

Abstract
Traumatic brain injury (s) is a leading cause of disability among young children and is associated with long-term cognitive deficits. These clinical findings have prompted an investigation of the hippocampus in an experimental model of trauma to the developing brain at postnatal day (p21). Previous studies using this model have revealed a progressive loss of neurons in the hippocampus as brain-injured animals mature to young adulthood. Here we determine if this hippocampal vulnerability is likewise reflected in altered neurogenesis and if the antioxidant glutathione peroxidase (GPx) modulates neurogenesis during maturation of the injured, immature brain. Male transgenic mice that overexpress glutathione peroxidase (GPx) and wildtype littermates were subjected to controlled cortical impact or sham surgery on p21. At two weeks postinjury, the numbers of proliferating cells and immature neurons within the subgranular zone were measured using Ki-67 and doublecortin, respectively. Bromodeoxyuridine (BrdU) was used to label dividing cells beginning two weeks postinjury. Survival (BrdU+) and neuronal differentiation (BrdU+/NeuN+) were then measured four weeks later with confocal microscopy. Two-way ANOVA revealed no significant interaction between genotype and injury. Subsequent analysis of the individual effects of injury and genotype, however, showed a significant reduction in subgranular zone proliferation (Ki-67) at two weeks postinjury (p=0.0003) and precursor cell survival (BrdU+) at six weeks postinjury (p=0.016) and a trend towards reduced neuronal differentiation (BrdU+/NeuN+) at six weeks postinjury (p=0.087). Overall, these data demonstrate that traumatic injury to the injured immature brain impairs neurogenesis during maturation and suggest that GPx cannot rescue this reduced neurogenesis.
Keywords: traumatic brain injury, dentate gyrus, hippocampus, glutathione peroxidase
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability among children, affecting more than 475,000 children annually in the United States alone (Langlois et al. 2004). Pediatric TBI results in a wide range of neurologic deficits (Mazzola and Adelson 2002) including cognitive dysfunction (Levin et al. 1997; Nybo et al. 2005). Although it has been suggested that the immature brain is resistant to damage secondary to TBI (Benz et al. 1999), recent clinical data highlight an age-dependent vulnerability. This is exemplified in studies showing that young children are more susceptible than older children to the cognitive sequelae of TBI (Adelson et al. 1997; Koskiniemi et al. 1995; Levin et al. 1992).
We have begun to explore the determinants of this vulnerability in an experimental model of traumatic injury to the immature murine brain (Pullela et al. 2006; Tong et al. 2002). We have found that traumatic injury to the frontoparietal cortex at p21, a time period that approximates the toddler-aged child (Yager and Thornhill 1997), results in predictable patterns of early irreversible hippocampal neuronal injury (Tong et al. 2002). Longer-term hippocampal vulnerability is evidenced by a reduction in the numbers of neurons in the hippocampus at two weeks postinjury with a further decline at six weeks postinjury when brain-injured animals reach young adulthood. Importantly, cognitive deficits emerge during this extended period of pathogenesis (Pullela et al. 2006). Together, these findings have prompted us to examine the hippocampus in more detail, focusing specifically on neurogenesis in the more chronically injured brain when cognitive deficits emerge.
The hippocampus is one of only two regions in the brain where new neurons are continually born throughout life (Eriksson et al. 1998). Neural precursor cells within the subgranular zone (SGZ) of the dentate gyrus produce immature neurons that migrate to the adjacent granule cell layer (GCL) where they mature and become functionally integrated into the hippocampal circuitry. Insults to the brain result in reduced SGZ neurogenesis and are associated with an impairment in hippocampal-dependent memory formation and synaptic plasticity (Nichols et al. 2005).
In the present study, we evaluate SGZ neurogenesis after TBI to the immature brain in the context of an altered antioxidant environment. Oxidative stress has been shown to be a determinant of hippocampal neurogenesis (Herrera et al. 2003; Zhang et al. 2005) and strategies to reduce oxidative stress (Cuppini et al. 2002), including administration of glutathione peroxidase (GPx) mimetics (Herrera et al. 2003), enhance neurogenesis. When compared to the adult brain, the immature brain appears to have a decreased antioxidant reserve in response to TBI, as demonstrated by its inability to upregulate GPx (Fan et al. 2003).
Here we examine the dependence of long-term hippocampal neurogenesis on GPx in the traumatized immature brain. We report a reduction in neurogenesis and show that increasing the antioxidant reserve through transgenic overexpression of GPx is unable to rescue neurogenesis. These data suggest that inadequate antioxidant reserves are not determinants in the decline of SGZ neurogenesis in the injured immature brain.
Materials and Methods
All experiments were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco, and carried out with the highest standards of animal care and housing according to the National Institutes of Health Guide for the care and use of laboratory animals. Heterozygous transgenic mice carrying 200 copies of the human intracellular GPx gene (GPx1, Mirochnitchenko et al. 1995) were bred to produce mixed litters of wildtype (WT) and GPx1-overexpressing (GPx Tg) mice. Genotype was identified from tail clippings taken on postnatal day (pnd) 14. PCR was performed on purified DNA using Platinum Supermix (Invitrogen, Carlsbad, CA) with the primer sequences ATG TGT GCT GCT CGG CTA GCG GC [5′-3′] (forward) and GCT GCA GGA ATT CGG GCG GG [5′-3′] (reverse). PCR products were separated on a 2% agarose gel containing ethidium bromide for UV visualization.
