

Abstract
Helicobacter pylori(H. pylori) infection has been linked to the development of chronic gastritis, duodenal ulcer disease, and gastric cancer. H. pylori- infected patients and animal models develop hypergastrinemia, chronic gastritis, and gastric atrophy. Since gastrin is an important regulator of gastric acid secretion and cell growth, H. pylori regulation of this hormone has been implicated in its pathogenesis. We investigated the effect of H. pylori infection on gastrin gene expression in mice and the effect of human isolates of the bacteria on gastrin transcription in a cell line. In addition to an increase in gastrin mRNA in H. pylori-infected mice, we found that the bacteria induced the endogenous human gastrin gene through MAP kinase-dependent signaling but not NFκB-dependent signaling. Moreover, activation of gastrin through MAPK signaling did not require CagA or VacA virulence factors. In transfection studies, we demonstrated that H. pylori-induction of the gastrin promoter thorough a GC-rich motif was mediated by inducible binding of Sp1 and Sp3 transcription factors. In conclusion, co-culturing live H. pylori bacteria with human cells is sufficient to induce gastrin gene expression.
Keywords: Sp1, Sp3, amanitin, SS1, CagA, MAP Kinase
Introduction
Helicobacter pylori (H. pylori) is a gastrointestinal pathogen that has been linked to the development of chronic gastritis, duodenal ulcer disease, and gastric cancer 1–4. Although the global average rate of infection is near 60%, most individuals infected with H. pylori remain asymptomatic. Nevertheless, gastric carcinoma remains the second leading cause of cancer deaths worldwide and H. pylori infection increases the risk of developing this disease3,5–7. However, the mechanisms by which H. pylori contribute to the development and progression of gastrointestinal pathology are unclear.
There is significant genetic diversity among H. pylori strains although bacteria that express the cytotoxin-associated gene A pathogenicity island (cagPAI) and virulence factors such as CagA are typically associated with more severe disease manifestations 5,6,8,9. The cagPAI encodes a type IV secretion system (T4SS), which translocates the CagA protein into the host cell where it is phosphorylated 8,9. In comparison to cagPAI mutant or deficient strains, wild type bacteria stimulate activation of transcription factors such as NFκB and AP1. CagPAI+ strains also show stronger activation of MAP kinase signaling pathways 6,10,11. Therefore host cell gene expression is modulated by components of the T4SS and possibly cagPAI encoded virulence factors such as CagA. Furthermore, genetic variations among H. pylori species might account for the highly variable consequences of H. pylori infection in humans 12
In addition to gastritis and gastric atrophy, hypergastrinemia is observed in a subset of H. pylori -infected patients 13–15. However, there are few studies examining H. pylori-regulation of human gastrin gene expression in vivo or in primary human G cells. Calam and coworkers showed that patients infected with H. pylori increased gastrin mRNA levels 16, while Sumii et al. reported no change in gastrin mRNA 17. Buchan and coworkers reported that H. pylori increases basal levels of gastrin in primary G cell cultures, but does not induce secretion, suggesting that H. pylori increases gastrin synthesis and possibly gene expression, but mRNA levels were not measured 18.
Similarly in several rodent models of H. pylori infection, the number of antral G cells increase coincident with elevated serum gastrin levels19,20. Antral gastrin is the primary hormonal regulator of gastric acid secretion. Furthermore, gastrin acts as a growth factor for gut-derived cell types. The essential role of gastrin in acid regulation and maintenance of gastric homeostasis suggests possible mechanisms by which H. pylori might alter gastrointestinal physiology by regulating the levels of gastrin. Since Helicobacter infection inhibits acid secretion 21, the observed hypergastrinemia might be in response to the hypochlorhydria. Indeed, direct inhibition of acid secretion by omeprazole is sufficient to stimulate gastrin gene expression in vivo 22. Prior studies suggested that Helicobacter colonizing the gastric antrum might create an alkaline pH sufficient to stimulate G cells through its production of urease and conversion of urea to ammonia 23. However, this mechanism was subsequently disproven by studies showing that H. pylori products rather than the live organism can stimulate gastrin release from cultured G cells 24,25. These results suggest that cellular components even from dead bacteria are sufficient to stimulate hormone release.
Recent studies by Kidd et al. demonstrate that multiple extracellular signals, e.g., pH, LPS, serotonin, β–adrenergic receptors and intracellular pathways, e.g., cAMP, ERKs, NFκb regulate gastrin release from primary G cells 26. However, the authors did not examine regulation of gastrin gene expression in response to these various conditions. Although suppression of somatostatin is the strongest inducer of gastrin release, we found that pro-inflammatory cytokines were able to stimulate gastrin release in the somatostatin null mouse demonstrating that extracellular signals other than somatostatin are capable of inducing gastrin secretion 27. Taken together, it is unclear whether the hypergastrinemia that occurs in H. pylori-infected individuals is due to hypochlorhydria, suppression of somatostatin, chronic gastritis, gastric atrophy or the direct induction of gastrin gene expression by the bacteria itself. Certainly several mechanisms are likely to contribute to the increase in plasma gastrin.
It has been shown using cell lines that oncogenic K-ras and EGF receptor ligands induce gastrin gene expression 28–30. Indeed, a gastrin EGF response element (gERE) was mapped to a GC-rich sequence within the first 240 bp of the gastrin promoter. This proximal promoter element binds the transcription factors Sp1, Sp3 and ZBP-89 31. Moreover, we showed previously that Mek1-dependent phosphorylation of Sp1 mediates EGF-activation of the gastrin promoter through this element 32. It has been established in numerous studies that H. pylori and possibly bacterial proteins such as CagA regulate gene expression through activation of signal transduction cascades that target specific transcription factors. Furthermore, a prior study established a MEK/ERK-dependant mechanism for the activation of both Sp1 and Sp3 in the H. pylori -mediated induction of the vascular endothelial growth factor-A (vegf-A) gene 33. Therefore, we tested the hypothesis that H. pylori regulate gastrin gene expression through specific DNA promoter elements that bind Sp1.
Methods
Helicobacter strains and cultures
The 26695 and SS1 H. pylori WT and mutant strains were generated as previously described 34. The J99 strain was obtained from ATCC. All H. pylori strains were grown on blood agar plates prior to inoculation of Brucella broth (DIFCO) supplemented with 10% heat-inactivated fetal bovine serum (FBS), Skirrow’s antibiotic and amphotericin B. H. pylori cultures were maintained by shaking in a gas exchange incubator under microaerophilc conditions at 37°C. The broth used for infection was cultured overnight. The presence of H. pylori was verified biochemically by catalase and urease tests as well as microscopic analysis for size, shape, and motility.
