

Abstract
Application of native chemical ligation logic to the case of an N-terminal proline is described. Two approaches were studied. One involved incorporation of a 3R-substituted thiyl-proline derivative. Improved results were obtained from a 3R-substituted selenol function, incorporated in the context of an oxidized dimer.
Introduction
The development and application of novel strategies for the convergent synthesis of homogeneous proteins represents a prominent challenge in synthetic chemistry. The seminal discovery, by Kent and co-workers, of native chemical ligation (NCL) marked a striking advance in the field of peptide synthesis.i According to this powerful logic, a peptide bearing a C-terminal aryl thioester (1) reacts with a second peptidyl fragment, 2, which is equipped with an N-terminal cysteine residue (Scheme 1, eq. 1). Following rate-determining trans-thioesterification, a rapid, irreversible S→N acyl transfer (3→4) generates the native amide bond.
Scheme 1.

Native Chemical Ligation (NCL).
Though originally limited to couplings that deliver a Cys residue at the site of ligation, the scope of the NCL technology was soon expanded, by Dawson and colleagues, via post-ligation metal-based desulfurization. In this way, NCL could, in the end, lead to ligations at Ala,ii Phe,iii and Seriv residues. While metal induced dethiylation served to enhance the range of NCL logic, there remained a significant need to accomplish RSH→RH conversion under conditions which are compatible with water as the solvent. The discovery, in our laboratory, of a high yielding, aqueous–compatible, SH specific, metal free dethiylation (MFD) method served to enhance, in a major way, the reach of NCL logic.v Through recourse to these MFD conditions, NCL was extended, in our laboratory and others, to include Ala,v Val,vi,vii Thr,viii Leu,ix,x and Lysxi,xii (Scheme 1, eq. 2). MFD conditions are both mild and highly selective, and may be employed in the presence of a range of peptide functionalities, including Met residues, Acm-protected Cys residues, and sensitive glycan domains.
Results and Discussion
Thio-Proline Ligation
We wondered about the possibility of expanding the menu of non-cysteine NCL inspired ligations to include the significant challenge of proline ligation.xiii It has been observed that C-terminal proline thioesters are relatively poor acyl donors, even in standard cysteine-based NCL settings.xiv The poor reactivity of the proline thioester functionality has been attributed to deactivation of the thioester by the proline amide carbonyl, rather than to steric factors. C-terminal proline p-nitrophenyl esters are somewhat more reactive, but these types of substrates are quite susceptible to hydrolysis under ligation conditions. In light of the inherent difficulties of accomplishing ligation at a C-terminal Pro residue, we explored an alternative strategy, whereby an appropriate thio-proline surrogate would reside at the N-terminus of a peptide, 8 (Scheme 1, eq. 3). Following thiol-mediated ligation, the resultant intermediate, 9, would be subjected to MFD to deliver the target peptide, 10, bearing a Pro residue at the site of ligation. The successful development of this strategy would represent an important expansion of the NCL paradigm, in that it would involve S→N acyl transfer at a secondary amine in the context of a ring system, and would be required to pass through a bridged tetrahedral intermediate (see 8 → 9).
Happily, our efforts along these lines were indeed successful, and we recently described reduction to practice of the envisioned thio-proline ligation protocol.xiii Interestingly, but not surprisingly, reaction viability was found to be highly dependent on the stereochemistry of the thio-Pro surrogate used. As shown in Scheme 2, only the trans-thioproline surrogate was found to be a viable participant in the ligation step. In the productive trans series, the large COP2 moiety is situated in a favorable exo position in the presumed tetrahedral S→N acyl transfer intermediate, while in the unproductive cis series, the COP2 group would need to occupy a hindered endo position.
Scheme 2.

Thio-Proline Ligation with Two Pro(SH) Diastereomers.a
aKey: (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5. P1: ALLVNSS−; P2: −WEPLN; Ar = 2-(ethyldithio)-phenyl; TCEP = tris(2-carboxyethyl)phosphine.
In a substrate scope study, we observed a strong correlation between steric bulk of the C-terminal amino acid residue and the quality of the reaction (Scheme 3). Thus, while ligation proceeded readily with relatively unhindered C-terminal Gly (14) or Gln (19) thioesters, reaction was significantly lower yielding with Phe (20) or Val (21) residues. Moreover, incorporation of a C-terminal Pro residue led to complete inhibition of ligation (22). In the Val–Pro ligation en route to 21, we observed that, while the trans-thioesterification step was rapid, the resulting thioester (cf. 18) was not adequately reactive to permit efficient S→N acyl transfer. In contrast, the Pro thioester substrate was not able to undergo the initiating transthio-esterification step. We attribute the failure of the Pro–Pro ligation to the deactivating nature of the peptide chain (P1) on the C-terminal Pro residue, a supposition which is supported by the comparatively facile formation of adduct 23 from peptide 12 and unsubstituted Pro amino acid, under standard coupling conditions.