Surgical procedures
On pnd 21, male GPx Tg and WT littermates were anesthetized with 1.25% 2,2,2-tribromoethanol (Avertin, Sigma, St. Louis, MO; 0.02 mL/g body weight). During both the surgical and postoperative recovery period, normal body temperature was maintained with a circulating-water heating pad.
Each animal was placed in a stereotaxic frame (Kopf, Tujunga, CA) for surgery. After a midline scalp incision, underlying soft tissues were reflected and a circular craniotomy, 5 mm in diameter, was made with a drill in the left parietal bone between bregma and lambda with the medial edge of the craniotomy 0.5 mm lateral to midline. Animals were then subjected to controlled cortical impact injury as we have previously described (Tong et al. 2002) using a convex impactor tip that was 3 mm in diameter and oriented perpendicular to the surface of the brain. The injury was generated using a velocity of 4 m/sec, 1 mm depth of penetration, and a sustained depression of 150 msec. Sham control animals underwent identical procedures but did not receive cortical impact. In both sham and injured animals, the scalp was closed with sutures and 1 ml of saline was injected subcutaneously to prevent dehydration during recovery. Animals were regularly observed until they had fully recovered from anesthesia.
GPx activity assay
To determine the activity of GPx1 in the brains of WT and GPx Tg after sham surgery or injury, WT (n= 5 shams; n= 6 injured) and GPx Tg (n= 4 shams; n= 3 injured) mice were anesthetized with 1 ml of 1.25% Avertin and transcardially perfused with 50 ml of isotonic saline. Brains were then dissected and separated into cortex, hippocampus, and thalamus. Tissue samples were homogenized in cold 0.1 M potassium phosphate buffer (pH 7.0) and then centrifuged at 12,000 rpm for 10 min. Ten microliters of the supernatant was used to determine the amount of protein in each sample using the Pierce BCA Protein Assay (Pierce, Rockford, IL). GPx activity was determined by spectrophotometric detection in a coupled test system in which glutathione reductase was used to regenerate oxidized glutathione (Fan et al. 2003; Flohe and Gunzler 1984). This reaction requires the oxidation of NADPH to NADP+ and the decreasing absorbance of NADPH was measured at an optical density of 340 nm. Activity was expressed as units per milligram of protein, where one unit is defined as 1 nmol NADPH oxidized per minute.
Markers of early neurogenesis – precursor cell proliferation and immature neurons
We have previously shown significant neuronal loss in the hippocampus at two weeks after TBI at pnd 21 (Pullela et al. 2006). To determine if TBI also influences precursor cell function at that same time point, a second group of WT (n= 3 shams; n= 4 injured) and GPx Tg (n= 5 shams; n= 6 injured) mice was euthanized two weeks postinjury. Mice were anesthetized as previously described and transcardially perfused with 50 ml of 10% buffered formalin. Brains were then dissected and postfixed in the same formalin solution for 72 hours. After fixation, brains were placed in a Mouse Brain Matrix (Harvard Aparatus, Natick, MA), which facilitated reproducible transverse sectioning of the whole brain. A 5 mm thick section of brain containing the dorsal hippocampus was obtained and placed into a plastic tissue holder and processed for paraffin embedding. Transverse sections were cut from the rostral face of each tissue block using a rotary microtome. Beginning at the rostral-most edge of the hippocampus, a series of five consecutive 5 μm sections was collected every 300 μm until the entire dorsal hippocampus had been sampled. The caudal boundary of the dorsal hippocampus was defined as the posterior commissure. A subset of these sections was reserved for Nissl staining to qualitatively assess injury.
To assess proliferating precursor cells and their progeny (immature neurons), sections were stained with antibodies against Ki-67 (DakoCytomation, Carpinteria, CA), a nuclear antigen expressed in all phases of the cell cycle except G0 (Kee et al. 2002), or doublecortin (DCx, Santa Cruz Biotechnology, Santa Cruz, CA), a tubulin-associated protein expressed in migrating neuroblasts (Englund et al. 2002). After deparaffinization and quenching of endogenous peroxidases (30 minutes in 0.3% hydrogen peroxide in 70% ethanol), sections were incubated in preheated 10 mM sodium citrate butter (pH 6.0) and boiled for 10 minutes using a microwave oven. Sections were then incubated in the sodium citrate buffer for an additional 20 minutes, washed in phosphate-buffered saline (PBS), and blocked for 30 minutes. Blocking was done with PBS containing 2% normal rabbit serum (NRS) for Ki-67 and 5% normal horse serum (NHS) for DCx. Sections were then incubated overnight at 4° C with the primary antibody (Ki-67 diluted 1:100 in PBS with 2% NRS and DCx diluted 1:500 in PBS with 5% NHS). After washing, sections were incubated for 60 minutes at room temperature with biotinylated secondary antibodies. Rabbit anti-rat IgG (Vector, Burlingame, CA) diluted 1:200 in PBS with 2% NRS was used for Ki-67 while anti-goat IgG (Vector) diluted 1:500 in 5% NHS was used for DCx. Biotinylated secondary antibodies were then detected using an ABC system (Vector) and diaminobenzidine as the chromogen. Sections were then counterstained with Gill's hematoxylin, dehydrated, and mounted.