In vivo H. pylori infection in mice
Six to eight week old C57BL/6 mice were orally inoculated once with 1 × 108 CFU of broth-cultured H. pylori, strain SS1, in 0.1 ml of broth. Control mice were not given bacteria. Mice were sacrificed 6 months after inoculation, and gastric tissue was collected for histological examination, PCR, and qRT-PCR analysis. Gastric RNA was harvested from mucosal scrapings of the whole stomach using TrIzol reagent (Invitrogen) according to the manufacturer's instructions. Purification of mRNA was accomplished using the QIAGEN’s RNeasy MiniKit and stored at 80°C. Generation of cDNAs was performed using the Invitrogen SuperScript First Strand kit and random hexamer primers.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated from AGS cells using Trizol reagent (Invitrogen) and DNAse treated (Roche). RNA quantitation was performed using the QuantiT RiboGreen RNA Assay Kit (Molecular Probes). Synthesis of cDNA was performed for 1 μg of mouse and human RNA using the I-script cDNA Synthesis Kit (Bio-Rad).
All qRT-PCR reactions were preformed in triplicate using the Bio-Rad I-cycler. The following primers were used to detect and quantify gene expression: mouse gastrin: (forward) ACACAACAGCCAACTATTC, (reverse) CAAAGTCCATCCATC CGTAG. Mouse GAPDH: (forward) TCAAGAAGGTGGTGAAGCAGG, (reverse) TATTATG GGGGTCTGGGATGG. Human gastrin (forward) CCCAGGCTCTCATCATCGAAGG, (reverse) GCCGAAGTCCATCCATCCATAGG. Human 18S rRNA: (forward) GATATGCTCAT GTGGTGTTG, (reverse) AATCTTCTTCAGTCGCTCCA 35. Human IL-8: (forward) TAGCAAAA TTGAGGCCAAGG, (reverse) AAACCAAGGCACAGTGGAAC.
Reactions using mouse RNA and primers were done at a volume of 20μl and contained the following: 1x reaction buffer, 5.5 mM MgCl2, 100nM of forward and reverse primers, 10nM fluorescein, 200μM dNTPs, SYBR green, and 0.025 units of Platinum Taq polymerase (Invitrogen). Amplification was performed under the following conditions; 3 min at 95°C, 35-40 cycles of 9s at 95°C and 1 min at 60°C, and 1 min at 55°C. Expression of mouse target genes was normalized to GAPDH.
Reactions using human primers were done at a volume of 20μl and contained the following: 1x reaction buffer, 1.5 mM MgCl2, 100nM of forward and reverse primers, 10nM fluorescein, 200uM dNTPs, SYBR green, and 0.025 U of Amplitaq Gold polymerase (Applied Biosystems). Reactions were performed with the following conditions: 50°C for 2 min and 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Gene expression was normalized to 18s rRNA. Expression of human target genes was normalized to 18s rRNA. The fold change for all reactions was calculated using the following equation .
Cell culture
AGS cells (human gastric adenocarcinoma) were grown in DMEM (Gibco- BRL) containing 10% FCS, 100 μg/ml penicillin, and 100 μg/ml streptomycin at 37°C with 5% CO2. The cells were serum starved for 48 h in serum-free HAMS/F12 media (Gibco) prior to any treatment or co-culture with H. pylori.
Plasmids and constructs
To generate the pGL3 gastrin reporter constructs, segments from -3.3 to -0.190 kb of the human gastrin promoter were ligated into the pGL3 basic luciferase vector (Promega). Gastrin promoter fragments from -2.3 to -0.19 kb were isolated by restriction digest from the -3.3 kb gastrin proximal promoter fragment. Site-directed mutations were introduced into the -0.240 kb construct using the QuickChange II Kit (Stratagene). Primers designed to introduce mutations were generated using the primer design application on the Stratagene website. All constructs were confirmed by sequence analysis. Several rounds of single mutation reactions were done to generate constructs with mutations in multiple sites. The activity of the Path Detect pAP1-Luc Cis-Reporter Plasmid (Stratagene) was used as a positive control for activation by H. pylori observed in the transfection experiments.
Transient transfections and luciferase reporter assays
AGS cells were seeded onto 12 or 24 well plates and grown to 50%–60% confluency prior to transfection. Plasmid DNA at concentrations of 0.5–1 μg/well were transfected using Fugene 6 (Roche) according to the manufacturer's protocol. The promoterless phRG-B Renilla luciferase reporter (Promega) served as a transfection control to normalize firefly luciferase activity. The cells were treated then harvested at 48 h after transfection. Firefly and Renilla luciferase were measured using the Dual Luciferase Assay Kit (Promega).
H. pylori co-culture with AGS cells
AGS cells were seeded at 50–75% confluency and cultured in serum-free media containing antibiotics for 48 h prior to H. pylori inoculation. H. pylori were quantified then resuspended in F12 media without antibiotics. The AGS cells were washed with PBS and maintained in serum-free and antibiotic-free media for the duration of the co-culture experiments with H. pylori.
Western blot analysis
Whole cell extracts were generated from AGS cells that were cultured in serum-free media 48h prior to treatment with chemical inhibitors and H. pylori co-culture. Cell pellets were lysed with M-PER Mammalian Protein Extraction Reagent (Pierce) containing complete protease inhibitor tablets (Roche) according to the manufacturers instructions. The protein concentration for each sample was determined using the bicinchoninic acid assay (Pierce Biochemicals). Protein lysates were resolved on 4–20% SDS- polyacrylamide gradientgels then transferred to Hybond-C Extra nitrocellulose membranes (Amersham). Membranes were blocked using KPL blocking solution (KPL, Inc, Gaithersburg, MD). Primary antibodies for GAPDH (Millipore), ERK1/2 (Cell Signaling), p38 (Cell Signaling), Sp1 (Santa Cruz Biotechnology), and Sp3 (Santa Cruz Biotechnology) were used at a concentration of 1:1000. Phosho-antibodies against p38 (Cell Signaling), ERK1/2 (Cell Signaling), and cJun (Cell Signaling) were used at a 1:500 concentration. Proteins were visualized using HRP-conjugated secondary antibodies and ECL reagent from the SuperSignal West Pico Chemiluminescent Kit (Thermo Fischer Scientific).
Electophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared using non-ionic detergent extraction from AGS cells following co-culture with H. pylori for various times. Oligonucleotide probes were designed according to regions of the human gastrin promoter. An AP1 binding oligo was designed using two consecutive API binding sites (TGCATCA x 2) and used as a positive control for H. pylori induced DNA binding. All oligos were flanked by BamHI and Bgl II sequences at the 5' and 3' ends respectively, annealed then labeled with [32P] γ-ATP using polynucleotide kinase (Roche). Gel shift assays were carried out at a final volume of 20 μl and contained 10mM Tris-HCl, 1mM EDTA, 1mM DTT, 5mM MgCl2, 1mM ZnCl2, 150mM KCl, 10% glycerol, and 300 ng polydI-dC. One microgram of antibodies against Sp1 or Sp3 antibodies (Santa Cruz Biotechnology) were used for gel shift assays to identify DNA binding proteins. One microliter of labeled probe at 30,000 cpm/μl was added to each reaction. DNA complexes were resolved on a 6% nondenaturing polyacrylamide gel containing the 45 mM Tris base, 45 mM boric acid, 1 mM EDTA (TBE) buffer.
