.032. Gfeller A, Dubugnon L, Liechti R, Farmer EE. Sci. Signal. 2010;3:cm3. doi: 10.1126/scisignal.3109cm3. Synthesis of a vinyl allene oxide model of prostanoid formation in corals: Corey EJ, Ritter K, Yus M, Najera C. Tetrahedron Lett. 1987;28:3547–3550.
- 45.A detailed investigation of this reaction was conducted in our laboratory, after the oxidation-initiated cyclization was employed in the synthesis of (+/−)-rocaglamide: Spencer WT, Levin ML, Frontier AJ. Org. Lett. 2011;13:414–417. doi: 10.1021/ol1027255.
- 46.(a) Brandsma L, Verkruijsse HD. Synthesis of Acetylenes, Allenes and Cumulenes. Amsterdam, The Netherlands: Elsevier Scientific Publishing Company; 1981. pp. 87–89. [Google Scholar]; (b) Leroux Y, Roman C. Tetrahedron Lett. 1973;14:2585–2586. [Google Scholar]; (c) Landor PD. In: The Chemistry of the Allenes. Landor SR, Landor PD, editors. Vol. 1. New York: Academic Press Inc.; 1982. pp. 155–159. [Google Scholar]; (d) Zimmer R. Synthesis. 1993:165–178. [Google Scholar]
- 47.Ishikawa T, Mizuta T, Hagiwara K, Aikawa T, Kudo T, Saito S. J. Org. Chem. 2003;68:3702–3705. doi: 10.1021/jo026592g. [DOI] [PubMed] [Google Scholar]
- 48.For recent reviews, see: Bellina F, Rossi R. Chem. Rev. 2010;110:1082–1146. doi: 10.1021/cr9000836. Johansson CCC, Colacot TJ. Angew. Chem. Int. Ed. 2010;49:676–707. doi: 10.1002/anie.200903424.
- 49.This ring-opening behavior also occurred in Magnus’ system when an enolate was unintentionally formed at the same position after a dissolving metal reduction (see reference 22.)
- 50.Comins DL, Dehghani A. Tetrahedron Lett. 1992:6299–6302. [Google Scholar]
- 51.Both Trost and our group observed complete hydrogenation selectivity on the concave face of a 2,3-didehydro-aglafolin derivative due to the overriding steric influence of the para-methoxyphenyl group. See reference 7.
- 52.(a) Bailey EJ, Barton DHR, Elks J, Templeton JF. J. Chem. Soc. 1962:1578. [Google Scholar]; (b) Gardner JN, Carlon FE, Gnoj O. J. Org. Chem. 1968;33:3294. doi: 10.1021/jo01272a055. [DOI] [PubMed] [Google Scholar]; (c) Gardner JN, Popper TL, Carlon FE, Gnoj O, Herzog HL. J. Org. Chem. 1968;33:3695. doi: 10.1021/jo01274a004. [DOI] [PubMed] [Google Scholar]
- 53.Adam W, Bialas J, Hadjiarapoglou L. Chem. Ber. 1991;124:2377–2377. [Google Scholar]
- 54.Amin S, Hecht SS, Hoffmann D. J. Org. Chem. 1981;46:2394–2398. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.