The number of proliferating precursor cells within the SGZ and immature neurons within the GCL were scored blinded to genotype and treatment. For each animal, counts were done on a series of consecutive sections, spaced 300 μm apart, beginning with the rostral-most section containing both the infra- and suprapyramidal blades of the dentate gyrus and ending once the posterior commissure was reached. Sections were visualized using a Nikon brightfield microscope and a 40× objective. The data are reported as the total sum of Ki-67- or DCx-positive cells in each animal.
Neurogenesis
To determine the effects of TBI on the survival and neuronal differentiation of newly born cells in the SGZ, a third group of WT (n= 4 shams; n= 6 injured) and GPx Tg (n= 6 shams; n= 7 injured) mice was administered 5-bromo-2′-deoxyuridine (BrdU, Sigma), a thymidine analog that is incorporated into the DNA of dividing cells and thus allows dividing cells and their progeny to be tracked. Beginning two weeks after injury (the same time point when Ki-67 and DCx were analyzed), mice were given seven consecutive daily intraperitoneal injections of BrdU (50 mg/kg diluted in saline). Three weeks after the last injection (six weeks postinjury), mice were anesthetized as described above and transcardially perfused with 50 ml of 4% paraformaldehyde. After postfixation in 4% paraformaldehyde for six hours, brains were cryoprotected in 30% sucrose for 48 hours and then sectioned into 50 μm sections using a sliding microtome. Floating sections were then double-stained with antibodies against BrdU and NeuN (a marker of mature neurons). Blocking steps were done in tris-buffered saline (TBS) containing 0.3% Triton X-100 (Sigma) and 3% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) while all antibody incubations were done in TBS with 0.3% Triton-X and 1% normal donkey serum. After washing in PBS, sections were blocked for 30 minutes at room temperature. Sections were then incubated overnight at 4° C with monoclonal mouse anti-NeuN (Chemikon, Temecula, CA, diluted 1:200). After washing, sections were then incubated for six hours at room temperature with the appropriate secondary fluorescent antibodies diluted at 1:200. Following this, sections were washed in TBS, postfixed for 10 minutes in 4% paraformaldehyde, washed for 10 minutes in saline, and then incubated in 3N hydrochloric acid for 30 minutes at 37°C. After further washing in TBS, sections were incubated with rat anti-BrdU (Oxford Biotechnology, Kidlington, Oxford, UK) diluted 1:100. Incubation with a secondary fluorescent antibody was done as described in the preceding step.
Confocal microscopy with a Nikon C1 confocal microscope was used to quantify surviving newly born cells (BrdU+) and newly born neurons (BrdU+/NeuN+). The observer was blinded to genotype and treatment. A z-stack at 60× magnification was obtained for every sixth section throughout the dorsal hippocampus (as defined above). Appropriate gain and black-level settings were obtained on control tissues stained with secondary antibodies alone. Upper and lower thresholds were set using a range indicator function to minimize data loss due to saturation. The total number of BrdU-positive cells within the dentate gyrus was scored. To quantify double-labeled cells, each BrdU-positive cell was manually examined in its full ‘z’ dimension and only those cells that were unambiguously associated with NeuN were scored as positive. Final counts were reported as the total sum of BrdU-positive or double-labeled cells in each animal.
Statistical analysis
Data were analyzed using two-way analysis of variance (ANOVA) with genotype (WT or GPx Tg) and treatment (sham or injury) as factors. Two-way ANOVA allowed us to test three null hypotheses for each experiment. The primary null hypothesis for each analysis was that there was no interaction between the two factors (genotype and treatment). When that null hypothesis proved correct (ie, the interaction between genotype and treatment was not significant), we could then individually test the null hypotheses that there was no effect of genotype and no effect of treatment on the study population. Significance was set at p<0.05.
Results
GPx activity is unchanged after TBI
We first measured GPx activity in the cortex, hippocampus, and thalamus of WT and GPx Tg animals subjected to either sham surgery or TBI. GPx activity was at least two-fold higher in each region of the brain in GPx Tg compared to WT littermates, validating this model of GPx overexpression. Two-way ANOVA revealed that only genotype had a significant effect upon GPx activity in the various brain regions assessed (p<0.0001). The effects of the interaction between genotype and treatment, as well as treatment alone, were not significant (p=0.40 and p=0.41, respectively). Thus, GPx activity remains unchanged in the immature WT and GPx transgenic brain after injury (Fig 1).
Figure 1. GPx activity 24 hours after sham surgery or TBI.
