

Abstract
P2X receptors are trimeric ATP-gated cation channels participating in diverse physiological processes. How ATP binding triggers channel opening remains unclear. Here the gating mechanism of a P2X receptor was studied by normal mode analysis and molecular dynamics (MD) simulations. Based on the resting-state crystal structure, a normal mode involving coupled motions of three β-strands (β1, β13, and β14) at the trimeric interface of the ligand-binding ectodomain and the pore-lining helix (TM2) in the transmembrane domain (TMD) was identified. The resulting widening of the fenestrations above the TMD and opening of the transmembrane pore produce known signatures of channel activation. In MD simulations, ATP was initially placed in the putative binding pocket (defined by four charged residues located in β1, β13 and β14) in two opposite orientations, with the adenine either proximal or distal to the TMD. In the proximal orientation, the triphosphate group extends outward to draw in the four charged residues, leading to closure of β13/β14 toward β1. The adenine ring, wedged between β1 and β13, acts as a fulcrum for the β14 lever, turning a modest closure around the triphosphate group into significant opening of the pre-TM2 loop. The motions of these β-strands are similar to those in the putative channel-activation normal mode. In the distal orientation, the ATP stabilizes the trimeric interface and the closure of the pre-TM2 loop, possibly representing desensitization. Our computational studies produced the first complete model, supported by experimental data, for how ATP binding triggers activation of a P2X receptor.
Keywords: antagonist design, ATP binding mode, channel-activation mechanism, ligand-gated ion channels
P2X receptors (P2XRs) are a family of ligand-gated nonselective cation channels activated by extracelluar ATP (1). Distributed throughout the human body, they participate in diverse physiological processes such as synaptic transmission, response to inflammation, and pain sensation, and thus are potential drug targets (2–7). Seven subtypes of P2XRs, P2XR1 to P2X7R, have been identified in mammals. They assemble to form functional homo- or hetero-trimeric channels (8), distinguishing them from the better-studied tetrameric and pentameric ligand-gated ion-channel families. The recent crystal structure of zebrafish P2X4 receptor (zfP2X4R) in the resting state represents a major advance in understanding the structure and function of P2XR ion channels (9). Each subunit of zfP2X4R contains two transmembrane helices (TM1 and TM2), which link the intracellular amino and carboxyl termini to the large ligand-binding ectodomain (Fig. 1). In the ectodomain, each subunit consists of 4 α-helices and 14 β-strands (β1–β14). β1 and β14 are connected to TM1 and TM2, respectively.
Fig. 1.

Sequence and structure of zfP2X4 receptor. (A) Sequence alignment of zfP2X4R and rat P2X2R (rP2X2R). The latter is usually used for electrophysiological studies. Secondary structures are labeled. Four charged residues (K70, K72, R298, and K316) in the putative ATP binding site and three pore-facing residues (L340, A344, and A347) are highlighted in red. (B) The trimeric structure of zfP2X4R in the resting state. β14 and TM2 of the front left subunit and β1 and TM2 of the front right subunit are in darker colors. The sidechains of the four charged residues are shown as spheres, as are the Cα atoms of the three pore-facing residues.
Studies based on mutagenesis have identified several highly conserved ectodomain residues as involved in ATP binding (10–18). The conserved residues include four cationic ones: K70 and K72 on β1, R298 on β13, and K316 on β14 (10, 19, 20). Aromatic and polar residues F188, T189, N296, and F297 were also implicated (12, 17). In the zfP2X4R crystal structure, these conserved residues line an open-jaw-shaped pocket, strongly suggesting the pocket as the ATP binding site (9). This putative binding site, located at the trimeric interface and approximately 45 Å from the transmembrane domain (TMD), is supported by intersubunit cross-linking between substituted cysteine residues (15, 21). Recently Jiang et al. (22) presented additional evidence by cross-linking 8-thiocyano-ATP (NCS-ATP) with single cysteine substitutions in the putative binding site of a P2X2R. NCS-ATP covalently labeled two previously unidentified positions L186 and N140 (corresponding to L191 and D145 in zfP2X4R; Fig. 1A), which resulted in distinct functional consequences. ATP-gated currents were potentiated by labeling at L186 but inhibited by labeling at N140.
