

Abstract
Mood disorders are highly heritable forms of major mental illness. A major breakthrough in elucidating the genetic architecture of mood disorders was anticipated with the advent of genome-wide association studies (GWAS). However, to date few susceptibility loci have been conclusively identified. The genetic etiology of mood disorders appears to be quite complex, and as a result, alternative approaches for analyzing GWAS data are needed. Recently, a polygenic scoring approach that captures the effects of alleles across multiple loci was successfully applied to the analysis of GWAS data in schizophrenia and bipolar disorder (BP). However, this method may be overly simplistic in its approach to the complexity of genetic effects. Data mining methods are available that may be applied to analyze the high dimensional data generated by GWAS of complex psychiatric disorders. We sought to compare the performance of five data mining methods, namely, Bayesian Networks (BN), Support Vector Machine (SVM), Random Forest (RF), Radial Basis Function network (RBF), and Logistic Regression (LR), against the polygenic scoring approach in the analysis of GWAS data on BP. The different classification methods were trained on GWAS datasets from the Bipolar Genome Study (2,191 cases with BP and 1,434 controls) and their ability to accurately classify case/control status was tested on a GWAS dataset from the Wellcome Trust Case Control Consortium. The performance of the classifiers in the test dataset was evaluated by comparing area under the receiver operating characteristic curves (AUC). BN performed the best of all the data mining classifiers, but none of these did significantly better than the polygenic score approach. We further examined a subset of SNPs in genes that are expressed in the brain, under the hypothesis that these might be most relevant to BP susceptibility, but all the classifiers performed worse with this reduced set of SNPs. The discriminative accuracy of all of these methods is unlikely to be of diagnostic or clinical utility at the present time. Further research is needed to develop strategies for selecting sets of SNPs likely to be relevant to disease susceptibility and to determine if other data mining classifiers that utilize other algorithms for inferring relationships among the sets of SNPs may perform better.
Keywords: data mining, Genome-Wide Association, Mood Disorders
Background
Previous research from family, twin and adoption studies shows that genetic factors play an important role in the etiology of mood disorders [1, 2]. With the advent of genome-wide association studies (GWAS), there was a great deal of enthusiasm that susceptibility loci for these disorders would be quickly identified. However, success has been limited and much of the heritability of these disorders remains unexplained. The genetic architecture of these disorders appears to be very complex and likely involves multiple genes from different molecular pathways acting independently and interactively. Current approaches for analyzing GWAS data which typically focus on testing one variant at a time may not be sufficient for capturing the complexity of these genetic effects [3–5].
Recently, a polygenic scoring approach was successfully applied to the analysis of GWAS data on schizophrenia and bipolar disorder (BP) [6]. The number of putative risk alleles associated with disorder in single SNP tests of a training GWAS dataset were weighted and summed to derive a polygenic score. The polygenic score was then found to be significantly associated with disorder status in another test GWAS dataset. This approach is of interest because it simultaneously considers the effects across multiple loci and reveals levels of association that are more compelling than tests of single SNPs. However, it may be overly simplistic as it merely sums these effects and does not adequately account for the complex relationships between putative risk alleles that may exist on the etiologic pathway to disease.
Data mining methods are available that may be better suited to analyzing the high dimensional data generated by GWAS of complex psychiatric disorders. Data mining is the process of extracting the complex relationships and correlations hidden in large data sets. It also includes computer modeling of learning processes and the discovery of new facts through observation and experimentation. There are different algorithms for carrying out data mining, and the prediction accuracy of these algorithms may vary [7, 8]. Several examples include self-organizing maps [9], decision tree [10], support vector machines [11], Bayesian network [12], neural networks [13], and genetic algorithms [14]. There are several software suites freely available for implementing these algorithms, including Weka [15] and GIST [16]. Recently data mining has been used in many domains of biomedicine, including protein classification [17], classification of cancer sub-types [18, 19], prediction of protein secondary and tertiary structure [20], text mining[21], and protein-protein interactions[22].
The goal of this study was to examine the application of various data mining methods to the analysis of GWAS data, and to evaluate their ability to predict disorder status. We further compared the performance of these methods against the recently reported polygenic scoring approach.