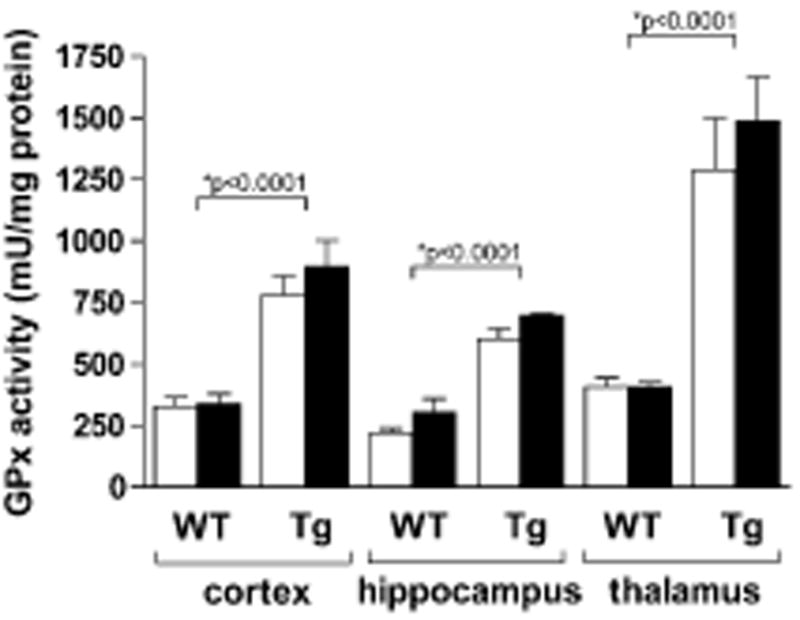
GPx activity from the ipsilateral cortex, hippocampus, and thalamus of sham and injured animals was measured using spectrophotometry. Two-way ANOVA revealed no interaction between genotype and injury, but genotype showed a significant difference in each brain region, with GPx Tg mice having at least two-fold activity compared to WTs. Injury had no effect on GPx activity. Open bars: sham animals; solid bars: brain-injured animals.
To determine if GPx overexpression influences weight gain during maturation, we compared body weights among the four experimental groups that received BrdU. No differences in body weight were noted at time of injury (p21, p=0.85), two weeks postinjury (time of first BrdU injection, p=0.97), and six weeks postinjury (time of euthanasia, p=0.71).
Gross cytoarchitecture is similar between GPx Tg and WT animals
There were no overt differences in sham-operated brains between genotypes. At two weeks postinjury, both genotypes showed prominent cortical tissue loss in the ipsilateral hemisphere (Fig 2). In some cases, this cortical lesion was associated with mild deformation of the underlying hippocampus.
Figure 2. Ipsilateral hemisphere two weeks postinjury.
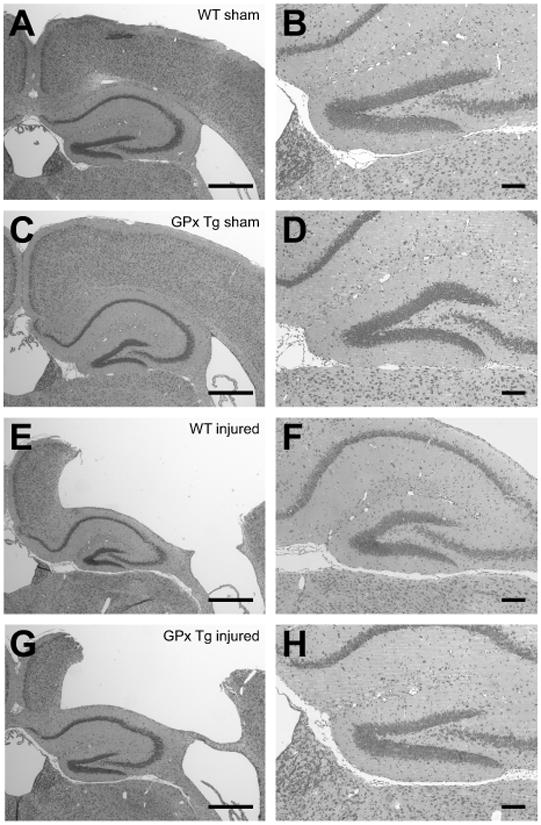
Nissl stains detail the gross pattern of injury from controlled cortical impact (E-H) compared to sham controls (A-D). TBI results in gross cortical tissue loss in both WT (E-F) and GPx Tg (G-H) animals with some mild deformation of the hippocampus. A-B, WT sham; C-D, GPx Tg sham; E-F, WT injured; G-H, GPx Tg injured. Scale bars: A, C, E, and G 500μm; B, D, F, and H 100μm.
TBI decreases hippocampal neurogenesis but GPx overexpression does not rescue neurogenesis
Ki-67 was first used to assess precursor cell proliferation. There was no significant interaction (p=0.21) between genotype and treatment on the numbers of proliferating precursor cells in the SGZ (Fig 3). Injury, but not genotype, had a significant effect (p=0.0003 and p=0.39, respectively), with injured animals in both groups showing lower numbers of proliferating precursor cells. These results indicate that traumatic injury to the immature brain results in a similar pattern of decreased SGZ precursor cell proliferation in WT and GPx Tg mice.
Figure 3. Precursor cell proliferation two weeks postinjury.