In several recent studies (9, 23–27), possible conformational changes in the ectodomain or TMD induced by ATP binding have been proposed. Based on the open-jaw shape of the putative ATP binding site revealed by the zfP2X4R crystal structure, Kawate et al. (9) suggested that the jaws close upon the bound agonist. By measuring the changes upon channel activation in accessibility of modifying agents to substituted cysteines in TM2, the channel gate has been located to the N-terminal portion of TM2 (23–25). This portion is the most constricted along the transmembrane pore in the zfP2X4R crystal structure. Jiang et al. (26) measured the rates of disulfide bond formation between two substituted cysteines, one at E63 and the other at R274 of P2X2R (corresponding to L64 in β1 and R280 in β12 of zfP2X4R, respectively), before and after ATP binding, and concluded that channel gating involved separation of these two positions. Recent experiments by Kawate et al. (27) suggested that permeant ions access the transmembrane pore via the lateral fenestrations [confirmed by Samways et al. (28)], and that the fenestrations expand upon channel activation. The fragmentary information involving different regions of P2XRs has advanced our understanding, but a complete mechanistic model, explaining how ATP binding triggers channel opening, is still lacking.
Here, based on normal mode analysis and molecular dynamics simulations, we developed a complete model, at the atomic level, for the gating mechanism of zfP2X4R. In this model, the bound ATP has its triphosphate group extended outward to draw in the four cationic residues K70, K72, R298, and K316, leading to closure of β13/β14 toward β1. The adenine ring, wedged between β1 and β13, acts as a fulcrum for the β14 lever, turning the modest closure around the triphosphate group into significant opening of the pre-TM2 loop and the N-terminal half of TM2. The proposed gating mechanism is validated by a host of experimental data (22–27), and provides insight for designing P2XR antagonists.
Results
Coupled Motions of Ectodomain and TMD.
Low-frequency normal modes of biomolecular systems have been identified with functional motions, e.g., gating motions of nicotinic acetylcholine receptors (29–32) and acid-sensing ion channel 1 (ASIC1) (33). Here we treated zfP2X4R by the elastic network model (34), in which each atom was elastically connected to neighboring atoms. The equilibrium conformation was taken as the resting structure (containing residues G32 to L361; see Fig. 1), in which the channel pore has narrow constrictions at positions L340 and A347 along TM2 (Fig. 2A).
Fig. 2.

Activated state generated by normal mode analysis. (A) and (B) HOLE images (47) of the TMD in the resting (TM2 in gray) and activated (TM2 in red, blue, and yellow) states. L340, A344, and A347 are shown as sticks. (C) Pore sizes in the resting (black curve) and activated (red curve) states. (D) Top view of TM2 in the resting and activated states. Cα atoms of L340 are shown as spheres. (E) Side view of the key secondary structures, including β1, β14, and TM2, in the resting and activated states. A347 is shown as a sphere to highlight the excessive and moderated bending, respectively, of TM2 in the resting and activated states. Cα atoms of K70, K316, and L340 are also shown as spheres. The coloring scheme in D and E are the same as in A and B. Note that the elastic network model produces only motional direction, not amplitude. The activated model shown here was generated by moving along the single mode, with an average amplitude of 1.6 Å for all but 10% of the most outlying atoms (which are prone to distortion in normal mode analysis).
We generated the 100 lowest-frequency modes, and searched for modes that exhibited both widening of the channel pore and closure motions around the putative ATP binding site. Two such modes were indeed found, and motions along the first of these modes produced the putative activated state, in which the constrictions at L340 and A347 are widened (Fig. 2 B and C). (The motional features putatively representing channel activation are largely shared by the second mode; Fig. S1.) The pore widening arises from the outward movement of the N-terminal half of TM2 (up to A347); the C-terminal half of TM2 moves along to reduce the significant bending around A347 seen in the resting structure (Fig. 2 D and E). In addition to the outward motion, TM2 undergoes upward translation along the helical axis, which contributes to the widening of the constriction formed by the L340 sidechains (Fig. 2E). At the same time, TM1 moves outward as a rigid body, and within the same subunit, the outward motions of TM2 and TM1 appear concerted (Fig. S2).
These motions of TM2 and TM1 are tightly coupled to motions of the ectodomain. In particular, β14 undergoes a clockwise rotation while β1 undergoes a counterclockwise rotation (side view; Fig. 2E). These opposite rotations have two effects. First, the upper ends of the two β-strands close up around the putative ATP binding site, as illustrated by a decrease in the distance between K316 and K70. Second, the lower ends of the two β-strands pull apart, “driving” the outward movement and upward translation of the TM2 N terminus and the rigid-body outward translation of TM1.
Validation of Putative Activated State by Substituted Cysteine Data.
In previous work (35, 36), we have shown that agonist-induced changes in accessibility of modifying agents to substituted cysteines are very valuable for validating structural models of ion-channel activation. Here we use modification and other literature data on substituted cysteines to interrogate our activated state for P2X4R.