Materials and Methods
Datasets
For this study, we used GWAS datasets on BP from the Bipolar Genome Studies (BiGS) Consortium [23] for the training dataset and from the Welcome Trust Case Control Consortium (WTCCC) [24] for the test dataset. The BiGS dataset consisted of two GWAS samples that have been reported on separately: the Genetic Association Information Network (GAIN) European American [25] and the Translational Genomics Research Institute (TGEN) (submitted) samples. The BP cases for both samples were collected by the National Institute of Mental Health Genetics Initiative Bipolar Disorder (NIMH-BP) Consortium [26]. Eligibility and assessment procedures for these cases have been described previously [27, 28]. Briefly, they were assessed with the Diagnostic Interview for Genetic Studies (DIGS) and this along with information from family informant data and medical records was used to assign diagnoses with best estimate procedures based on DSM-III-R or DSM-V criteria [29]. All cases were unrelated Caucasians and had a diagnosis of either bipolar I disorder (BPI) or schizoaffective disorder, bipolar subtype (SABP). The controls for the two GWAS samples came from a separate recruitment effort [30]. All control subjects completed a psychiatric questionnaire and those endorsing a history of BP, psychosis or major depression were excluded. Controls were matched by ethnicity, age and sex to the BP cases. The BP cases and controls in both GWAS samples were genotyped using the Affymetrix 6.0 array. Strict quality control (QC) measures were applied to the resulting data including dropping subjects with missing data rate ≥ 1%, and dropping SNPs with minor allele frequency < 1%, missing data rate ≥ 5%, and Hardy-Weinberg Equilibrium p-value < 10−6 among the controls. Principal component analysis with Eigenstrat was used to identify evidence of population stratification in the sample and to remove subjects who were outliers in terms of ancestral background. We combined the GAIN-EA and TGEN samples into one dataset consisting of 2,191 cases with BP and 1,434 controls genotyped on a total of 673,715 SNPs.
The testing dataset was obtained from the Wellcome Trust Case Control Consortium (WTCCC). Cases were ascertained from multiple sites around the United Kingdom (UK) and assessed using semi-structured lifetime diagnostic psychiatric interviews (in most cases the Schedules for Clinical Assessment in Neuropsychiatry). All cases had a diagnosis of a bipolar related disorder by Research Diagnostic Criteria. A common set of 3000 controls were obtained from the 1958 British Cohort study and selected from UK blood donors. The cases and controls were all Caucasian of European descent and were genotyped on the Affymetrix 500K Mapping Array [31]. QC procedures were applied to the genotyped data and included dropping subjects with missing data rate ≥ 5%, and dropping SNPs with quality score < 90%, minor allele frequency < 1%, missing data rate ≥ 5%, and Hardy-Weinberg Equilibrium p-value < 10−6 among controls. After QC, the WTCCC sample consisted of 1868 cases and 2996 controls with genotype data on 397,333 SNPs. To ensure a common set of SNPs across the training and testing datasets, we imputed the WTCCC dataset using phased haplotype data from HapMap I & II release 24 [32] as the reference panel. We used the program BEAGLE [33] to verify the orientation of the WTCCC dataset in the positive strand and then to generate most likely genotype probabilities for autosomal SNPs in the cases and controls. We retained for downstream analyses only those SNPs with a minor allele frequency ≥ 1%, R2 > 0.3, and HWE p-value > 1×10−6 among the controls. For computational efficiency, we selected a random subset of 1000 bipolar I cases and 1000 controls for the testing dataset.
Feature Selection
To render the comparisons of the different classifiers more computationally feasible, we explored two different approaches for selecting a subset of SNPs to build the case/control prediction models (i.e., feature selection). The key parameters of these different approaches are described in Table 1. First, we used PLINK [34] to perform allelic tests of association between each SNP and case/control status in the training dataset, and then used the results to carry out the two different approaches for feature selection. We used the LD-based clumping procedure in PLINK to prune the number of SNPs to a relatively uncorrelated set of the most significantly associated SNPs. We examined two different combinations of p-value and r2 parameters for guiding the pruning. We refer to these as the Whole Genome 1 (WG1) and Whole Genome 2 (WG2) sets. In the second approach, we focused only on a set of approximately 13,000 genes that prior experimental work [35] has suggested are expressed in the brain. We hypothesized that genes expressed in the brain are most likely to be relevant to a psychiatric disorder like BP. We extracted all SNPs within these genes plus/minus 30 kb in our combined GWAS dataset and performed the LD-based clumping procedure using the same two combinations of p-value and r2 parameters as above. We refer to these as the Brain Expressed 1 (BE1) and Brain Expressed 2 (BE2) sets.
Table 1.
Parameters used to define four sets of SNP feature sets
| Clumped Sets | p-value | r2 | SNPs |
|---|---|---|---|
| Whole Genome 1 (WG1) | < 0.01 | 0.25 | 3,514 |
| Whole Genome 2 (WG2) | < 0.05 | 0.25 | 14,634 |
| Brain Expressed 1 (BE1) | < 0.01 | 0.25 | 1,252 |
| Brain Expressed 2 (BE2) | < 0.05 | 0.25 | 5,366 |
Statistical Analyses
We trained five different data mining classifiers on the training dataset using the program WEKA and the four sets of SNPs described in Table 1. The five data mining methods were Bayesian networks (BN), support vector machine (SVM), Random Forest (RF), Radial Basis Function network (RBF), and logistic regression (LR). Each of these methods takes a different approach for modeling the relationship between covariates and provides a predicted probability of the outcome. In our case, the covariates were the SNP genotypes and the outcome was BP case versus control status. We assessed how well the trained models predicted case/control status in the testing dataset. We used the default parameters in WEKA for training and testing.