Chemical inhibitors
Inhibitors were added to cells 30 min prior to co-culture with H. pylori. The following inhibitors were used: α-amanitin (Sigma), PD98050 (Cell Signaling Technology), ammonium pyrrolidinedithiocarbamate (APDT, Calbiochem), SB203580 (Calbiochem), and SP600125 (Calbiochem).
Statistics
Data were compared using the Mann-Whitney U test. The mean ± standard error of the mean (SEM) are shown. A P value of <0.05 (*) was considered significant.
Results
H. pylori Infection Induces Gastrin Gene Expression in Mice
In human subjects and animal models, H. pylori infection is associated with chronic gastritis and hypergastrinemia. However, increases in plasma gastrin and the population of G-cells have not been consistently correlated with changes in gastrin gene expression 16,17. Therefore, we studied gastrin gene expression in a mouse model of H. pylori infection. C57BL/6 mice were infected with the mouse-adapted SS1 strain of H. pylori and harvested after 6 months. Persistence of the bacterial infection was confirmed by PCR amplification of the bacterial 16S gene. Using quantitative PCR, a 10-fold increase in gastrin mRNA was observed in mice infected with H. pylori when compared to uninfected controls (Fig. 1). The observed increase in gastrin mRNA expression suggested that infection by H. pylori correlated with an increase in gastrin gene expression. Since a direct effect of the bacteria on the G cell cannot be established in vivo we utilized a human AGS gastric cell line to characterize the effect of H. pylori on gastrin gene expression.
Figure 1. H. pylori Stimulate Gastrin Gene Expression in Infected Mice.
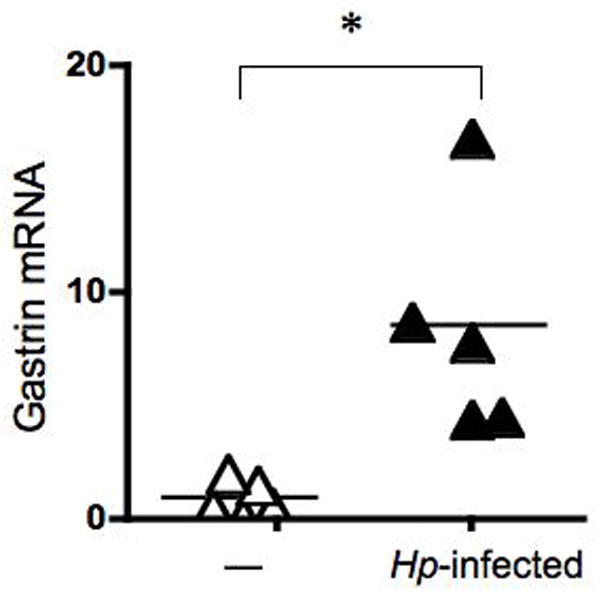
Mice were infected with H. pylori SS1and analyzed six months after infection. qRT-PCR analysis of gastrin normalized to GAPDH (Gastrin mRNA) was performed using RNA isolated from the stomachs of infected and uninfected (—) mice (N = 5 mice). The mean is indicated by a bar. *P < 0.05.
Gastrin mRNA Induction Requires Live Bacteria, But Not CagA
We examined the ability of H. pylori to regulate endogenous gastrin gene expression was examined in AGS cells by qRT-PCR. Cells were co-cultured with the 26695 H. pylori strain, which is a wild type, human isolate 36. The endogenous human gastrin gene was maximally induced 6 to 7-fold by 9 h (Fig. 2A) with the maximal 100 m.o.i. (Fig. 2B).
Figure 2. H. pylori Stimulate Expression of Gastrin in AGS Cells.
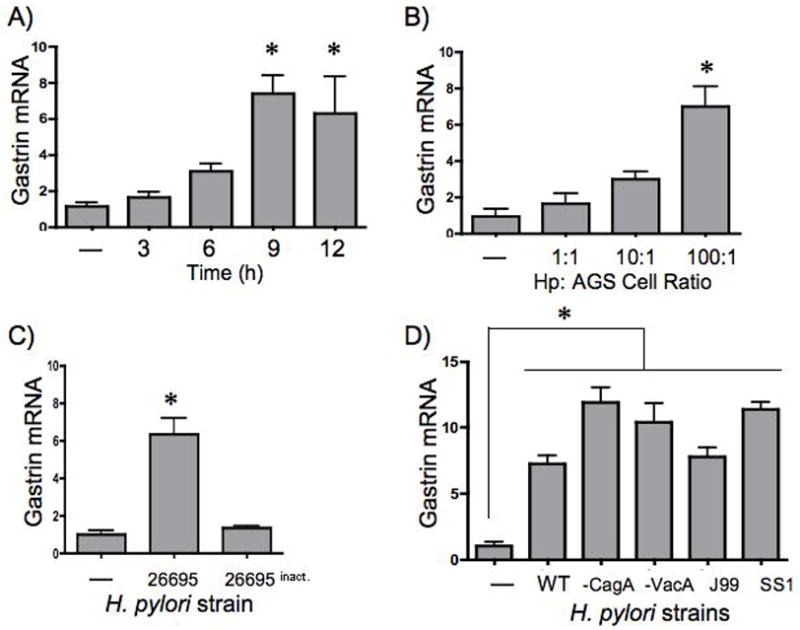
AGS cells were infected with H. pylori strain 26695 (a human isolate) 48h after serum starvation. The cells were cultured with bacteria at a m.o.i of 100 to 1 at various times (A) or with increasing amounts of bacteria (m.o.i) for 9h (B). All subsequent H. pylori co-culture experiments were performed at 100 to 1 m.o.i. for 9h. AGS cells were co-cultured with live or inactive (formalin fixed, heat-treated) bacteria (C). qRT-PCR analyses of gastrin and 18S rRNA transcripts were performed on total RNA and the amount of gastrin mRNA was normalized to 18S rRNA (Gastrin mRNA). All samples were compared to the untreated sample which was set to 1. The mean fold change + SEM is shown for three independent experiments. *P < 0.05 compared to untreated cells was considered significant. (D) AGS cells were co-cultured with the wild type H. pylori strain 26695 (WT), or isotypes mutants of 26695 that were null for CagA (-CagA) or VacA (-VacA). Two additional human Hp isolates were also used, the wild type J99 strain and the mouse-adapted SS1 strain. qRT-PCR analyses of gastrin and 18S rRNA transcripts were performed. Gastrin mRNA was normalized to the 18S transcript. The mean fold change + SEM three experiments is shown. *P< 0.05 compared to untreated cells was considered significant.
After establishing the optimal conditions for gastrin induction we investigated whether live bacteria were required for the effect. AGS cells were co-cultured with live H. pylori or bacteria that was heat-inactivated and formalin fixed. Indeed, we found that killed bacteria were unable to induce gastrin mRNA expression (Fig. 2C). Similar results were observed using the SS1 strain under the same conditions (data not shown).