Modification of substituted cysteines has placed the channel gate at the N-terminal portion of TM2 (23–25). Upon channel activation, P2X2R residues V48, I332, N333, T336, A337, L338, T339, I341, V343, S345, and F346 have increased accessibility (23, 24). In P2X4R, the corresponding residues are V50, L340, N341, A344, G345, L346, A347 L349, L351, N353, and V354. In Fig. 3A, we show the differences in solvent accessible surface area of these residues between the putative activated state and the resting state, along with the changes in modification rates of these positions upon channel activation. It can be seen that these 11 residues all have increased solvent accessible surface areas in the putative activated state, similar to those indicated by the cysteine modification data. Of the 11 residues (Fig. S2), L340, A344, A347 are pore-facing; V50 is in the small intersubunit TM1-TM2 interface (facing I336); N341 projects into that interface; G345 and N353 are in the intrasubunit TM1-TM2 interface; L346 and L349 project into the spacing between TM1 of the same subunit and TM2 of an adjacent subunit; and L351 and V354 face the wide intracellular vestibule. The increases in solvent accessibility for V50 and N341 in particular are an indication of the separation between the transmembrane helices of neighboring subunits, due to the concerted outward motions of each subunit. However, although the motions within each subunit are apparently concerted, the extent of the motion of the outer TM1 is greater than that of the inner TM2, giving rise to the increased solvent accessibility for G345 and N353.
Fig. 3.

Validation of putative activated state against experimental data. (A) Changes in solvent accessibility of TMD residues upon channel activation, either from substituted cysteine modification rates by Ag+ (23, 24) or from normal mode analysis (NMA). Blue bars are ln (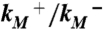 ), where
), where 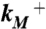 and
and 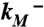 are modification rate constants in the presence and absence of ATP. Red bars are differences in solvent accessible surface area (ΔSASA, in Å2 and shown as scaled by a factor of 2) of residues between the activated and resting states. ΔSASA was calculated by mutating each residue to cysteine in the two states and taking the difference in accessible surface area for the sidechain only. Given the sequence differences between zfP2X4R and rP2X2R, it is perhaps not very meaningful to quantify the comparison. Nevertheless, after excluding L340, the correlation between the experimental and calculation data has an R2 of 0.58. Note that a change in the motional amplitude used for generating the activated model changes the magnitudes of ΔSASAs for the individual residues but not their signs and rankings, so the correlation with experimental data is preserved. (B) The lateral fenestration in the resting and activated states. D61, Q329, and I336 are shown as spheres in darker colors, which are visible only when the spheres are exposed.
are modification rate constants in the presence and absence of ATP. Red bars are differences in solvent accessible surface area (ΔSASA, in Å2 and shown as scaled by a factor of 2) of residues between the activated and resting states. ΔSASA was calculated by mutating each residue to cysteine in the two states and taking the difference in accessible surface area for the sidechain only. Given the sequence differences between zfP2X4R and rP2X2R, it is perhaps not very meaningful to quantify the comparison. Nevertheless, after excluding L340, the correlation between the experimental and calculation data has an R2 of 0.58. Note that a change in the motional amplitude used for generating the activated model changes the magnitudes of ΔSASAs for the individual residues but not their signs and rankings, so the correlation with experimental data is preserved. (B) The lateral fenestration in the resting and activated states. D61, Q329, and I336 are shown as spheres in darker colors, which are visible only when the spheres are exposed.
Recent data of Kawate et al. (27) and Samways et al. (28) support the idea that permeant ions access the transmembrane pore via the lateral fenestrations. Kawate et al. (27) found that the cysteine substitution of a P2X2R residue, I328, whose counterpart (I336) in P2X4R is located at the bottom of the lateral fenestration (Fig. 3B), is inaccessible to a bulky modifying agent in the absence of ATP. Accompanied by ATP, the agent rapidly reacts with I328C, initially potentiating the ATP-activated current (perhaps by stabilizing the activated state while occluding one of the three lateral fenestrations) but eventually blocking the current completely. As Fig. 3B shows, the lateral fenestration widens significantly in our activated state, and I336 becomes more exposed. The widening of the fenestration results from the pulling apart of the lower ends of β14 and β1.
Moreover, Kawate et al. (27) found that a disulfide bond between cysteine substitutions of P2X2R E59 and Q321 (corresponding to P2X4R D61 and Q329, located at the top of the lateral fenestration; Fig. 3B) inhibits the ATP-activated current; breaking the disulfide bond by a reducing agent results in a significant increase in the current. In our activated state, the distance between D61 and Q329 of P2X4R increases as the lower ends of β1 and β14 pull apart, thus explaining the increase in ATP-activated current when the two positions are freed from a disulfide bond. Kawate et al. further observed that a disulfide bond between P2X2R P89C and F291C (corresponding to P2X4R P95 and F299) potentiates the ATP-activated current. The observation could be explained by noting that this disulfide bond tightens the trimeric interface, at a location just above the four conserved cationic residues. This disulfide bond in effect accentuates the conformation changes induced by ATP binding. Explanation of other experimental observations will be deferred to Discussion.
ATP Binding Modes.