We used PLINK to implement the polygenic score approach for predicting case/control status using the four sets of SNPs described in Table 1. We calculated polygenic scores for the subjects in the testing dataset by taking the weighted sum of the number of “risk” alleles at each of the SNPs in the set, where the weight was based on the odds ratio for association of the SNP with case/control status in the training dataset. We then used R [36] to fit a logistic regression model with the polygenic score as a covariate and examined how well the model predicted case/control status in the testing dataset.
We compared the performance of the data mining and polygenic scoring classifiers in the testing dataset using area under the curve (AUC) analyses. In the AUC analyses, the sensitivity of the classifier was graphed against the value “1- specificity” of predicting case/control status at different thresholds of the predicted probabilities from the model, and the area under the resulting curve was calculated. The AUC can be interpreted as the probability that a classifier will correctly predict the case from a randomly chosen pair of cases and controls. The greater the AUC, the better is the performance of the classifier. Figure 1 shows the overall workflow for these analyses.
Figure 1.
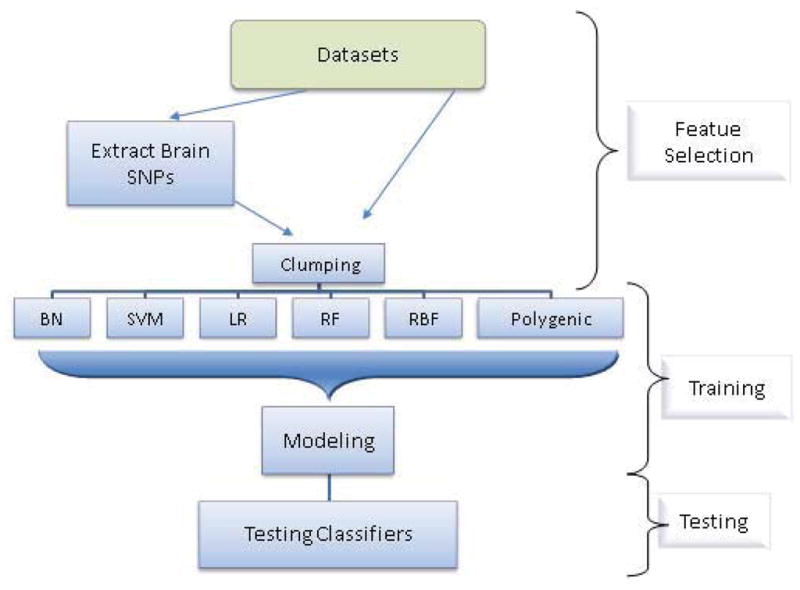
Overall workflow design of the study
Results
Figure 2 shows the results of applying the six classifiers to the testing dataset using the whole genome SNP sets. With the WG1 set of 3,514 SNPs, the BN performed the best of the five data mining classifiers. However, none of the data mining classifiers performed substantially better than the simple polygenic score classifier. The results were essentially the same with the set of 14,634 SNPs in WG2. Interestingly, the LR classifier could not be successfully fitted with the larger set of SNPs. Figure 3 shows the results of applying the six classifiers to the test dataset using the brain expressed SNP sets. Again, the BN performed slightly better than the other data mining classifiers, but none of these did better than the polygenic score classifier. All the classifiers performed worse with the smaller number of SNPs in the brain expressed set compared to the whole genome set.
Figure 2.

Comparisons of the area under the receiver operating characteristic curves for prediction with the data mining and polygenic score approaches in the testing dataset using the two whole genome SNP sets. (NA: not applicable due to computational burden)
Figure 3.

Comparisons of the area under the receiver operating characteristic curves for prediction with the data mining and polygenic score approaches in the testing dataset using the two brain expressed SNP sets.
Recent findings with the polygenic score approach have suggested that it performs better as even more liberal p-value thresholds for including SNPs in the score calculation are used. A report on schizophrenia [30] showed that the best AUC was achieved at p-value thresholds as high as 0.5. In order to determine if this was true with our BP datasets, we examined how the polygenic score approach performed in predicting case/control status in the testing dataset using p-value thresholds of 0.1 and 0.5 without clumping in the whole genome SNPs. The results are shown in Figure 4. For comparison, the results of the polygenic score approach using BE1, BE2, WG1, WG2 sets are shown again here. As in the previous report on schizophrenia, the performance of the polygenic score approach improved as more SNPs were included in the score calculation with more liberal p-value thresholds. Notably, we were unable to test the data mining classifiers using these larger sets of SNPs because of computational burden.