Scheme 3.

Thio-Proline Ligation: Substrate Scope.a
aKey: (a) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5. P1: ALLVNSS−; P2: −WEPLN; Ar = 2-(ethyldithio)-phenyl.
Thio-Proline Ligation: Application to a Possible Synthesis of Homogeneous Erythropoietin
A long-term effort in our laboratory is devoted to accomplishing the chemical synthesis of homogeneous erythropoietin (EPO).xv Found in nature as a 165-residue protein bearing four sites of glycosylation, EPO is the primary regulator of erythropoiesis and is widely prescribed for the treatment of anemia. EPO represents a particularly compelling synthetic target, due to its challenging structure and therapeutic relevance. Moreover, at least in principle, successful realization of our mission would enable access, for the first time, to homogeneous erythropoietins containing defined, but variable, glycoforms at the conserved glycosylation sites. Such access could set the stage for a combined chemistry/glycobiology program to learn more about why nature glycosylates so many of its important proteins.
In the context of our EPO synthetic effort, we recently sought to assemble the bis-glycosylated hEPO(79–166) fragment, 24.xvi In a retrosynthetic sense, we envisioned organizing the target glycopeptide into two long polypeptide segments (26 and 28) and two shorter glycopeptide domains (25 and 27). These would be iteratively merged through a series of thiol-assisted ligations, to generate the full glycopeptide backbone, 29, bearing three extraneous sulfur functionalities. Finally, global MFD would deliver the target EPO glycopeptide, 24. The success of this particular proposed retrosynthesis would be predicated on our ability to achieve efficient Pro ligation between polypeptide, 26, bearing the trans-thio-Pro surrogate at its N-terminus, and the glycopeptide fragment, 25, incorporating a C-terminal ortho-thiophenolic ester and a Gln residue.
In the event, the projected sequence of ligations did proceed as expected. Thus, Cys-mediated ligation between 27 and 28, followed by Fmoc removal and thiazolidine ring opening generated the ligation acceptor corresponding to hEPO(125–166). We were pleased to observe that, under our optimized conditions, the hoped-for Pro ligation between 25 and 26 could be achieved in a modest but serviceable isolated yield (23%). The two glycopeptide fragments were subsequently merged to generate 29, bearing three erstwhile thiol groups (in red). Finally, three-fold dethiolation was achieved under our standard MFD conditions, to afford the target system, 24. The successful realization of this challenging synthetic route serves to further demonstrate the complexity-building potential of our newly developed thio-proline ligation protocol, coupled with our mild and selective MFD conditions while exploiting our recently developed O-mercapatouryl rearrangement to reveal, in site, the required thioester.
Proline Ligation via a Selenol Surrogate
Though the thioproline de-thiylation sequence had been applied to fairly complex settings, we were nonetheless mindful of its potential limitations. Thus, as we have shown (Scheme 3), ligation efficiency is severely compromised by the presence of a bulky amino acid on the C-terminal coupling partner. In an effort to expand the scope of our Pro ligation method, we took note of some recent methodological advances that have focused on overcoming issues apparently arising from steric congestion in ligation chemistry. In particular, Durek and Alewood have described the conversion of thioesters to selenoesters as a means to form highly reactive C-terminal ligation partners.xvii Similarly, Dawson and colleagues have reported the ligation and selective deselenization of peptides feature N-terminal selenocysteine residues.xviii
Thus, in an effort to enhance the rate of intramolecular acyl transfer in the pseudo-proline ligation, we wondered about the feasibility of ligating peptides via N-terminal prolines containing C4 selenol functionality. We hypothesized that the increased nucleophilicity of the selenol would lend itself to more rapid trans-esterification. The intermediate selenium ester, 31, might well be expected to display increased reactivity relative to the analogous thioester, and could conceivably accommodate productive Se→N transfer even in the presence of bulky C-terminal residues.xix,xx In the course of these investigations, we would also explore the relative rate and efficiency of deselenization of a secondary selenol (32→10), compared to desulfurization of a secondary thiol. It was anticipated that, owing to the ease with which divalent selenium compounds are oxidized, the selenoproline moiety might require presentation as a dimer, which would be reductively activated to reveal its selenol function. Moreover, for widespread convenience, the capacity to integrate such residue into a peptide sequence through solid phase peptide synthesis (SPPS) techniques would be most helpful.