No structure has so far been determined for any ATP-bound P2XR. Although the closure of the upper ends of β1 and β14 in two adjacent subunits generated by the normal mode analysis is suggestive of an ATP-induced conformational change, it is of great interest to directly observe the configuration of the bound ATP, its interactions with the binding site, and the induced conformational change of the receptor. Therefore we carried out explicit-solvent molecular dynamics (MD) simulations of the ATP binding process. To reduce the system size and hence speed up the simulations, we included just the ectodomain of zfP2X4R (residues Y57 to N334).
We wanted to place the ATP at a reasonable location and orientation around the resting zfP2X4R structure to start the MD simulations. We took advantage of the information that the four cationic residues, K70 and K72 on β1, R298 on β13, and K316 on β14, at the trimeric interface are likely to line the ATP binding site (10, 11, 14, 16, 19, 20), and ran the AutoDock program (37) by restraining the ATP to a 30-Å box centered at the center of mass of the four-charge cluster. The two positions, L191 and D145 in zfP2X4R, to which NCS-ATP can cross-link are located on opposite sides of the four-charge cluster, suggesting that ATP can bind the receptor in two opposite orientations (22). Thus, after clustering the docked ATP configurations in the putative binding pocket, two orientations of the ATP, with the adenine ring either proximal or distal to the TMD, were selected to start separate MD simulations. In each trajectory, three ATP molecules with threefold symmetric configurations were placed into the trimeric zfP2X4R structure. As reference, simulations of the apo form of zfP2X4R were also carried out.
After 35-ns simulations, the ATP molecules with proximal adenine underwent significant changes in both location and internal conformation, whereas the molecules with distal adenine underwent much less adjustments (Fig. S3). In both orientations, the oxygen atoms of the triphosphate group form stable salt bridges with K70, K72, R298, and K316 (Fig. 4). Accompanying the salt-bridge formation, the upper ends of β1 in one subunit and β14 in the adjacent subunit close up (Fig. 5 A and B). However, the similarity of the conformational changes induced by the ATP molecules in opposite orientations ends here; the rest of the receptor responds very differently to the two ATP binding modes.
Fig. 4.

Two alternative ATP binding modes. (A) Proximal adenine. (B) Distal adenine. Salt bridges between the triphosphate group and the four charged residues are shown by dashed lines. In A, the carbonyl oxygen of G294 forms a hydrogen bond with ATP N6 atom. The side chain of A292 (with Cα shown as a sphere) is poised to form an additional hydrogen bond upon mutation to serine.
Fig. 5.

ATP-induced conformational changes of zfP2X4R in MD simulations. (A) ATP in proximal orientation. (B) ATP in distal orientation. β1 and β14 are shown in both side view (Upper) and top view (Lower). One ATP molecule and β13 in the top front left subunit are also shown in the side view. The β-strands in the resting state are shown in gray as reference. K70 and K316 are shown as spheres to highlight the closure between β1 and β14 at the top; F327 is shown as spheres to indicate whether the β14 C termini move apart.
In the simulations with proximal adenine, the triphosphate group extends outward and the adenine ring wedges between β1 and β13, with N6 forming a hydrogen bond with the backbone carbonyl oxygen of G294 (Fig. 4A). N6 is approximately 4 Å away from Cβ of A292; an A292S mutation could result in an additional hydrogen bond with N6. Interestingly, Petrenko et al. (38) recently identified the corresponding residue, S275, in P2X3R as directly interacting with ATP, and the S275A mutation reduced ATP potency.
When ATP is bound in the proximal orientation, the lower ends of β1 and β14 pull apart (Fig. 5A). The distances between Cα atoms in neighboring subunits at the F327 position (near the C terminus of β14) increase by approximately 3 Å, an outward expansion suggestive of pore open. In contrast, no significant change in F327 Cα distances is seen in the simulations with distal adenine (Fig. 5B). In this case, ATP binding appears to stabilize the lower part of the ectodomain in the closed conformation, suggestive of desensitization.
It is interesting that the receptor responds to the two ATP binding modes so differently. Because the triphosphate group forms similar salt-bridge interactions with the four-charge cluster, the different conformational changes of the receptor can likely be attributed to the different positions of the adenine ring. Below the four-charge cluster, the ridge at the trimeric interface narrows significantly. The proximal adenine, wedged between β1 and β13, acts as a fulcrum for the β14 lever, turning a modest closure around the triphosphate group into significant opening of the pre-TM2 loop. In contrast, when the adenine is in the distal position, no such fulcrum exists and the bound ATP merely serves to tighten the trimeric interface and stabilize the closed conformation.
Concordance of Activation Models from Normal Mode Analysis and MD Simulations.