In H. pylori-infected patients, there is significant variability in the clinical outcome that is often associated with the genetic variations between H. pylori strains. Similarly, laboratory studies have indicated that cagPAI+ and CagA+ strains influence the ability of H. pylori to regulate gene expression and activate specific signal transduction pathways. Therefore, we investigated whether there were differences among H. pylori strains that might influence their ability to induce the gastrin gene. AGS cells were co-cultured with three wild type strains 26695, J99 as well as the SS1 strain, all of which contain complete cagPAI37, despite the prevailing view that the SS1 cagPAI is dysfunctional. Moreover, we examined gastrin gene expression following co-culture with 26695 isogenic mutants that lacked the CagA and vacuolating toxin A (VacA) gene with AGS cells. All of the H. pylori induced gastrin mRNA expression. Higher levels were observed with the CagA- and VacA-deficient strains as well as the SS1 strain (Fig. 2D). Since the VacA-deficient strain showed levels of gastrin induction comparable to those of the CagA-deficient and SS1 strains, we concluded that the increased activation of gastrin mRNA is not influenced directly by either CagA or VacA but may be the result of other bacterial proteins.
Induction of Gastrin Requires Activation of MAPK Signaling
It has been shown previously that H. pylori activate multiple signaling pathways including MAP kinase, NFκB, and PI3K 38. Therefore, AGS cells were treated with chemical inhibitors of the ERK, JNK and p38 MAP kinases (MAPK) to identify the signal transduction pathways activated by H. pylori. AGS cells were pre-treated for 30 min with inhibitors then co-cultured with H. pylori for 9 h (Fig. 3A). ERK and p38 inhibitors completely blocked gastrin induction by H. pylori while the JNK inhibitor blocked H. pylori induction by 50%. Although H. pylori clearly activate NFκB signaling 39,40, inhibiting this pathway with APDT did not block induction of the gastrin gene. To confirm that the inhibitors blocked the activation of ERK, cJun and p38 MAPKs, western blots were performed and demonstrated that the doses of the inhibitors were sufficient to block activation of these signaling pathways (Fig. 3C). Moreover, the dose of APDT inhibited NFκB-mediated induction of IL-8 gene expression (Fig. 3B) 41. Thus we concluded that H. pylori induction of the human gastrin gene is mediated by MAPK rather than through NFκB signaling.
Figure 3. MAP Kinase Inhibitors Block H. pylori -induced Gastrin Expression.
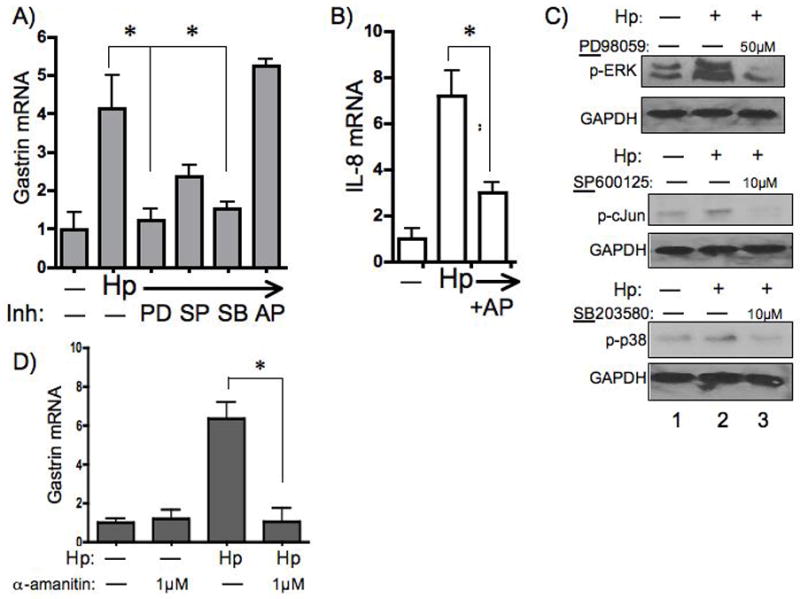
AGS cells were treated with vehicle alone (—) or with chemical inhibitors of ERK, JNK and p38 MAP Kinases respectively (PD98059: 50μM, SP600125: 10μM, SB203580: 10μM) or NFκB (APDT: 10μM) signaling 30 min prior to co-culture with the H. pylori 26695 strain (Hp). qRT-PCR analysis of gastrin transcripts (A) or IL-8 transcripts (B) were normalized to 18S rRNA. The mean fold change + SEM three experiments is shown *P< 0.05 compared to untreated (—) cells. Western blots were performed on whole cell extracts prepared from cells cultured without (—) or with (+) H. pylori ± the indicated inhibitors (C). (D) AGS cells were treated without (—) or with 1μM α-amanitin prior to co-culture with the H. pylori 26695 strain. Gastrin mRNA normalized to 18S rRNA transcripts was determined by qRT-PCR of total RNA. The mean fold change + SEM for three experiments is shown. *P < 0.05 compared to untreated cells.
H. pylori Stimulates Transcriptional Initiation of Gastrin mRNA
To determine if the induction of gastrin by H. pylori was due to transcriptional initiation, AGS cells were pre-treated with the RNA polymerase inhibitor α-amanitin for 30 min prior to co-culturing with the bacteria. The basal expression of gastrin was not affected by 0.1 μM, 0.5 μM, or 1 μM of α-amanitin (data not shown). However, treatment with 1 μM α-amanitin completely blocked H. pylori -induced gastrin gene expression when the cells were co-cultured with the 26695 strain (Fig. 3D). Similar results were obtained using the SS1 strain (data not shown). Therefore, we concluded that H. pylori induce gastrin by stimulating transcriptional initiation rather than by increasing mRNA stability.
H. pylori Stimulates Gastrin Expression Through GC-rich DNA Elements
To identify the H. pylori -responsive element within the human gastrin promoter, we generated a series of luciferase constructs representing the sequences between − 3.3 kb to − 190 bp of the gastrin promoter (Fig. 4A). The basal activity of each reporter construct is shown as relative light units and indicated that the highest basal activity was within the first 240 bp of the promoter as previously reported (Fig. 4B)42. Transient transformants of AGS cells expressing the various gastrin reporters were co-cultured with H. pylori. We observed a significant induction of promoter activity for all gastrin promoter constructs from −3.3 to −0.24 kb (Fig. 4C). However, when the 43 bp region between −240 bp and −190 bp was deleted, there was a 30% decrease in promoter activation suggesting the presence of an H. pylori responsive element contained within this region. AGS cells expressing an AP1 luciferase reporter were significantly induced when co-cultured with H. pylori. This induction is consistent with prior studies demonstrating the ability of H. pylori to activate target genes through AP1 DNA elements. To validate that the decrease in gastrin promoter activation was due to an H. pylori -specific effect, transfected cells were also treated with PMA (Fig. 4D). There was no difference in PMA-induced promoter activity between the -240 bp and -190 bp promoter constructs suggesting that the region contained an H. pylori -specific response element. A significant reduction in the PMA- induced activity of the -2.3 kb reporter did not show a difference in Hp-induced activity. Therefore we concluded that this DNA segment contained a regulatory region associated with induction by PMA and not H. pylori
Figure 4. H. pylori Stimulate the Proximal Gastrin Promoter.