The first objective was to synthesize the previously unknown trans-selenoproline, 37.xxi Beginning with commercially available pyrrolidine 33, Mitsunobu inversion provided the cis-iodo proline derivative, 34, in high yield (Scheme 6).xxii Nucleophilic displacement with selenobenzoic acidxxiii generated 35 in 84% yield. This intermediate displayed the key diagnostic 13C resonance of ~200 ppm for a selenocarboxylate.xxiv Removal of the benzoate and saponification of the methyl ester occurred in concert to provide, upon aqueous work-up, N-Boc selenoproline dimer 36, in 79% yield. Finally, cleavage of the Boc group under acidic conditions afforded “oxidatively dimerized” 37, in near quantitative yield.
Scheme 6.

Synthesis of trans-seleno-Pro, 37.
aKey: (a) PPh3, DIAD, CH3I, THF, 0 °C → 23 °C, 88–92% yield; (b) BzSeH, DIPEA, DMF, 60 °C, 84%; (c) K2CO3, aq. MeOH, 79%; (d) HCl/CH2Cl2, 95%.
With trans-seleno-Pro (37) in hand, we first examined the ligation in the context of a single amino acid elongation (Scheme 7). The ligation between 38 and 37 was conducted at pH 7.4 to 7.6, in degassed buffer in the presence of an aromatic thiol (4-mercaptophenylacetic acid, MPAA). As previously described by Dawson,xviii MPAA serves to desymmetrize the selenium dimer, liberating small amounts of selenol to participate in the opening ligation. Moreover, MPAA further activates the thioester through trans-thioesterification. Under these conditions, peptide 38 was consumed within 3 hours, generating adduct 40 in 92% yield. We note that, due to the oxidative sensitivity of the presumed selenol, standard degassing by passing a stream of nitrogen through the reaction was ineffective. Instead, the buffered solution was degassed according to the freeze-pump-thaw protocol.
Scheme 7.

Seleno-Proline Ligation.
In order to gain access to appropriate quantities of material for further studies, we directed our attention to the incorporation of a “selenoproline dimer” into a peptide through SPPS techniques. For optimal reaction efficiency, standard SPPS protocols require the use of excess amounts of each amino acid. However, in order to synthesize a dimeric peptide, we would require submolar quantities of the selenoproline. In the event, we were pleased to find that our target dimeric peptides could be smoothly prepared through the use of 0.55 eq. of the dimer, by employing extended reaction times in the coupling step. Through recourse to this approach, we were able to readily gain access to our target peptide, 43, in high yield (see Supporting Information for details).
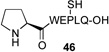
We next sought to evaluate the relative efficiency of deselenation compared to dethiolation in model peptidyl systems, bearing N-terminal seleno-Pro and thio-Pro residues, respectively. As outlined in Table 1, peptide 41, bearing an N-terminal thio-Pro surrogate, underwent dethiolation at 37 °C to generate 42 afer 90 min, in 83% yield (entry 1). No dethiolation was observed at lower temperatures. By contrast, as shown in entry 2, peptide 43 rapidly underwent TCEP-induced deselenation at both room temperature (15 min, 92% yield) and 5 °C (30 min, 88% yield). We next examined the deselenation of a selenosulfide-Pro, which represents the penultimate product of selenoproline ligation. We were pleased to find that the reduction proceeded within 90 minutes to deliver 42 in excellent yield (entry 3). Finally, peptide 45, incorporating both seleno- and thio-Pro residues, was subjected to reducing conditions. As expected on the basis of the Dawson precedent, the seleno- functionality was smoothly and selectively removed, to deliver adduct 46 in 90% yield (entry 4). This finding serves to establish the potential to selectively remove secondary selenide in the presence of an unprotected thiol moiety in the proline series.
Table 1.