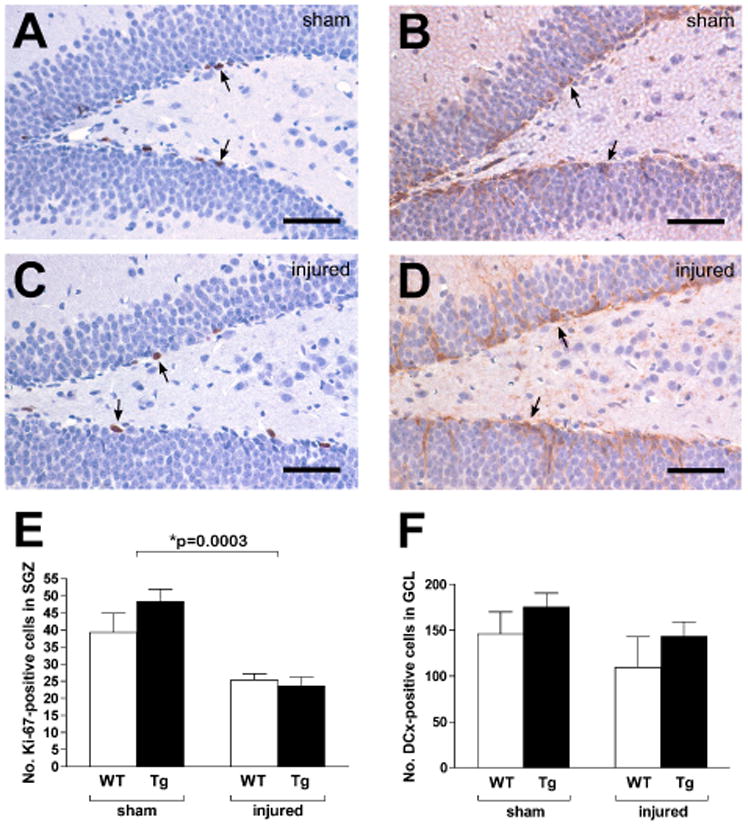
Ki-67 immunohistochemistry was used to assess precursor cell proliferation in the subgranular zones (SGZ) of sham (A, WT) and injured (C, GPx Tg) mice. Quantification of Ki-67+ cells (arrows) revealed a significant reduction in injured animals relative to sham controls (E, p=0.0003). Early neuronal differentiation within the granule cell layer (GCL) was assessed with DCx immunohistochemistry in sham (B, WT) and injured (D, GPx Tg) animals. There was no interaction between genotype and injury (p=0.92). In contrast to Ki-67, both injury and genotype had no effect on DCx numbers (p=0.13 and p=0.16), respectively). Scale bars: 100μm.
DCx was next used to measure early neuronal differentiation within the GCL. As with Ki-67, the interaction between genotype and treatment on DCx-positive cell numbers was not significant (p=0.92). However, the number of immature neurons two weeks postinjury was also not affected by injury or genotype (p=0.13 and p=0.16, respectively, Fig 3)
We next evaluated survival and neuronal differentiation of newly born cells. There was no significant interaction (p=0.56) between genotype and treatment on numbers of surviving (BrdU+) precursor cells in the SGZ (Fig 4). Injury, but not genotype, however, had a significant effect (p=0.016 and p=0.31, respectively), with injured animals in both groups showing lower numbers of surviving precursor cells. As a final marker of neurogenesis, we analyzed the number of newly born (BrdU-positive) cells that differentiated into mature neurons (BrdU- and NeuN-positive cells). The pattern was similar to what we found with BrdU-positive cell counts, with injury showing a trend toward significance (p=0.087), but no effect of genotype or the interaction between genotype and treatment (p=0.71 and p=0.37, respectively). When the percentages of newly born cells that had differentiated into mature neurons were compared (WT sham 63.2±4.9%; WT injury 72.1±3.5%; GPx Tg sham 63.8±8.6%; GPx Tg injury 62.4±9.4%), we found no differences between genotypes or injury groups (interaction: p=0.17; genotype: p=0.22; injury: p=0.31), indicating that neither injury nor GPx overexpression affected the relative percentage of newly born cells that become neurons.
Figure 4. Proliferating cell survival and neuronal differentiation.
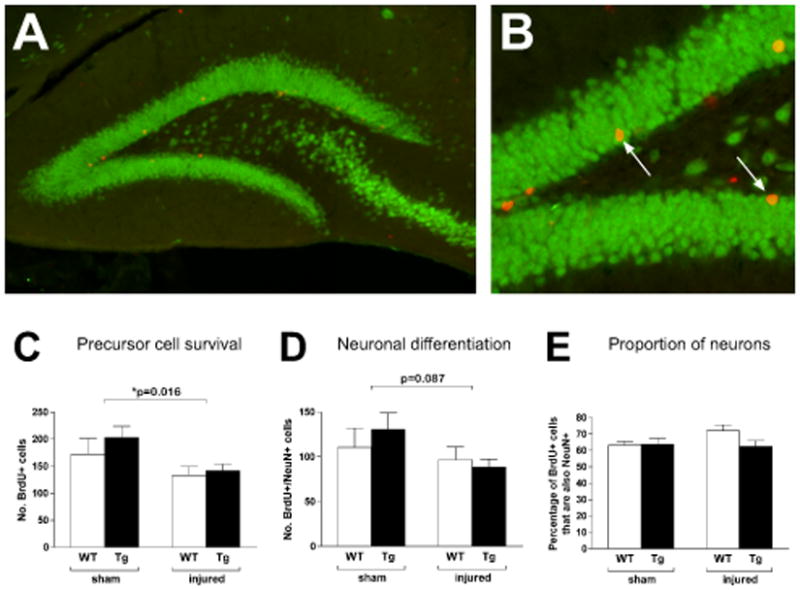
BrdU was administered beginning two weeks postinjury for seven consecutive days. Animals were euthanized three weeks after the final injection (six weeks postinjury) and assessed for proliferating cell survival (BrdU+, red) and neuronal differentiation (double-labelling with NeuN, green) within the dentate gyrus. The majority of BrdU+ cells were located in the subgranular zone (A, GPx Tg sham) where double-labeled cells could also be found (B, arrows). As with Ki-67, the interaction between genotype and injury had no effect on proliferating cell survival (p=0.56) but injury resulted in a significant reduction (p=0.016, C). A similar pattern was seen with neuronal differentiation, with injury trending towards an effect (p=0.087, D). Thus, while injury reduces proliferating cell survival and possibly neuronal differentiation, GPx overexpression is unable to rescue those changes. No differences were seen in the percentage of BrdU+ cells that differentiated into neurons (E). Scale bars: A 100μm; B 50μm.