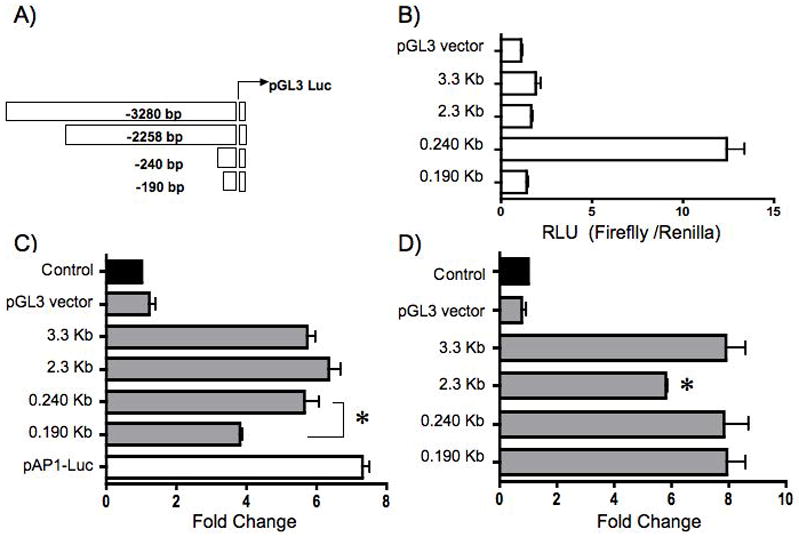
AGS cells were transiently transfected with the empty vector (pGL3), a series of gastrin luciferase constructs, or an AP1 luciferase reporter (pAP1-Luc). A schematic representation of the gastrin luciferase constructs is shown (A). The basal activity of the gastrin constructs is shown as relative light units (RLU) of the firefly normalized to the Renilla luciferase activity (B). Cells were co-cultured with H. pylori (C) or 100 nM PMA (D) 48h post-transfection. The mean fold change + SEM for three separate experiments was performed in triplicate. The filled bar represents the baseline expression in RLU for each construct set to 1 and fold changes were the ratio of the stimulated activity compared to basal expression of each construct.
Upon examination of the region between −240 bp and −190 bp, we identified two cytosine-rich sites were identified as putative H. pylori responsive elements (C2AC4=GC-rich) (Fig.5A, Site1). However, since the −190 bp construct retained the ability to be induced by H. pylori (4-fold), additional downstream promoter sequences containing a GC-richmotif at −120 bp and the reverse complement at −74 bp were also evaluated (Fig. 5A, sites 2 and 3).
Figure 5. H. pylori Stimulate the Gastrin Promoter Through GC-rich DNA Elements.
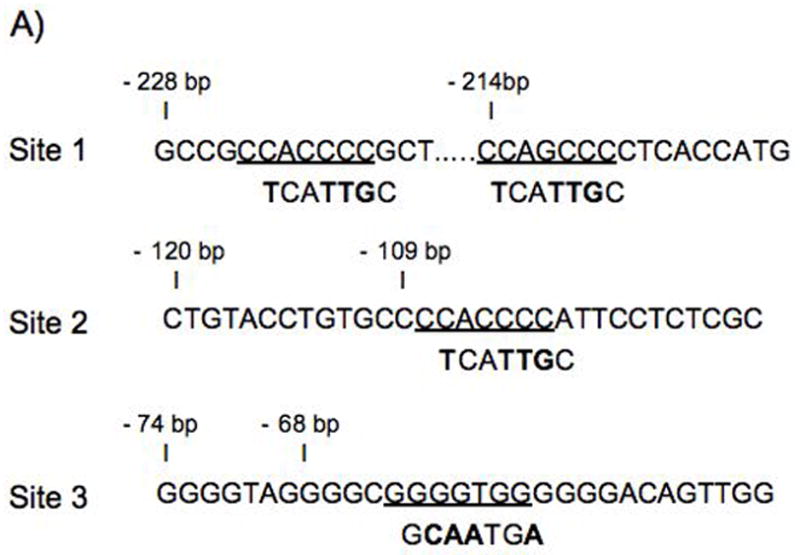
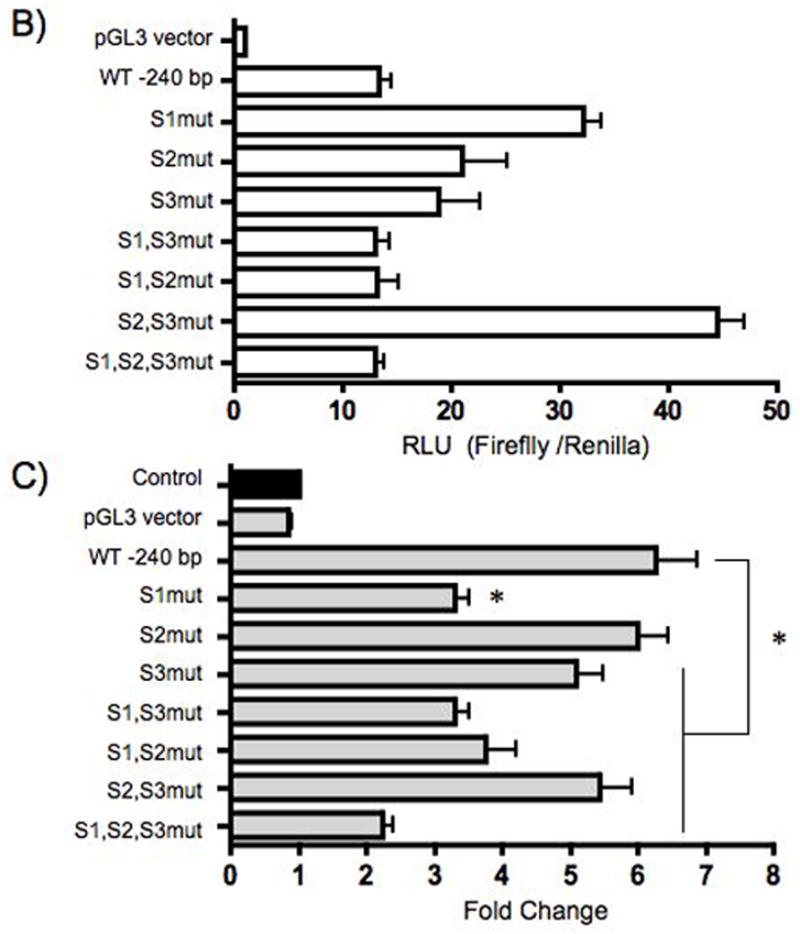
AGS cells were transiently transfected with the wild type (WT) 240 bp gastrin luciferase reporter or the reporter containing point mutations at the indicated sites (S1,2,3). Altered nucleotides are shown in bold font (A). The basal activity of gastrin constructs is shown as relative light units (RLU) of firefly to Renilla activity (B). The cells were co-cultured with H. pylori 48 h post-transfection (C). The mean fold change + SEM for three separate experiments was performed in triplicate. The filled bar represents the baseline expression in RLU for each construct set to 1 and fold changes were the ratio of the stimulated activity compared to basal expression.