Comparison of Proline Desulfurization and Deselenation.a
| Entry | Peptide | Product | Conditions |
|---|---|---|---|
| 1 | 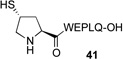 |
A, 90 min, 83% B, 48 h, NR |
|
| 2 | 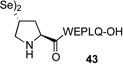 |
C, 15 min, 92% D, 30 min, 88% |
|
| 3 | 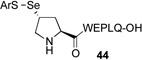 |
E, 90 min, 91% | |
| 4 | 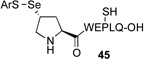 |
 |
E, 90 min, 90% |
Key: (A) VA-044, TCEP, t-BuSH, aq. CH3CN, 37 °C; (B) VA-044, TCEP, t-BuSH, aq. CH3CN, 5°C; (C) TCEP, 6 M Gn•HCl, 200 mM NaH2PO4, pH 5– 6, 23°C; (D) TCEP, 6 M Gn•HCl, 200 mM NaH2PO4, pH 5–6, 5°C; (E) DTT (Dithiothreitol), 6 M Gn•HCl, 200 mM NaH2PO4, then TCEP, pH 5–6.
We now sought to probe the scope of this protocol by examining the coupling of 43 with a range of peptides with significant complexity at the C-terminus. The results of this study are presented in Table 2. As shown, peptides 38 and 48, bearing C-terminal Gly and Ala residues, respectively, underwent rapid ligation to generate, following deselenation, 47 and 48 in high yield (entries 1 and 2). The more sterically demanding Phe-containing peptide, 50, required a longer ligation time, but after 10h, the resultant peptide was reduced to deliver 51 in good yield (80%). A significant improvement in reaction efficiency was observed with peptide 52, presenting a challenging Val residue at its C-terminus. As described above (Scheme 3), this peptide was quite resistant to ligation with the analogous thio-Pro bearing peptide, proceeding to only ~15% conversion within 23h. However, 52 underwent ligation with the more reactive seleno-Pro peptide, 43, to generate, following reduction, a 66% yield of adduct 53 within 12h. This finding confirms our hypothesis that the enhanced nucleophilicity of the selenoester intermediate does, indeed, enable a more facile Se→N acyl transfer. Unfortunately, the selenol derivative was not sufficiently reactive to overcome the deactivating effects of the proline amide carbonyl in peptide 54. Thus, only ca. 5% of product 55 is currently obtainable, even at elevated temperatures.
Table 2.
Selenoproline Ligation: Substrate Scope.a
 | |||
|---|---|---|---|
| Entry | C-terminal Peptide |
Product | Time / Yield |
| 1 | H-ALLVNSSGPWEPLQ-OH 47 |
4.5 h, 84% | |
| 2 | 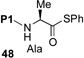 |
H-ALLVNSSAPWEPLQ-OH 49 |
4.5 h, 88% |
| 3 | 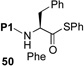 |
H-ALLVNSSFPWEPLQ-OH 51 |
10 h, 80% |
| 4 | 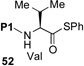 |
H-ALLVNSSVPWEPLQ-OH 53 |
12 h, 66% |
| 5 | 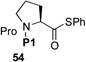 |
H-ALLVNSSPPWEPLQ-OH 55 |
24 h, NR |
Key: (a) MPAA, Ligation Buffer (6 M Gn•HCl, 200 mM NaH2PO4), pH 7.4–7.6, 23 °C; (b) DTT, Ligation Buffer; then TCEP, pH 5.0–6.0, 23 °C. P1: ALLVNSS−; P2: −WEPLQ.
Conclusion
In summary we have described herein recent advances from our laboratory in proline ligation. We have developed a two-step ligation protocol involving a thio-assisted proline coupling, followed by a mild and high-yielding MFD. This transformation is particularly well suited to ligations wherein the C-terminal residue is relatively unhindered. We have further demonstrated the utility of this method in synthesizing complex systems, through application to the synthesis of a doubly glycosylated fragment of hEPO. Most importantly, we have developed a second-generation ligation protocol, featuring a seleno-proline amino acid surrogate. The selenol functionality has been found to be readily removed following ligation under mild conditions. Moreover, our seleno-proline mediated ligation appears to be well suited to more sterically demanding systems and is thus likely to be of value in building complex glycans.
Supplementary Material
Scheme 4.

Synthesis of hEPO(79–166) Glycopeptide, 24.a
aKey: (a) 1. 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5, 67%; 2. piperidine, DMSO, 61%; 3. 0.2M MeONH2, 60%; (b) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, pH 7.5, 23%; (c) 6 M Gn•HCl, 100 mM NaH2PO4, 50 mM TCEP, 200 mM MPAA, pH 7.8, 40%; (d) TCEP, VA-044, tBuSH. Ar = 2-(ethyldithio)-phenyl; R = CH2CH2CO2Et; VA-044 = 2,2’-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride
Scheme 5.