Discussion
This is the first study to assess hippocampal neurogenesis after TBI to the immature brain. We focused on the time period spanning two to eight weeks postinjury, which parallels the progressive loss of neurons in the hippocampus (Pullela et al. 2006). We found that TBI reduces proliferation and survival of SGZ precursor cells. Moreover, GPx overexpression does not alter these injury-induced changes in hippocampal neurogenesis. That neurogenesis is not rescued by increased GPx activity suggests that oxidative events are not the key determinants of reduced neurogenesis after traumatic injury to the immature brain.
Neurogenesis after TBI
A number of elegant studies have examined neurogenesis within the first week after traumatic injury to the adult brain, when apoptosis and neuronal death are at their peak (McIntosh et al. 1998; Sato et al. 2001). What has emerged from this collective effort is that brain injury increases markers of neurogenesis in both the SGZ and subventricular zone after either controlled cortical impact (Braun et al. 2002; Dash et al. 2001; Kernie et al. 2001; Yoshimura et al. 2003) or fluid percussion injury (Chirumamilla et al. 2002; Emery et al. 2005; Gaulke et al. 2005; Rice et al. 2003). Interestingly, Dash et al. (Dash et al. 2001) examined BrdU expression within the dentate gyrus and found that while the number of BrdU-positive cells peaked at three days postinjury, BrdU expression returned to baseline by two weeks postinjury. These findings suggest that increased neurogenesis may only be a transient phenomenon in the injured adult brain. Such a response may reflect a reparative response (Picard-Riera et al. 2004) or may lead to aberrant reorganization of the brain, as illustrated in a seizure model where newly born neuronal precursors show altered migration with subsequent aberrant integration into the hippocampal circuitry (Parent et al. 1997).
Although the effects of TBI on the immature brain are poorly understood, several studies have characterized the patterns of neuronal degeneration and loss as well as changes in plasticity within the hippocampus (Adelson et al. 2001; Adelson et al. 1996; Bittigau et al. 2004; Bittigau et al. 1999; Card et al. 2005; Tong et al. 2002). While these studies employed different models of TBI, each demonstrates that the hippocampus, and in particular subregion CA3 and the dentate gyrus, are vulnerable to TBI and that this vulnerability is seen as early as one day postinjury. These findings are similar to what is seen in the adult brain, where neurons in both CA3 (Baldwin et al. 1997; Grady et al. 2003; Nawashiro et al. 1995) and the dentate hilus (Lowenstein et al. 1992) are particularly vulnerable.
The present study examines hippocampal neurogenesis after TBI to the immature brain beginning two weeks postinjury. We have previously shown that the injured immature brain undergoes a prolonged period of pathogenesis as demonstrated by expansion of the cortical lesion and hippocampal neuronal loss (Pullela et al. 2006). Here we show that neurogenesis is impaired during this dynamic period of secondary pathogenesis. We observed a reduction in the numbers of proliferating SGZ precursor cells as well as the subsequent survival and neuronal differentiation of the progeny of proliferating cells. It remains unclear, however, whether these reductions in survival and neuronal differentiation were simply due to a smaller population of proliferating cells at the time of BrdU administration or whether these processes were also individually altered by TBI. As designed, our methods did not allow for direct quantitative comparisons between the number of Ki67-positive cells at two weeks postinjury and precursor cell survival at six weeks postinjury. In addition, we did not address the potential for newly born cells to differentiate into non-neuronal cell types, such as astrocytes and microglia. Astrocytic and microglial proliferation are known components of the pathogenesis of TBI (Igarashi et al. 2001; Kernie et al. 2001; Tong et al. 2002) so it remains to be elucidated how the proliferation of such cell populations are affected in the injured immature brain.
Recently, Sun et al. (Sun et al. 2007) examined the long-term fate of proliferating cells in the hippocampus after fluid percussion injury to the adult rat brain. Injured animals showed significantly higher levels of newly-born cell survival at both five days and ten weeks postinjury when compared to shams. While the time points, animal model, and injury model of this study do not correspond with ours, these findings suggest the possibility that the adult and immature brains respond differently to traumatic injury. Further examination of the time course of injury in both ages is needed.
Modulators of hippocampal neurogenesis
A number of factors have been implicated in the regulation of hippocampal neurogenesis in both normal and disease states, including neurotransmitters, growth hormones, glucocorticoids, inflammation, and oxidative stress (Herrera et al. 2003; Nichols et al. 2005; Zhang et al. 2005). The latter is of particular interest in the setting of pediatric trauma (Bauer and Fritz 2004; Potts et al. 2006). TBI generates reactive oxygen and nitrogen species via several mechanisms, including excitotoxicity, mitochondrial dysfunction, neutrophil infiltration, heme degradation, and induction of nitric oxide synthase, xanthine oxidase, and the cyclooxygenase pathway (Hall and Braughler 1993; Lewen et al. 2000). In contrast to the adult brain, the immature brain is unable to upregulate the antioxidant GPx in response to TBI (Fan et al. 2003). There is also clinical evidence that head-injured children have a reduced antioxidant reserve after TBI (Bayir et al. 2002).