Figure 4.

Comparisons of the area under the receiver operating characteristic curves for the polygenic score approach under different p-value thresholds
Discussion
We compared several novel approaches for analyzing high dimensional GWAS data that test multiple SNPs simultaneously in order to detect relevant associations that might be missed by more conventional approaches which test each SNP individually. In particular, we tested five different data mining classifiers, including BN, SVM, RF, RBF and LR, and compared their performance against a recently developed polygenic score approach that simply takes a weighted sum of risk alleles across a number of SNPs as a predictor.
A recent report [6] has shown that the polygenic score approach can be used to effectively capture the association effects of multiple loci with complex psychiatric disorders. A subsequent paper examined in more detail the performance characteristics of this polygenic approach [37]. We hypothesized that data mining approaches, which have been developed specifically to analyze high-dimensional data, may provide a useful alternative for analyzing GWAS data. Several studies in recent years have reported on various successful uses of data mining methods for the analysis of genetic data [38, 39]. These studies have used SNPs from select groups of targeted genes for the classification of data and to predict the susceptibility to disease. To our knowledge, the current study is the first to examine the performance of applying these data mining approaches in the context of GWAS.
Contrary to our expectations, we found that none of the data mining classifiers did any better than the relatively simplistic polygenic score approach. We had reasoned that because of their performance characteristics the data mining classifiers would be able to detect complex interaction patterns between the SNP predictors and thereby better predict disease status. However, either the data mining classifiers that we used in this study failed to detect the complex interaction patterns between SNPs as we anticipated, or these complex interaction patterns were not present in the GWAS data. The data mining classifiers suffered from the additional limitation of not being able to analyze larger sets of predictor SNPs due to computational burden. As has been shown in previous studies, we observed that the polygenic score approach performed incrementally better with larger sets of SNPs. This may be due to the fact that psychiatric disorders like bipolar disorder are highly polygenic and the increased numbers of SNPs are tagging a greater proportion of this polygenic component. With more efficient algorithms for fitting the data mining classifiers, it is possible they may also be applied to analyze GWAS data with greater success. A common challenge for all the classifiers we tested is the problem of appropriate feature selection. Although we found that the prediction improved with increasing numbers of SNPs included in the classifier models, it is conceivable that we could do even better if we were able to identify and include only the most etiologically relevant sets of SNPs. We sought to realize such improvements by focusing on SNPs within genes that are expressed solely in the brain, guided by the notion that such SNPs would be relevant for psychiatric disorders. This strategy was not successful here, but other strategies that utilize alternative functional annotations for the SNPs merit further investigation. Another limitation of these data mining approaches is that they are often “black boxes” in terms of understanding how the selected features interact with one another. Therefore it is difficult to draw inferences about the etiologic relationship between the risk features and disease susceptibility.
It is important to note that while we have used the ability of these different methods to classify disease status based on SNP genotypes as a means of comparing their performance, none of them are sufficiently accurate to be used as a tool for prediction in clinical settings. The hope is that with the further development of analytic algorithms for capturing complex patterns in high dimensional data, the use of larger GWAS samples sizes for training the prediction models, and new strategies for using functional information to select the appropriate SNP features, we will successfully develop classifiers that are ultimately clinical useful.