Seleno-Proline Ligation.
ACKNOWLEDGMENT
This work was supported by NIH grant CA28824 (S.J.D.). SDT is grateful to Weill Cornell for an NIH postdoctoral fellowship (CA062948). Special thanks to Dr. George Sukenick, Hui Fang, and Sylvi Rusli of SKI's NMR core facility for spectroscopic assistance and to Dr. Karthik Iyer and Dr. Jennifer Stockdill for helpful discussions.
Footnotes
ASSOCIATED CONTENT
General experimental procedures, including spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- i.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- ii.Yan LZ, Dawson PE. J Am Chem Soc. 2001;123:526. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- iii.Crich D, Banerjee A. J Am Chem Soc. 2007;129:10064. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]
- iv.Okamoto R, Kajihara Y. Angew Chem Int Edit. 2008;47:5402. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
- v.Wan Q, Danishefsky SJ. Angew Chem Int Edit. 2007;46:9248. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- vi.Chen J, Wan Q, Yuan Y, Zhu JL, Danishefsky SJ. Angew Chem Int Ed. 2008;47:8521. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Haase C, Rohde H, Seitz O. Angew Chem Int Edit. 2008;47:6807. doi: 10.1002/anie.200801590. [DOI] [PubMed] [Google Scholar]
- viii.Chen J, Wang P, Zhu JL, Wan Q, Danishefsky SJ. Tetrahedron. 2010;66:2277. doi: 10.1016/j.tet.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ix.Tan ZP, Shang SY, Danishefsky SJ. Angew Chem Int Edit. 2010;49:9500. doi: 10.1002/anie.201005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- x.Harpaz Z, Siman P, Kumar KSA, Brik A. Chembiochem. 2010;11:1232. doi: 10.1002/cbic.201000168. [DOI] [PubMed] [Google Scholar]
- xi.Yang RL, Pasunooti KK, Li FP, Liu XW, Liu CF. J Am Chem Soc. 2009;131:13592. doi: 10.1021/ja905491p. [DOI] [PubMed] [Google Scholar]
- xii.Kumar KSA, Haj-Yahya M, Olschewski D, Lashuel HA, Brik A. Angew. Chem Int Edit. 2009;48:8090. doi: 10.1002/anie.200902936. [DOI] [PubMed] [Google Scholar]
- xiii.Shang SY, Tan ZP, Dong SW, Danishefsky SJ. J Am Chem Soc. 2011;133:10784. doi: 10.1021/ja204277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiv.Pollock SB, Kent SBH. Chem Commun. 2011;47:2342. doi: 10.1039/c0cc04120c. [DOI] [PubMed] [Google Scholar]
- xv.(a) Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJJ. Am. Chem. Soc. 2009;131:5424. doi: 10.1021/ja808704m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yuan Y, Chen J, Wan Q, Tan Z, Chen G, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5432. doi: 10.1021/ja808705v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kan C, Trzupek JD, Wu B, Wan Q, Chen G, Tan Z, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5438. doi: 10.1021/ja808707w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvi.Dong S, Shang S, Tan Z, Danishefsky SJ. Isr. J. Chem. 2011;51:968. doi: 10.1002/ijch.201100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvii.Durek T, Alewood PF. Angew Chem Int Ed Engl. 2011;50:12042. doi: 10.1002/anie.201105512. [DOI] [PubMed] [Google Scholar]
- xviii.Metanis N, Keinan E, Dawson PE. Angew Chem Int Edit. 2010;49:7049. doi: 10.1002/anie.201001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.Pearson RG, Sobel H, Songstad J. J Am Chem Soc. 1968;90:319. [Google Scholar]
- xx.Glass RS. Phosphorus Sulfur. 1998;136:159. [Google Scholar]
- xxi.Caputo R, DellaGreca M, de Paola I, Mastroianni D, Longobardo L. Amino Acids. 2010;38:305. doi: 10.1007/s00726-009-0251-x. [DOI] [PubMed] [Google Scholar]
- xxii.Shangguan N, Joullie M. Tetrahedron Lett. 2009;50:6748. doi: 10.1016/j.tetlet.2009.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxiii.Knapp S, Darout E. Org Lett. 2005;7:203. doi: 10.1021/ol047889z. [DOI] [PubMed] [Google Scholar]
- xxiv.Koketsu M, Nada F, Hiramatsu S, Ishihara H. J Chem Soc Perk T 1. 2002:737. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.