We propose that the conformational changes of the receptor observed in the MD simulations with proximal adenine represent channel activation. These changes, induced by the ATP binding, are very similar to those in the putative channel-activation normal mode (Fig. S4). In both studies, β14 undergoes clockwise rotation and β1 undergoes counterclockwise rotation, such that the upper ends of these β-strands close up and the lower ends pull apart. The two studies are also complementary: In the MD simulations the closure of the β14 and β1 upper ends are seen to be induced by interactions of the ATP triphosphate group with the four-charge cluster; in the normal mode analysis, the separation of the β14 and β1 lower ends are seen to drive the opening of the channel pore. Further confirmation for the latter action will require MD simulations of the full-length protein.
Discussion
Based on normal mode analysis and molecular dynamics simulations, we have developed a complete model, at the atomic level, for the gating mechanism of a P2X4 receptor. Our gating model is validated by some key experimental data, and as discussed below, explains other experimental observations on channel activity. Our computational studies provide insight for designing P2XR antagonists and contribute to the mechanistic understanding of ligand-gated ion channels in general.
Inter- and Intrasubunit Coupling During Channel Activation.
Coupled motions between the subunits in the ectodomain, between the ectodomain and the TMD, and between the transmembrane helices of the TMD are expected to be essential for transmitting the action of ATP binding to affect channel opening. Mutations in these junctures have been observed to result in perturbed channel activity. Below we discuss these mutations in light of our activation model.
Jiang et al. (26) introduced cysteines at E63 and R274 of P2X2R (corresponding to L64 in β1 and R280 in β12 of P2X4R, respectively) and found that they formed a disulfide bond in the absence of ATP. The presence of ATP reduced the rate of disulfide bond formation, indicating that ATP binding might induce the separation of the adjacent subunits at the level of these two positions. Jiang et al. suggested that this separation is transmitted to the TMD to open the channel. This suggestion is consistent with our gating model. A crucial motion in this model is the pulling apart of the lower ends of β1 and β14. The lower end of β14 forms a β-sheet with β11 and β12, and therefore they undergo concerted motion—as β14 pulls away from β1 so does β12.
In the zfP2X4R structure, Y57 in the post-TM1 loop forms a hydrogen bond with D267 in the β11-β12 loop of the same subunit. This interaction may be important for coupling the ectodomain with the TMD (6); the corresponding Y55C and D261A mutants of P2X2R lost response to ATP (11, 39). In our activated structure, the hydrogen bond between Y57 and D267 is maintained, contributing to the concerted outward motions of TM1 and TM2 within the same subunit (Fig. S5). Losing the hydrogen bond would weaken the intrasubunit TM1-TM2 coupling and possibly impair channel function.
In zfP2X4R, V50 and I336 are in the small intersubunit TM1-TM2 interface (Fig. S2). In our activated state, this interface increases in spacing due to the concerted outward motions of each subunit. The separation between V50 and I336 explains the inhibition of the ATP-activated current by a disulfide bond between the corresponding positions (V48 and I328) in P2X2R (39). The intersubunit disulfide bonds would limit the separation of the subunits in our activation model.
Two Opposite ATP Orientations.
Our MD simulations showed that ATP can bind P2XR in two different orientations. While the triphosphate group interacts with a four-charge cluster, the adenine can be either below or above the charge cluster, i.e., either proximal or distal to the TMD. The proximal adenine acts as a fulcrum, leading to conformational changes that open the channel, but with the distal adenine, the closed conformation is stabilized.
The two different ATP orientations were suggested by the observation that NCS-ATP can cross-link with both L186C and N140C of P2X2R (22), placing the adenine in either the proximal or distal position. But importantly, our simulation results with the two opposite ATP orientations can explain the distinct functional consequences of labeling at L186C and at N140C. NCS-ATP labeling at L186C probably results in conformational changes similar to those induced by an ATP molecule bound in the proximal orientation, thus priming the receptor for “proximal” binding of two other ATP molecules at the neighboring trimeric interfaces, leading to the observed potentiation effect. In contrast, NCS-ATP labeling at N140C is akin to binding an ATP molecule in the distal orientation, leading to the observed inhibition effect.
The occurrence of two opposite orientations for ATP binding is supported by the fact that diadenosine polyphosphates (e.g., Ap4A), in which two adenines are linked by a chain of (e.g., four) phosphates, are potent agonists for P2XRs (40). It is likely that the two adenines of Ap4A bind to opposite sides of the tetraphosphate group. The proximal adenine acts as the fulcrum for transmitting the ligand-binding action to the TMD; the second adenine, on the distal side, where the binding pocket widens (Fig. 4), may form additional interactions with the receptor.
Our MD simulations showed that “distal” binding of ATP stabilizes the receptor in the closed conformation. We further speculate that this binding mode corresponds to channel desensitization. Suppose that ATP in the proximal orientation has faster but weaker binding than in the distal orientation. ATP molecules would first bind in the proximal orientation and then exchange into the distal orientation. Consequently the receptor would first be activated and then become desensitized. A desensitization mechanism resulting from exchange of agonist orientations seems very intriguing.