Acknowledgments
The authors express their profound appreciation to the families who participated in this project, and to the many clinicians who facilitated the referral of participants to the study. Data and biomaterials for the NIMH samples were collected as part of 10 projects that participated in the NIMH Bipolar Disorder Genetics Initiative. From 1991 to 1998, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, U01 MH46282—John Nurnberger, MD, PhD, Marvin Miller, MD and Elizabeth Bowman, MD; Washington University, St Louis, MO, U01 MH46280—Theodore Reich, MD, Allison Goate, PhD and John Rice, PhD; Johns Hopkins University, Baltimore, MD U01 MH46274—J Raymond DePaulo Jr, MD Sylvia Simpson, MD, MPH and Colin Stine, PhD; NIMH Intramural Research Program, Clinical Neurogenetics Branch, Bethesda, MD—Elliot Gershon, MD, Diane Kazuba, BA and Elizabeth Maxwell, MSW. From 1999 to 2003, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, R01 MH59545—John Nurnberger, MD, PhD, Marvin J Miller, MD, Elizabeth S Bowman, MD, N Leela Rau, MD, P Ryan Moe, MD, Nalini Samavedy, MD, Rif El-Mallakh, MD (at University of Louisville), Husseini Manji, MD (at Wayne State University), Debra A Glitz, MD (at Wayne State University), Eric T Meyer, MS, Carrie Smiley, RN, Tatiana Foroud, PhD, Leah Flury, MS, Danielle M Dick, PhD and Howard Edenberg, PhD; Washington University, St Louis, MO, R01 MH059534, John Rice, PhD, Theodore Reich, MD, Allison Goate, PhD and Laura Bierut, MD; Johns Hopkins University, Baltimore, MD, R01 MH59533—Melvin McInnis, MD, J Raymond DePaulo Jr, MD, Dean F MacKinnon, MD, Francis M Mondimore, MD, James B Potash, MD, Peter P Zandi, PhD, Dimitrios Avramopoulos and Jennifer Payne; University of Pennsylvania, PA, R01 MH59553—Wade Berrettini, MD, PhD; University of California at Irvine, CA, R01 MH60068—William Byerley, MD and Mark Vawter, MD; University of Iowa, IA, R01 MH059548—William Coryell, MD and Raymond Crowe, MD; University of Chicago, Chicago, IL, R01 MH59535—Elliot Gershon, MD, Judith Badner, PhD, Francis McMahon, MD, Chunyu Liu, PhD, Alan Sanders, MD, Maria Caserta, Steven Dinwiddie, MD, Tu Nguyen, Donna Harakal; University of California at San Diego, CA, R01 MH59567—John Kelsoe, MD, Rebecca McKinney, BA; Rush University, IL, R01 MH059556—William Scheftner, MD, Howard M Kravitz, DO, MPH, Diana Marta, BA, Annette Vaughn-Brown, MSN, RN and Laurie Bederow, MA; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis J McMahon, MD, Layla Kassem, PsyD, Sevilla Detera-Wadleigh, PhD, Lisa Austin, PhD, Dennis L Murphy, MD. From 2003–2007, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, R01 MH59545, John Nurnberger, M.D., Ph.D., Marvin J. Miller, M.D., Elizabeth S. Bowman, M.D., N. Leela Rau, M.D., P. Ryan Moe, M.D., Nalini Samavedy, M.D., Rif El-Mallakh, M.D. (at University of Louisville), Husseini Manji, M.D. (at Johnson and Johnson), Debra A. Glitz, M.D. (at Wayne State University), Eric T. Meyer, Ph.D., M.S. (at Oxford University, UK), Carrie Smiley, R.N., Tatiana Foroud, Ph.D., Leah Flury, M.S., Danielle M. Dick, Ph.D (at Virginia Commonwealth University), Howard Edenberg, Ph.D.; Washington University, St. Louis, MO, R01 MH059534, John Rice, Ph.D, Theodore Reich, M.D., Allison Goate, Ph.D., Laura Bierut, M.D. K02 DA21237; Johns Hopkins University, Baltimore, M.D., R01 MH59533, Melvin McInnis, M.D., J. Raymond DePaulo, Jr., M.D., Dean F. MacKinnon, M.D., Francis M. Mondimore, M.D., James B. Potash, M.D., Peter P. Zandi, Ph.D, Dimitrios Avramopoulos, and Jennifer Payne; University of Pennsylvania, PA, R01 MH59553, Wade Berrettini, M.D., Ph.D.; University of California at San Francisco, CA, R01 MH60068, William Byerley, M.D., and Sophia Vinogradov, M.D.; University of Iowa, IA, R01 MH059548, William Coryell, M.D., and Raymond Crowe, M.D.; University of Chicago, IL, R01 MH59535, Elliot Gershon, M.D., Judith Badner, Ph.D., Francis McMahon, M.D., Chunyu Liu, Ph.D., Alan Sanders, M.D., Maria Caserta, Steven Dinwiddie, M.D., Tu Nguyen, Donna Harakal; University of California at San Diego, CA, R01 MH59567, John Kelsoe, M.D., Rebecca McKinney, B.A.; Rush University, IL, R01 MH059556, William Scheftner, M.D., Howard M. Kravitz, D.O., M.P.H., Diana Marta, B.S., Annette