To determine whether the GC-rich elements were required for H. pylori induction, point mutations were introduced to disrupt protein binding at these elements (Fig. 5A). The results showed that mutating the GC-richelements within site 1 or sites 2 and 3 together increased the basal activity compared to that of the wild type reporter (Fig. 5B). The introduction of other point mutations did not alter basal activity. Mutations of site 1 reduced gastrin promoter activation when H. pylori were co-cultured with cells transfected with WT or mutant reporters (Fig. 5C). This result was comparable to the decrease in activity observed with a deletion between − 240 bp and −190 bp (~30%). In addition, decreased activation was also observed with mutation of site 3 alone and mutation of site 1 with either site 2 or 3. There was no effect on the activation gastrin reporters when site 2 alone was mutated. The mutation of sites 2 and 3 together caused a slight decrease in H. pylori induced activity. However the mutation of sites 1, 2, and 3 together resulted in the greatest suppression in H. pylori -activation (from 6.5 to 2-fold, Fig. 5C). Therefore we concluded that the GC-richelements between -240 and -190 were required for maximal activation of the gastrin promoter by H. pylori.
H. pylori Induce Sp1/Sp3 Protein Binding to GC-rich DNA Elements
Next, we identified the nuclear proteins binding to the specific GC-rich sites by electrophoretic mobility shift assay. We determined that the two major complexes binding these elements were transcription factors Sp1 and Sp3 (Fig. 6). We observed increased protein binding activity in extracts from H. pylori treated cells to the -228 and -120 probes (Fig. 6, lanes 1, 2, 5 and 6). However, Sp1 binding to the -74 element remained unchanged while Sp3 binding decreased with H. pylori treatment (Fig. 6, lanes 9 and 10). By contrast, AP1 protein binding increased consistent with prior studies (Fig. 6, lanes 13 and 14)43,44.
Figure 6. H. pylori Stimulate Binding of Sp1 and Sp3 to the GC-rich DNA Elements.
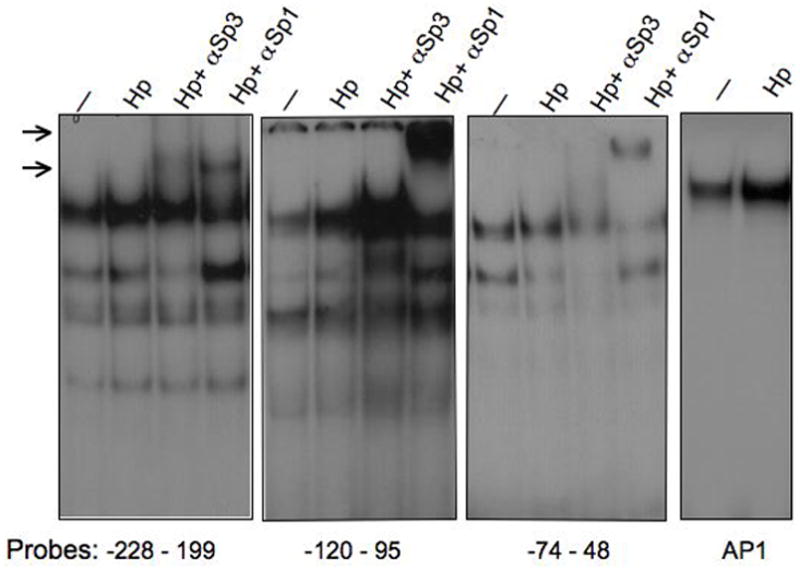
EMSA reactions were carried out with 4μg of nuclear extracts from AGS cells that were untreated (—) or co-cultured with H. pylori 26695 strain for 3 h (Hp). Labeled probes represent GC-rich DNA elements of the human gastrin promoter and a consensus AP1 binding sequence. The samples were incubated with antibodies against Sp3 or Sp1 as indicated. Arrows indicate the supershifted Sp1 and Sp3 DNA complexes.
Discussion
In this study, we demonstrated that the increase in gastrin mRNA initiated by an H. pylori infection is due to transcriptional initiation. We used the AGS cell line to show that gastric cells can respond directly to the bacteria alone. It has been widely reported that the number of G cells and the release of gastrin increases during H. pylori infection in human subjects and various animal models 19,45,46. Indeed, we also observed that gastrin gene expression was higher in H. pylori-infected mice when compared to uninfected mice. Yet, few studies have actually examined whether the increase is related to induction of gastrin gene expression as opposed to an increase in secretion of the hormone. Moreover, the cell line experiments were important to perform in order to distinguish between a direct induction of human gastrin gene expression by bacteria versus receptor-mediated activation by pro-inflammatory cytokines. Separating these two events is impossible to achieve in vivo in human subjects or mouse models prompting our use of a human cell line and human isolates of H. pylori. While nearly pure rat G cell cultures have been used to study gastrin release 26 transfecting these primary cells with various constructs still remains problematic.
We found that H. pylori alone effectively induced the endogenous human gastrin gene. Furthermore, no induction was observed when the cells were co-cultured with dead bacteria, which underscores the requirement for viable bacteria. H. pylori virulence factors have been associated with more severe clinical outcomes for infected patients and the presence or absence of these factors can alter the regulatory affect on cells as well 10,47,48. Specifically, H. pylori strains that lack CagA tend to induce less inflammation and are less commonly associated with the development of ulcers and adenocarcinoma 49,50.