The potential for distinct functional consequences of the two alternative binding modes offers opportunities for designing P2XR antagonists. Antagonists could result from modifications of ATP that would favor binding in the distal orientation over the proximal orientation. In view of the 40% to 50% sequence identities between their ectodomains (6), the seven P2XR subtypes may respond differently to these modifications, possibly resulting in subtype-selective antagonists.
Other Ligand-Gated Ion Channels.
It is interesting to compare the gating mechanism developed here with those for other ligand-gated ion channels. The TMD of ASIC1 is similar in architecture to that of P2XRs, comprised of two transmembrane helices from each subunit in a trimer (41). The patterns of TM2 substituted cysteine modification rates in the closed and open states (42) are also similar to those of P2X2R (24), both ruling out significant TM2 rotation during pore opening. The atomistic details revealed by the present computational studies on P2XR activation might be instructive for ASIC1.
Like P2XRs, the ligand-binding site of nicotinic acetylcholine receptors is located at the oligomeric interface, and MD simulations (35, 43) suggest that a long β-strand (β10) is involved in transmitting the action of ligand binding to the TMD. However, in P2XRs, the pivotal β-strand (β14) is directly connected to the pore-lining helix, whereas in acetylcholine receptors the pivotal β-strand spatially separated from the pore-lining helix (M2) by several loops.
Ionotropic glutamate receptors differ from P2XRs in key respects, including a ligand-binding site located within a two-lobed domain of each subunit, and lobe closure as pivotal in transmitting the action of ligand binding to the TMD. But similar to what is seen here for P2X4R, the pore-lining helix (M3) and an outer helix (M1) of the same subunit undergo concerted motions (36).
In summary, our computational studies produced a complete model for how ATP binding leads to P2XR channel activation. The detailed gating mechanism explains a host of experimental observations and provides insight for designing antagonists.
Methods
Normal Mode Analysis.
The starting structure of zfP2X4R was taken from the Brookhaven Protein Data Bank entry 3H9V (9), with missing Arg137 and other missing sidechains built and hydrogen atoms added. Normal mode analysis was done by the elNémo program (44). Elastic connections were made between each atom and its neighbors within a 12-Å cutoff. Of the 100 lowest-frequency modes, the 10 leading to the largest widening of the channel pore ranged in mode numbers (sorted in ascending eigenvalues) from 61 to 97. Of these 10, two, modes 82 and 85, were also among the 10 modes with the largest closure motions around the putative ATP binding site. We chose mode 82 as representing channel activation. The original mode lacked threefold symmetry. The displacement of the subunit with the largest pore widening was replicated to the other two subunits to generate the symmetric model for the activated state. This model is similar to mode 85 in many respects. Both are soft modes, in the sense that the collectivity, at approximately 0.45 in each case, is relatively high (collectivity at 1 corresponds to a mode in which the motions of all the atoms have the same amplitude). Although it would be possible to generate models for the activated state by combining two or more modes, there was no reason to suggest that these models would be superior to the model represented by the single modes of 82 and 85.
MD Simulations.
Parallel simulations of the ectodomain in the apo form and with ATP molecules bound in two different orientations were carried out. The starting pose of the ATP molecules was generated by AutoDock Vina (37). Each system was neutralized with sodium and chloride ions at a 0.1 M concentration and solvated by TIP3P water molecules. The total numbers of atoms in the three systems were approximately 182,000, and the dimensions of the simulation boxes were 118 × 118 × 128 Å3. The simulations were carried out by the NAMD program version 2.7b3 (45). Charmm22 all-hydrogen parameters were used (46). Periodic boundary conditions were applied and the particle mesh Ewald method was used to treat long-range electrostatic interactions. The simulations were run for 35 ns.
The configurations of the three ATP molecules with distal adenine were stable during the 35-ns simulations. In contrast, the three ATP molecules with proximal adenine behaved differently after 10 ns: One moved relatively little from the initial pose; one had the adenine leaving the binding pocket; and one had the adenine inserted between β1 and β13 and the triphosphate group turned outward. We focused on the last ATP configuration. From the snapshot at 18.6 ns, the conformation of the subunit with β13 wrapping this ATP was replicated to the other two subunits to produce a symmetrized trimer. The new trimer was stable in a 10-ns simulation (Cα root-mean-square-deviation at approximately 1.5 Å) and was assumed to model the activated state.
Repeat simulations with ATP molecules initially in the proximal orientations were also carried out. The “activated” configuration was reproduced.
Supplementary Material
ACKNOWLEDGMENT.
This work was supported by National Institutes of Health Grant GM58187.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1119546109/-/DCSupplemental.