Vaughn-Brown, M.S.N., R.N., and Laurie Bederow, M.A.; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis J. McMahon, M.D., Layla Kassem, Psy.D., Sevilla Detera-Wadleigh, Ph.D, Lisa Austin, Ph.D, Dennis L. Murphy, M.D.; Howard University, William B. Lawson, M.D., Ph.D., Evarista Nwulia, M.D., and Maria Hipolito, M.D. Control subjects from the National Institute of Mental Health Schizophrenia Genetics Initiative (NIMH-GI), data and biomaterials are being collected by the “Molecular Genetics of Schizophrenia II” (MGS-2) collaboration. The investigators and coinvestigators are: ENH/Northwestern University, Evanston, IL, MH059571, Pablo V. Gejman, M.D. (Collaboration Coordinator; PI), Alan R. Sanders, M.D.; Emory University School of Medicine, Atlanta, GA,MH59587, Farooq Amin, M.D. (PI); Louisiana State University Health Sciences Center; New Orleans, Louisiana, MH067257, Nancy Buccola APRN, BC, MSN (PI); University of California-Irvine, Irvine, CA,MH60870, William Byerley, M.D. (PI); Washington University, St. Louis, MO, U01, MH060879, C. Robert Cloninger, M.D. (PI); University of Iowa, Iowa, IA,MH59566, Raymond Crowe, M.D. (PI), Donald Black, M.D.; University of Colorado, Denver, CO, MH059565, Robert Freedman, M.D. (PI); University of Pennsylvania, Philadelphia, PA, MH061675, Douglas Levinson M.D. (PI); University of Queensland, Queensland, Australia, MH059588, Bryan Mowry, M.D. (PI); Mt. Sinai School of Medicine, New York, NY,MH59586, Jeremy Silverman, Ph.D. (PI). Genome-wide SNP genotyping of the NIMH samples was performed through the Genetic Association Information Network under the direction of the Bipolar Genetics Studies Collaboration. The Principal Investigators and Co-Investigators were: University of California San Diego, La Jolla, CA, John R. Kelsoe, M.D. (PI), Tiffany A. Greenwood, Ph.D., Paul D. Shilling, Ph.D., Caroline Nievergelt, Ph.D.; Scripps Research Institute, La Jolla, CA: Nicholas Schork, Ph.D. (PI), Erin N. Smith, Ph.D., Cinnamon Bloss, Ph.D.; Indiana University, Bloomington, IN, John Nurnberger, M.D. (PI), Howard J. Edenberg, Ph.D., Tatiana Foroud, Ph.D.; University of Chicago, Chicago, IL, Elliot Gershon, M.D. (PI), Chunyu Liu, Ph.D., Judith A. Badner, Ph.D.; Rush University Medical Center, Chicago, IL, William A. Scheftner, M.D.; Howard University, Washington, DC, William B. Lawson, M.D. (PI), Evaristus A. Nwulia, M.D., Maria Hipolito, M.D.; University of Iowa, Iowa City, IA, William Coryell, M.D. (PI); Washington University, St. Louis, MO, John Rice, Ph.D. (PI); University of California San Francisco, San Francisco, CA, William Byerley, M.D. (PI); National Institute of Mental Health, Bethesda, MD, Francis McMahon, M.D. (PI), Thomas G. Schulze, M.D.; University of Pennsylvania, Philadelphia, PA, Wade Berrettini, M.D., Ph.D. (PI); Johns Hopkins University, Baltimore, MD, James B. Potash, M.D. (PI), Peter P. Zandi, Ph.D., Pamela Belmonte Mahon, PhD; University of Michigan, Ann Arbor, MI, Melvin G. McInnis, M.D. (PI), Sebastian Zöllner, Ph.D.; Translation Genomic Research Institute, Phoenix, AZ, David Craig, Ph.D. (PI), Szabolics Szelinger. Data and biomaterials for the subjects in the Wellcome Trust Case-Control Consortium were collected by: University of Aberdeen, Foresterhill, Aberdeen, UK, Gerome Breen, David St Clair; Birmingham University, Birmingham, UK, Sian Caesar, Katherine Gordon-Smith, Lisa Jones; Cardiff University, Cardiff, UK, Christine Fraser, Elaine K Green, Detelina Grozeva, Marian L Hamshere, Peter A Holmans, Ian R Jones, George Kirov, Valentina Moskvina, Ivan Nikolov, Michael C O’Donovan, Michael J Owen, Nick Craddock; The Institute of Psychiatry, King’s College, London, UK, David A Collier, Amanda Elkin, Anne Farmer, Richard Williamson, Peter McGuffin; Royal Victoria Infirmary, Newcastle upon Tyne, UK, Allan H Young, I Nicol Ferrier; Supported in part by R01 MH079799 (Smoller)
Contributor Information
Mehdi Pirooznia, Email: mpirooz1@jhmi.edu, School of Medicine, Johns Hopkins University, Baltimore, MD 21287, USA.
Fayaz Seifuddin, Email: fseifud1@jhmi.edu, School of Medicine, Johns Hopkins University, Baltimore, MD 21287, USA.
Jennifer Judy, Email: jtoolan@jhsph.edu, School of Public Health, Johns Hopkins University, Baltimore, MD 21205, USA.