The bacterial virulence factors, CagA and VacA were not required for an increase in gastrin mRNA, but rather other yet to be determined components of the live bacteria were required to induce gastrin through activation of MAPK signaling. Indeed, Helicobacter lipopolysaccharides have recently been shown to activate the ERK MAPK 51; while CagA and VacA activate host genes through the NFκB pathway 40,52. Further Cag3 has recently been shown to be a non- CagA, non-VacA protein component of the T4S system that possibly activates a distinctive signal transduction pathway 53. Therefore we concluded that Helicobacter stimulates gastrin through a general bacterial factor, such as an outer membrane protein, or lipopolysaccharide as opposed to a virulence factor expressed from a pathogenicity island not present in all bacteria. Helicobacter LPS stimulation of gastrin release from primary G cells was partially inhibited by the ERK inhibitor PD98059 supporting LPS as one candidate 26. Documenting that H. pylori stimulated transcriptional initiation permitted mapping of the DNA elements mediating the induction. We found that a CCACCC (GC-rich) binding motif, mediated gastrin promoter activation. More importantly, we showed that H. pylori-induced MAPK signaling induced binding of transcription factors Sp1 and Sp3 to the GC-rich element at −109 bp upstream from the transcription start site of the human gastrin gene, but not at the −68 GC-rich element previously shown to mediate EGF responsiveness 54. Although the −68 GC-rich element binds Sp1, the induction of gastrin gene transcription by EGF through ERK signaling is achieved by phosphorylation of Sp1 31 rather than inducible binding of Sp1 to the −68 GC-rich site (gERE) 32. Activation of the EGF receptor by H. pylori has also been reported for immortalized human gastric cells 44. H. pylori induction of gastrin gene expression and secretion might retrospectively explain the observations from the 1960’s of duodenal ulcer patients exhibiting parietal cell hyperplasia and enhanced acid secretion 55,56. Furthermore, in some colorectal cancers, mutations in Ki-ras, which signals through ERK kinase, were found in tumors with elevated levels of gastrin 30. Thus collectively, our findings demonstrate that H. pylori can directly induce gastrin gene expression and reiterates the idea that activation of MAPK signaling is the primary proximal signaling mechanism for induction of gastrin gene expression, an event that might also impact the ability of the bacteria to promote transformation.
Footnotes
Disclosures
This work was supported by Public Health Service grant R01-DK45729 to JLM. Core facility support was provided by Public Health Service grant P30 DK34933. The authors have no competing financial interests.
References
- 1.Peterson WL. Helicobacter pylori and peptic ulcer disease. N Engl J Med. 1991;324:1043–1048. doi: 10.1056/NEJM199104113241507. [DOI] [PubMed] [Google Scholar]
- 2.Blaser MJ, Parsonnet J. Parasitism by the "slow" bacterium Helicobacter pylori leads to altered gastric homeostasis and neoplasia. J Clin Invest. 1994;94:4–8. doi: 10.1172/JCI117336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsonnet J, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267–1271. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- 4.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 5.Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 6.Naumann M, et al. Activation of activator protein 1 and stress response kinases in epithelial cells colonized by helicobacter pylori encoding the cag pathogenicity island. J Biol Chem. 1999;274:31655–31662. doi: 10.1074/jbc.274.44.31655. [DOI] [PubMed] [Google Scholar]
- 7.Herrera V, Parsonnet J. Helicobacter pylori and gastric adenocarcinoma. Clin Microbiol Infect. 2009;15:971–976. doi: 10.1111/j.1469-0691.2009.03031.x. [DOI] [PubMed] [Google Scholar]
- 8.Peek RM., Jr Helicobacter pylori strain-specific modulation of gastric mucosal cellular turnover: implications for carcinogenesis. J Gastroenterol. 2002;37 (Suppl 13):10–16. doi: 10.1007/BF02990093. [DOI] [PubMed] [Google Scholar]
- 9.Odenbreit S, et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 10.Keates S, et al. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Biol Chem. 2001;276:48127–48134. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- 11.Wessler S, et al. Helicobacter pylori activates the histidine decarboxylase promoter through a mitogen-activated protein kinase pathway independent of pathogenicity island-encoded virulence factors. J Biol Chem. 2000;275:3629–3636. doi: 10.1074/jbc.275.5.3629. [DOI] [PubMed] [Google Scholar]
- 12.Atherton JC, et al. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 13.Calam J, Gibbons A, Healey ZV, Bliss P, Arebi N. How does Helicobacter pylori cause mucosal damage? Its effect on acid and gastrin physiology. Gastroenterology. 1997;113:S43–49. doi: 10.1016/s0016-5085(97)80010-8. discussion S50. [DOI] [PubMed] [Google Scholar]
- 14.Chittajallu RS, Dorrian CA, Neithercut WD, Dahill S, McColl KE. Is Helicobacter pylori associated hypergastrinaemia due to the bacterium's urease activity or the antral gastritis? Gut. 1991;32:1286–1290. doi: 10.1136/gut.32.11.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levi S, et al. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet. 1989;1:1167–1168. doi: 10.1016/s0140-6736(89)92752-9. [DOI] [PubMed] [Google Scholar]
- 16.Gibbons AH, Legon S, Walker MM, Ghatei M, Calam J. The effect of gastrin-releasing peptide on gastrin and somatostatin messenger RNAs in humans infected with Helicobacter pylori. Gastroenterology. 1997;112:1940–1947. doi: 10.1053/gast.1997.v112.pm9178686. [DOI] [PubMed] [Google Scholar]
- 17.Sumii M, et al. Expression of antral gastrin and somatostatin mRNA in Helicobacter pylori-infected subjects. Am J Gastroenterol. 1994;89:1515–1519. [PubMed] [Google Scholar]
- 18.Richter-Dahlfors A, Heczko U, Meloche RM, Finlay BB, Buchan AM. Helicobacter pylori-infected human antral primary cell cultures: effect on gastrin cell function. Am J Physiol. 1998;275:G393–401. doi: 10.1152/ajpgi.1998.275.3.G393. [DOI] [PubMed] [Google Scholar]
- 19.Rieder G, Merchant JL, Haas RH. pylori cag-Type IV secretion system facilitates corpus colonization to induce precancerous conditions in mongolial gerbil. Gastroenterology. 2005;128:1229–1242. doi: 10.1053/j.gastro.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 20.Zavros Y, Rieder G, Ferguson A, Merchant JL. Gastritis and hypergastrinemia due to Acinetobacter lwoffii in mice. Infect Immun. 2002;70:2630–2639. doi: 10.1128/IAI.70.5.2630-2639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waghray M, et al. Interleukin-1beta Promotes Gastric Atrophy Through Suppression of Sonic Hedgehog. Gastroenterology. 2009;138:562–572. doi: 10.1053/j.gastro.2009.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brand SJ, Stone D. Reciprocal regulation of antral gastrin and somatostatin gene expression by omeprazole-induced achlorhydria. J Clin Invest. 1988;82:1059–1066. doi: 10.1172/JCI113662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chittajallu RS, Neithercut WD, Macdonald AM, McColl KE. Effect of increasing Helicobacter pylori ammonia production by urea infusion on plasma gastrin concentrations. Gut. 1991;32:21–24. doi: 10.1136/gut.32.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beales I, et al. Effect of Helicobacter pylori products and recombinant cytokines on gastrin release from cultured canine G cells. Gastroenterology. 1997;113:465–471. doi: 10.1053/gast.1997.v113.pm9247465. [DOI] [PubMed] [Google Scholar]