Interest in GPx as a modifier of injury-related events in the immature brain arose from findings in models of neonatal hypoxia/ischemia. Transgenic mice that overexpressed the antioxidant superoxide dismutase fared surprisingly worse than WTs (Ditelberg et al. 1996). The poorer outcome was subsequently attributed to an elevation in hydrogen peroxide levels secondary to the increased superoxide dismutase activity. Importantly, outcomes are improved after neonatal hypoxia/ischemia in mice that overexpress GPx (Sheldon et al. 2004).
We have shown that GPx activity does not increase in response to traumatic injury to the immature brain (Fan et al. 2003). In the present study, we determined if increased GPx activity would rescue neurogenesis. Hippocampal neurogenesis in the adult brain is impaired by oxidative stress (Herrera et al. 2003; Zhang et al. 2005) and enhanced by antioxidant agents such as tocopherols (Cuppini et al. 2002) and ebselen (Herrera et al. 2003), a GPx mimetic. Conversely, we found decreased neurogenesis in the injured immature brain despite increased GPx activity. Such findings raise a question regarding the potential factors that contribute to the decline in hippocampal neurogenesis. Perhaps the oxidative stress that has been well documented in the acutely-injured brain is not a prevalent factor during later stages of pathogenesis. Alternatively, decreased neurogenesis may occur independently of antioxidant status and reflect other factors that are unique to the extended period of pathogenesis that has been seen in the injured immature brain (Pullela et al. 2006). Ongoing inflammation, for example, could influence neurogenesis. In preliminary studies, we have shown that inflammation, characterized in part by microglial activation and the influx of leukocytes occurs over an extended period of time in the injured immature brain (Tsuro-Aoyagi et al. 2006). Inflammation has been linked to decreased neurogenesis (Nichols et al. 2005) and as such may be a determinant of neurogenesis in the injured immature brain.
Acknowledgments
This research was supported by NIH grant NS050159.
We would like to thank Dr. Charles McCulloch for his invaluable assistance with the statistical analysis, Kelly Fishman and Jennifer Baure for their technical assistance, and Dr. Oleg Mirochnitchenko for his generous gift of GPx transgenic breeding pairs.
References
- Adelson PD, Dixon CE, Robichaud P, Kochanek PM. Motor and cognitive functional deficits following diffuse traumatic brain injury in the immature rat. J Neurotrauma. 1997;14(2):99–108. doi: 10.1089/neu.1997.14.99. [DOI] [PubMed] [Google Scholar]
- Adelson PD, Jenkins LW, Hamilton RL, Robichaud P, Tran MP, Kochanek PM. Histopathologic response of the immature rat to diffuse traumatic brain injury. J Neurotrauma. 2001;18(10):967–976. doi: 10.1089/08977150152693674. [DOI] [PubMed] [Google Scholar]
- Adelson PD, Robichaud P, Hamilton RL, Kochanek PM. A model of diffuse traumatic brain injury in the immature rat. J Neurosurg. 1996;85(5):877–884. doi: 10.3171/jns.1996.85.5.0877. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Gibson T, Callihan CT, Sullivan PG, Palmer E, Scheff SW. Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. J Neurotrauma. 1997;14(6):385–398. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- Bauer R, Fritz H. Pathophysiology of traumatic injury in the developing brain: an introduction and short update. Exp Toxicol Pathol. 2004;56(1-2):65–73. doi: 10.1016/j.etp.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51(5):571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Benz B, Ritz A, Kiesow S. Influence of age-related factors on long-term outcome after traumatic brain injury (TBI) in children: A review of recent literature and some preliminary findings. Restor Neurol Neurosci. 1999;14(2-3):135–141. [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Felderhoff-Mueser U, Ikonomidou C. Apoptotic neurodegeneration in the context of traumatic injury to the developing brain. Exp Toxicol Pathol. 2004;56(1-2):83–89. doi: 10.1016/j.etp.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H, Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C, Asikainen I, Kaste M, Sarna S, Sato M, Fukushi Y, Ishizawa S, Okinaga S, Muller RM, Shibahara S. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol. 1999;45(6):724–735. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. adulthood. In the groups of patients with moderate and mild brain injuries such a relationship was not found between age or pre-injury education and outcome. [DOI] [PubMed] [Google Scholar]
- Braun H, Schafer K, Hollt V. BetaIII tubulin-expressing neurons reveal enhanced neurogenesis in hippocampal and cortical structures after a contusion trauma in rats. J Neurotrauma. 2002;19(8):975–983. doi: 10.1089/089771502320317122. [DOI] [PubMed] [Google Scholar]
- Card JP, Santone DJ, Jr, Gluhovsky MY, Adelson PD. Plastic reorganization of hippocampal and neocortical circuitry in experimental traumatic brain injury in the immature rat. J Neurotrauma. 2005;22(9):989–1002. doi: 10.1089/neu.2005.22.989. [DOI] [PubMed] [Google Scholar]
- Chirumamilla S, Sun D, Bullock MR, Colello RJ. Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J Neurotrauma. 2002;19(6):693–703. doi: 10.1089/08977150260139084. [DOI] [PubMed] [Google Scholar]