Pamela B. Mahon, Email: pbelmon2@jhmi.edu, School of Medicine, Johns Hopkins University, Baltimore, MD 21287, USA
James B. Potash, Email: jpotash@jhmi.edu, School of Medicine, Johns Hopkins University, Baltimore, MD 21287, USA
Peter P. Zandi, Email: pzandi@jhsph.edu, School of Public Health, Johns Hopkins University, Baltimore, MD 21205, USA.
References
- 1.Todd RD, Botteron KN. Child Adolesc Psychiatr Clin N Am. Vol. 11. WB Saunders; Philadelphia, PA: 2002. Etiology and genetics of early-onset Mood Disorders. Genetic contributions to early-onset psychopathology; pp. 449–518. [DOI] [PubMed] [Google Scholar]
- 2.Merikangas KR, Low NC. The epidemiology of mood disorders. Curr Psychiatry Rep. 2004;6:411–421. doi: 10.1007/s11920-004-0004-1. [DOI] [PubMed] [Google Scholar]
- 3.Sklar P, Smoller JW, Fan J, Ferreira MA, Perlis RH, Chambert K, VL, et al. Whole-genome association study of bipolar disorder. Mol Psychiatry. 2008;13:558–569. doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W, et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry. 2009;14:755–763. doi: 10.1038/mp.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferreira MA, O'Donovan MC, Meng YA, Jones IR, DM, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarca AL, V, Carey J, Chen XW, Romero R, Draghici S. Machine learning and its applications to biology. PLoS Comput Biol. 2007;3:e116. doi: 10.1371/journal.pcbi.0030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma KL. Machine learning to boost the next generation of visualization technology. IEEE Comput Graph Appl. 2007;27:6–9. doi: 10.1109/mcg.2007.129. [DOI] [PubMed] [Google Scholar]
- 9.Yan A. Application of self-organizing maps in compounds pattern recognition and combinatorial library design. Comb Chem High Throughput Screen. 2006;9:473–480. doi: 10.2174/138620706777698562. [DOI] [PubMed] [Google Scholar]
- 10.Sachs GS. Decision tree for the treatment of bipolar disorder. J Clin Psychiatry. 2003;64(Suppl 8):35–40. [PubMed] [Google Scholar]
- 11.Byvatov E, Schneider G. Support vector machine applications in bioinformatics. Appl Bioinformatics. 2003;2:67–77. [PubMed] [Google Scholar]
- 12.Yousef M, Nebozhyn M, Shatkay H, Kanterakis S, Showe LC, Showe MK. Combining multi-species genomic data for microRNA identification using a Naive Bayes classifier. Bioinformatics. 2006;22:1325–1334. doi: 10.1093/bioinformatics/btl094. [DOI] [PubMed] [Google Scholar]
- 13.Tsodyks M, Gilbert C. Neural networks and perceptual learning. Nature. 2004;431:775–781. doi: 10.1038/nature03013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamshidi M. Tools for intelligent control: fuzzy controllers, neural networks and genetic algorithms. Philos Transact A Math Phys Eng Sci. 2003;361:1781–1808. doi: 10.1098/rsta.2003.1225. [DOI] [PubMed] [Google Scholar]
- 15.Frank E, Hall M, Trigg L, Holmes G, Witten IH. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479–2481. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
- 16.Pavlidis P, Wapinski I, Noble WS. Support vector machine classification on the web. Bioinformatics. 2004;20:586–587. doi: 10.1093/bioinformatics/btg461. [DOI] [PubMed] [Google Scholar]
- 17.Sonego P, Pacurar M, Dhir S, Kertesz-Farkas A, Kocsor A, Gaspari Z, Leunissen JA, Pongor S. A Protein Classification Benchmark collection for machine learning. Nucleic Acids Res. 2007;35:D232–236. doi: 10.1093/nar/gkl812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Statnikov A, Wang L, Aliferis CF. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinformatics. 2008;9:319. doi: 10.1186/1471-2105-9-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng Y, Li W, Liu Y. A hybrid approach for biomarker discovery from microarray gene expression data for cancer classification. Cancer Inform. 2007;2:301–311. [PMC free article] [PubMed] [Google Scholar]
- 20.Cozzetto D, Tramontano A. Advances and pitfalls in protein structure prediction. Curr Protein Pept Sci. 2008;9:567–577. doi: 10.2174/138920308786733958. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez-Esteban R. Biomedical text mining and its applications. PLoS Comput Biol. 2009;5:e1000597. doi: 10.1371/journal.pcbi.1000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugaya N, Ikeda K. Assessing the druggability of protein-protein interactions by a supervised machine-learning method. BMC Bioinformatics. 2009;10:263. doi: 10.1186/1471-2105-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahon PB, Payne JL, MacKinnon DF, Mondimore FM, Goes FS, Schweizer B, Jancic D, Coryell WH, Holmans PA, Shi J, Knowles JA, Scheftner WA, Weissman MM, Levinson DF, DePaulo JR, Jr, Zandi PP, Potash JB. Genome-wide linkage and follow-up association study of postpartum mood symptoms. Am J Psychiatry. 2009;166:1229–1237. doi: 10.1176/appi.ajp.2009.09030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007 Jun 7;447(7145):661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manolio TA, Rodriguez LL, Brooks L, Abecasis G, Ballinger D, et al. New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat Genet. 2007;39:1045–1051. doi: 10.1038/ng2127. [DOI] [PubMed] [Google Scholar]
- 26.National Institute of Mental Health Genetic Initiative of Bipolar Disorder (NIMH-BP) Consortium. http://www.nimh.nih.gov/index.shtml.