References
- 1.Khakh BS, Alan North R. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442:527–532. doi: 10.1038/nature04886. [DOI] [PubMed] [Google Scholar]
- 2.Sim JA, et al. Altered hippocampal synaptic potentiation in P2X4 knock-out mice. J Neurosci. 2006;26:9006–9009. doi: 10.1523/JNEUROSCI.2370-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook SP, Vulchanova L, Hargreaves KM, Elde R, McCleskey EW. Distinct ATP receptors on pain-sensing and stretch-sensing neurons. Nature. 1997;387:505–508. doi: 10.1038/387505a0. [DOI] [PubMed] [Google Scholar]
- 4.Surprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol. 2009;71:333–359. doi: 10.1146/annurev.physiol.70.113006.100630. [DOI] [PubMed] [Google Scholar]
- 5.Jarvis MF, Khakh BS. ATP-gated P2X cation-channels. Neuropharmacology. 2009;56:208–215. doi: 10.1016/j.neuropharm.2008.06.067. [DOI] [PubMed] [Google Scholar]
- 6.Browne LE, Jiang L-H, North RA. New structure enlivens interest in P2X receptors. Trends Pharmacol Sci. 2010;31:229–237. doi: 10.1016/j.tips.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young MT. P2X receptors: Dawn of the post-structure era. Trends Biochem Sci. 2010;35:83–90. doi: 10.1016/j.tibs.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang L-H, et al. Subunit arrangement in P2X receptors. J Neurosci. 2003;23:8903–8910. doi: 10.1523/JNEUROSCI.23-26-08903.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawate T, Michel JC, Birdsong WT, Gouaux E. Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature. 2009;460:592–598. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ennion S, Hagan S, Evans RJ. The role of positively charged amino acids in ATP recognition by human P2X1 receptors. J Biol Chem. 2000;275:29361–29367. doi: 10.1074/jbc.M003637200. [DOI] [PubMed] [Google Scholar]
- 11.Jiang L-H, Rassendren F, Surprenant A, North RA. Identification of amino acid residues contributing to the ATP-binding site of a purinergic P2X receptor. J Biol Chem. 2000;275:34190–34196. doi: 10.1074/jbc.M005481200. [DOI] [PubMed] [Google Scholar]
- 12.Roberts JA, Evans RJ. ATP binding at human P2X1 receptors. J Biol Chem. 2004;279:9043–9055. doi: 10.1074/jbc.M308964200. [DOI] [PubMed] [Google Scholar]
- 13.Roberts JA, Evans RJ. Contribution of conserved polar glutamine, asparagine and threonine residues and glycosylation to agonist action at human P2X1 receptors for ATP. J Neurochem. 2006;96:843–852. doi: 10.1111/j.1471-4159.2005.03593.x. [DOI] [PubMed] [Google Scholar]
- 14.Wilkinson WJ, Jiang L-H, Surprenant A, North RA. Role of ectodomain lysines in the subunits of the heteromeric P2X2/3 receptor. Mol Pharmacol. 2006;70:1159–1163. doi: 10.1124/mol.106.026658. [DOI] [PubMed] [Google Scholar]
- 15.Marquez-Klaka B, Rettinger J, Bhargava Y, Eisele T, Nicke A. Identification of an intersubunit cross-link between substituted cysteine residues located in the putative ATP binding site of the P2X1 receptor. J Neurosci. 2007;27:1456–1466. doi: 10.1523/JNEUROSCI.3105-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zemkova H, et al. Role of aromatic and charged ectodomain residues in the P2X4 receptor functions. J Neurochem. 2007;102:1139–1150. doi: 10.1111/j.1471-4159.2007.04616.x. [DOI] [PubMed] [Google Scholar]
- 17.Roberts JA, et al. Cysteine substitution mutagenesis and the effects of methanethiosulfonate reagents at P2X2 and P2X4 receptors support a core common mode of ATP action at P2X receptors. J Biol Chem. 2008;283:20126–20136. doi: 10.1074/jbc.M800294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodnar M, et al. Amino acid residues constituting the agonist binding site of the human P2X3 receptor. J Biol Chem. 2011;286:2739–2749. doi: 10.1074/jbc.M110.167437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans RJ. Structural interpretation of P2X receptor mutagenesis studies on drug action. Br J Pharmacol. 2010;161:961–971. doi: 10.1111/j.1476-5381.2010.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev. 2011;63:641–683. doi: 10.1124/pr.110.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marquez-Klaka B, Rettinger J, Nicke A. Inter-subunit disulfide cross-linking in homomeric and heteromeric P2X receptors. Eur Biophys J. 2009;38:329–338. doi: 10.1007/s00249-008-0325-9. [DOI] [PubMed] [Google Scholar]