- 25.Beales IL, Calam J. Helicobacter pylori increases gastrin release from cultured canine antral G-cells. Eur J Gastroenterol Hepatol. 2000;12:641–644. doi: 10.1097/00042737-200012060-00011. [DOI] [PubMed] [Google Scholar]
- 26.Kidd M, Hauso O, Drozdov I, Gustafsson BI, Modlin IM. Delineation of the chemomechanosensory regulation of gastrin secretion using pure rodent G cells. Gastroenterology. 2009;137:231–241. 241 e231–210. doi: 10.1053/j.gastro.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Zavros Y, et al. Treatment of Helicobacter Gastritis With Interleukin-4 Requires Somatostatin. Proc Natl Acad Sci. 2003;100:12944–12949. doi: 10.1073/pnas.2135193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ford MG, Valle JD, Soroka CJ, Merchant JL. EGF receptor activation stimulates endogenous gastrin gene expression in canine G cells and human gastric cell cultures. J Clin Invest. 1997;99:2762–2771. doi: 10.1172/JCI119466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Godley-Merchant J, Brand SJ. Regulation of the gastrin promoter by epidermal growth factor and neuropeptides. Proc Natl Acad Sci USA. 1989;86:3036–3040. doi: 10.1073/pnas.86.9.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakata H, Wang SL, Chung DC, Westwick JK, Tillotson LG. Oncogenic ras induces gastrin gene expression in colon cancer. Gastroenterology. 1998;115:1144–1153. doi: 10.1016/s0016-5085(98)70085-x. [DOI] [PubMed] [Google Scholar]
- 31.Merchant JL, Du M, Todisco A. Sp1 phosphorylation by Erk 2 stimulates DNA binding. Biochem Biophys Res Commun. 1999;254:454–461. doi: 10.1006/bbrc.1998.9964. [DOI] [PubMed] [Google Scholar]
- 32.Chupreta S, Du M, Todisco A, Merchant JL. EGF stimulates gastrin promoter through activation of Sp1 kinase activity. Am J Physiol Cell Physiol. 2000;278:C697–708. doi: 10.1152/ajpcell.2000.278.4.C697. [DOI] [PubMed] [Google Scholar]
- 33.Strowski MZ, et al. Helicobacter pylori stimulates host vascular endothelial growth factor-A (vegf-A) gene expression via MEK/ERK-dependent activation of Sp1 and Sp3. FASEB J. 2004;18:218–220. doi: 10.1096/fj.03-0055fje. [DOI] [PubMed] [Google Scholar]
- 34.Akopyants NS, Eaton KA, Berg DE. Adaptive mutation and cocolonization during Helicobacter pylori infection of gnotobiotic piglets. Infect Immun. 1995;63:116–121. doi: 10.1128/iai.63.1.116-121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neuvians TP, et al. Standardization strategy for quantitative PCR in human seminoma and normal testis. J Biotechnol. 2005;117:163–171. doi: 10.1016/j.jbiotec.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Tomb JF, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1998;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 37.Thompson LJ, et al. Chronic Helicobacter pylori infection with Sydney strain 1 and a newly identified mouse-adapted strain (Sydney strain 2000) in C57BL/6 and BALB/c mice. Infect Immun. 2004;72:4668–4679. doi: 10.1128/IAI.72.8.4668-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer-ter-Vehn T, Covacci A, Kist M, Pahl HL. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J Biol Chem. 2000;275:16064–16072. doi: 10.1074/jbc.M000959200. [DOI] [PubMed] [Google Scholar]
- 39.Takeshima E, et al. NF-kappaB activation by Helicobacter pylori requires Akt-mediated phosphorylation of p65. BMC Microbiol. 2009;9:36–44. doi: 10.1186/1471-2180-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Takeshima E, et al. Helicobacter pylori VacA activates NF-kappaB in T cells via the classical but not alternative pathway. Helicobacter. 2009;14:271–279. doi: 10.1111/j.1523-5378.2009.00683.x. [DOI] [PubMed] [Google Scholar]
- 41.Bhattacharyya A, et al. Mitogen-activated protein kinases and nuclear factor-kappaB regulate Helicobacter pylori-mediated interleukin-8 release from macrophages. Biochem J. 2002;368:121–129. doi: 10.1042/BJ20020555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merchant JL, Demediuk B, Brand SJ. A GC-rich element confers epidermal growth factor responsiveness to transcription from the gastrin promoter. Mol Cell Biol. 1991;11:2686–2696. doi: 10.1128/mcb.11.5.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding SZ, et al. Helicobacter pylori and mitogen-activated protein kinases mediate activator protein-1 (AP-1) subcomponent protein expression and DNA-binding activity in gastric epithelial cells. FEMS Immunol Med Microbiol. 2008;53:385–394. doi: 10.1111/j.1574-695X.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashktorab H, et al. Transactivation of the EGFR by AP-1 is induced by Helicobacter pylori in gastric cancer. Am J Gastroenterol. 2007;102:2135–2146. doi: 10.1111/j.1572-0241.2007.01400.x. [DOI] [PubMed] [Google Scholar]
- 45.Kudo T, et al. Pattern of transcription factor activation in Helicobacter pylori-infected Mongolian gerbils. Gastroenterology. 2007;132:1024–1038. doi: 10.1053/j.gastro.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konturek SJ, et al. Implication of gastrin in cyclooxygenase-2 expression in Helicobacter pylori infected gastric ulceration. Prostaglandins Other Lipid Mediat. 2001;66:39–51. doi: 10.1016/s0090-6980(01)00142-3. [DOI] [PubMed] [Google Scholar]
- 47.Pillinger MH, et al. Helicobacter pylori stimulates gastric epithelial cell MMP-1 secretion via CagA-dependent and -independent ERK activation. J Biol Chem. 2007;282:18722–18731. doi: 10.1074/jbc.M703022200. [DOI] [PubMed] [Google Scholar]
- 48.Keates S, et al. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol. 1999;163:5552–5559. [PubMed] [Google Scholar]
- 49.Nomura AM, Perez-Perez GI, Lee J, Stemmermann G, Blaser MJ. Relation between Helicobacter pylori cagA status and risk of peptic ulcer disease. Am J Epidemiol. 2002;155:1054–1059. doi: 10.1093/aje/155.11.1054. [DOI] [PubMed] [Google Scholar]
- 50.Blaser MJ, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Research. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 51.Yokota S, Okabayashi T, Rehli M, Fujii N, Amano K. Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect Immun. 2010;78:468–476. doi: 10.1128/IAI.00903-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamb A, et al. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–1249. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinto-Santini DM, Salama NR. Cag3 is a novel essential component of the Helicobacter pylori Cag type IV secretion system outer membrane subcomplex. J Bacteriol. 2009;191:7343–7352. doi: 10.1128/JB.00946-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merchant JL, Shiotani A, Mortensen E, Shumaker D, Abraczinskas D. EGF stimulation of the human gastrin promoter requires Sp1. J Biol Chem. 1995;270:6314–6319. doi: 10.1074/jbc.270.11.6314. [DOI] [PubMed] [Google Scholar]
- 55.Crean GP, Marshall MW, Rumsey RD. Parietal cell hyperplasia induced by the administration of pentagastrin (ICI 50,123) to rats. Gastroenterology. 1969;57:147–155. [PubMed] [Google Scholar]
- 56.Card WI, Marks IN. The relationship between the acid output of the stomach following "maximal" histamine stimulation and the parietal cell mass. Clin Sci. 1960;19:147–163. [PubMed] [Google Scholar]