- 27.Dick DM, Foroud T, Flury L, Bowman ES, Miller MJ, Rau NL, et al. Genomewide linkage analyses of bipolar disorder: a new sample of 250 pedigrees from the National Institute of Mental Health Genetics Initiative. Am J Hum Genet. 2003 Jul;73(1):107–14. doi: 10.1086/376562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kassem L, Lopez V, Hedeker D, Steele J, Zandi P, McMahon FJ. Familiality of polarity at illness onset in bipolar affective disorder. Am J Psychiatry. 2006 Oct;163(10):1754–9. doi: 10.1176/ajp.2006.163.10.1754. [DOI] [PubMed] [Google Scholar]
- 29.Nurnberger JI, Jr, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, et al. Diagnostic interview for genetic studies. Rationale, unique features, and training. NIMH Genetics Initiative. Arch Gen Psychiatry. 1994 Nov;51(11):849–59. doi: 10.1001/archpsyc.1994.03950110009002. [DOI] [PubMed] [Google Scholar]
- 30.Sanders AR, Duan J, Levinson DF, Shi J, He D, Hou C, et al. No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am J Psychiatry. 2008 Apr;165(4):497–506. doi: 10.1176/appi.ajp.2007.07101573. [DOI] [PubMed] [Google Scholar]
- 31.Affymetrix 500K Mapping Array. http://www.affymetrix.com/browse/products.jsp?productId=131459&navMode=34000&navAction=jump&aId=productsNav.
- 32.HapMap Homepage. http://hapmap.ncbi.nlm.nih.gov/
- 33.Browning BL, Yu Z. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am J Hum Genet. 2009;85:847–861. doi: 10.1016/j.ajhg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson MB, Kawasawa YI, Mason CE, Krsnik Z, Coppola G, Bogdanovic D, Geschwind DH, Mane SM, State MW, Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The R Project for Statistical Computing. http://www.r-project.org/
- 37.Wray NR, Middeldorp CM, Birley AJ, Gordon SD, Sullivan PF, Visscher PM, Nyholt DR, Willemsen G, de Geus EJ, Slagboom PE, Montgomery GW, Martin NG, Boomsma DI. Genome-wide linkage analysis of multiple measures of neuroticism of 2 large cohorts from Australia and the Netherlands. Arch Gen Psychiatry. 2008;65:649–658. doi: 10.1001/archpsyc.65.6.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nat Genet. 2005;37:435–440. doi: 10.1038/ng1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang LC, Hsu SY, Lin E. A comparison of classification methods for predicting Chronic Fatigue Syndrome based on genetic data. J Transl Med. 2009;7:81. doi: 10.1186/1479-5876-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR. A primer on learning in Bayesian networks for computational biology. PLoS Comput Biol. 2007;3:e129. doi: 10.1371/journal.pcbi.0030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SM, Abbott PA. Bayesian networks for knowledge discovery in large datasets: basics for nurse researchers. J Biomed Inform. 2003;36:389–399. doi: 10.1016/j.jbi.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 42.Rodin A, Mosley TH, Jr, Clark AG, Sing CF, Boerwinkle E. Mining genetic epidemiology data with Bayesian networks application to APOE gene variation and plasma lipid levels. J Comput Biol. 2005;12:1–11. doi: 10.1089/cmb.2005.12.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Boyle NM, Palmer DS, Nigsch F, Mitchell JB. Simultaneous feature selection and parameter optimisation using an artificial ant colony: case study of melting point prediction. Chem Cent J Oct. 2008;29(2):21. doi: 10.1186/1752-153X-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duan KB, Rajapakse JC, Wang H, Azuaje F. Multiple SVM-RFE for gene selection in cancer classification with expression data. IEEE Trans Nanobioscience Sep. 2005;4(3):228–34. doi: 10.1109/tnb.2005.853657. [DOI] [PubMed] [Google Scholar]