- 22.Jiang R, et al. Agonist trapped in ATP-binding sites of the P2X2 receptor. Proc Natl Acad Sci USA. 2011;108:9066–9071. doi: 10.1073/pnas.1102170108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, Chang T-H, Silberberg SD, Swartz KJ. Gating the pore of P2X receptor channels. Nat Neurosci. 2008;11:883–887. doi: 10.1038/nn.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M, Kawate T, Silberberg SD, Swartz KJ. Pore-opening mechanism in trimeric P2X receptor channels. Nat Commun. 2010;1:44. doi: 10.1038/ncomms1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kracun S, Chaptal V, Abramson J, Khakh BS. Gated access to the pore of a P2X receptor. J Biol Chem. 2010;285:10110–10121. doi: 10.1074/jbc.M109.089185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang R, et al. A putative extracellular salt bridge at the subunit interface contributes to the ion channel function of the ATP-gated P2X2 receptor. J Biol Chem. 2010;285:15805–15815. doi: 10.1074/jbc.M110.101980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawate T, Robertson JL, Li M, Silberberg SD, Swartz KJ. Ion access pathway to the transmembrane pore in P2X receptor channels. J Gen Physiol. 2011;137:579–590. doi: 10.1085/jgp.201010593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samways DSK, Khakh BS, Dutertre S, Egan TM. Preferential use of unobstructed lateral portals as the access route to the pore of human ATP-gated ion channels (P2X receptors) Proc Natl Acad Sci USA. 2011;108:13800–13805. doi: 10.1073/pnas.1017550108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng X, Lu B, Grant B, Law RJ, McCammon JA. Channel opening motion of α7 nicotinic acetylcholine receptor as suggested by normal mode analysis. J Mol Biol. 2006;355:310–324. doi: 10.1016/j.jmb.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 30.Taly A, et al. Implications of the quaternary twist allosteric model for the physiology and pathology of nicotinic acetylcholine receptors. Proc Natl Acad Sci USA. 2006;103:16965–16970. doi: 10.1073/pnas.0607477103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samson AO, Levitt M. Inhibition mechanism of the acetylcholine receptor by alpha-neurotoxins as revealed by normal-mode dynamics. Biochemistry. 2008;47:4065–4070. doi: 10.1021/bi702272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu F, Hummer G. Gating transition of pentameric ligand-gated ion channels. Biophys J. 2009;97:2456–2463. doi: 10.1016/j.bpj.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang H, et al. Inherent dynamics of the acid-sensing ion channel 1 correlates with the gating mechanism. PLoS Biol. 2009;7:e1000151. doi: 10.1371/journal.pbio.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tirion MM. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys Rev Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 35.Yi M, Tjong H, Zhou H-X. Spontaneous conformational change and toxin binding in α7 acetylcholine receptor: Insight into channel activation and inhibition. Proc Natl Acad Sci USA. 2008;105:8280–8285. doi: 10.1073/pnas.0710530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong H, Zhou H-X. Atomistic mechanism for the activation and desensitization of an AMPA-subtype glutamate receptor. Nat Commun. 2011;2:354. doi: 10.1038/ncomms1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrenko N, Khafizov K, Tvrdonova V, Skorinkin A, Giniatullin R. Role of the ectodomain serine 275 in shaping the binding pocket of the ATP-gated P2X3 receptor. Biochemistry. 2011;50:8427–8436. doi: 10.1021/bi200812u. [DOI] [PubMed] [Google Scholar]
- 39.Jiang L-H, Rassendren F, Spelta V, Surprenant A, North RA. Amino acid residues involved in gating identified in the first membrane-spanning domain of the rat P2X2 receptor. J Biol Chem. 2001;276:14902–14908. doi: 10.1074/jbc.M011327200. [DOI] [PubMed] [Google Scholar]
- 40.Wildman SS, Brown SG, King BF, Burnstock G. Selectivity of diadenosine polyphosphates for rat P2X receptor subunits. Eur J Pharmacol. 1999;367:119–123. doi: 10.1016/s0014-2999(98)00976-5. [DOI] [PubMed] [Google Scholar]
- 41.Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. 2009;460:599–604. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li T, Yang Y, Canessa CM. Outlines of the pore in open and closed conformations describe the gating mechanism of ASIC1. Nat Commun. 2011;2:399. doi: 10.1038/ncomms1409. [DOI] [PubMed] [Google Scholar]
- 43.Cheng X, Wang H, Grant B, Sine SM, McCammon JA. Targeted molecular dynamics study of C-loop closure and channel gating in nicotinic receptors. PLoS Comput Biol. 2006;2:e134. doi: 10.1371/journal.pcbi.0020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suhre K, Sanejouand Y-H. ElNémo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:610–614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 47.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MSP. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph. 1996;14:354–360. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.