- Cuppini R, Ciaroni S, Cecchini T, Ambrogini P, Ferri P, Cuppini C, Ninfali P, Del Grande P. Tocopherols enhance neurogenesis in dentate gyrus of adult rats. Int J Vitam Nutr Res. 2002;72(3):170–176. doi: 10.1024/0300-9831.72.3.170. [DOI] [PubMed] [Google Scholar]
- Dash PK, Mach SA, Moore AN. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res. 2001;63(4):313–319. doi: 10.1002/1097-4547(20010215)63:4<313::AID-JNR1025>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM. Brain injury after perinatal hypoxiaischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res. 1996;39(2):204–208. doi: 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- Emery DL, Fulp CT, Saatman KE, Schutz C, Neugebauer E, McIntosh TK. Newly born granule cells in the dentate gyrus rapidly extend axons into the hippocampal CA3 region following experimental brain injury. J Neurotrauma. 2005;22(9):978–988. doi: 10.1089/neu.2005.22.978. [DOI] [PubMed] [Google Scholar]
- Englund U, Bjorklund A, Wictorin K. Migration patterns and phenotypic differentiation of long-term expanded human neural progenitor cells after transplantation into the adult rat brain. Brain Res Dev Brain Res. 2002;134(1-2):123–141. doi: 10.1016/s0165-3806(01)00330-3. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4(11):1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Fan P, Yamauchi T, Noble LJ, Ferriero DM. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma. 2003;20(5):437–445. doi: 10.1089/089771503765355513. [DOI] [PubMed] [Google Scholar]
- Flohe L, Gunzler WA. Assays of glutathione peroxidase. Methods Enzymol. 1984;105:114–121. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- Gaulke LJ, Horner PJ, Fink AJ, McNamara CL, Hicks RR. Environmental enrichment increases progenitor cell survival in the dentate gyrus following lateral fluid percussion injury. Brain Res Mol Brain Res. 2005;141(2):138–150. doi: 10.1016/j.molbrainres.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady MS, Charleston JS, Maris D, Witgen BM, Lifshitz J. Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological estimation. J Neurotrauma. 2003;20(10):929–941. doi: 10.1089/089771503770195786. [DOI] [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Free radicals in CNS injury. Res Publ Assoc Res Nerv Ment Dis. 1993;71:81–105. [PubMed] [Google Scholar]
- Herrera DG, Yague AG, Johnsen-Soriano S, Bosch-Morell F, Collado-Morente L, Muriach M, Romero FJ, Garcia-Verdugo JM. Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. Proc Natl Acad Sci U S A. 2003;100(13):7919–7924. doi: 10.1073/pnas.1230907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi T, Huang TT, Noble LJ. Regional vulnerability after traumatic brain injury: gender differences in mice that overexpress human copper, zinc superoxide dismutase. Exp Neurol. 2001;172(2):332–341. doi: 10.1006/exnr.2001.7820. [DOI] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods. 2002;115(1):97–105. doi: 10.1016/s0165-0270(02)00007-9. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Erwin TM, Parada LF. Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J Neurosci Res. 2001;66(3):317–326. doi: 10.1002/jnr.10013. [DOI] [PubMed] [Google Scholar]
- Koskiniemi M, Kyykka T, Nybo T, Jarho L. Long-term outcome after severe brain injury in preschoolers is worse than expected. Arch Pediatr Adolesc Med. 1995;149(3):249–254. doi: 10.1001/archpedi.1995.02170150029004. [DOI] [PubMed] [Google Scholar]
- Langlois J, Rutland-Brown W, Thomas K. Traumatic brain injury in the United States: Emergency department visits, hospitalizations, and deaths. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2004. [Google Scholar]
- Levin HS, Aldrich EF, Saydjari C, Eisenberg HM, Foulkes MA, Bellefleur M, Luerssen TG, Jane JA, Marmarou A, Marshall LF, et al. Severe head injury in children: experience of the Traumatic Coma Data Bank. Neurosurgery. 1992;31(3):435–443. doi: 10.1227/00006123-199209000-00008. discussion 443-434. [DOI] [PubMed] [Google Scholar]
- Levin HS, Song J, Scheibel RS, Fletcher JM, Harward H, Lilly M, Goldstein F. Concept formation and problem-solving following closed head injury in children. J Int Neuropsychol Soc. 1997;3(6):598–607. [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17(10):871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12(12):4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzola CA, Adelson PD. Critical care management of head trauma in children. Crit Care Med. 2002;30(11 Suppl):S393–401. doi: 10.1097/00003246-200211001-00003. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ. The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Appl Neurobiol. 1998;24(4):251–267. doi: 10.1046/j.1365-2990.1998.00121.x. [DOI] [PubMed] [Google Scholar]
- Mirochnitchenko O, Palnitkar U, Philbert M, Inouye M. Thermosensitive phenotype of transgenic mice overproducing human glutathione peroxidases. Proc Natl Acad Sci U S A. 1995;92(18):8120–8124. doi: 10.1073/pnas.92.18.8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawashiro H, Shima K, Chigasaki H. Selective vulnerability of hippocampal CA3 neurons to hypoxia after mild concussion in the rat. Neurol Res. 1995;17(6):455–460. [PubMed] [Google Scholar]
- Nichols NR, Agolley D, Zieba M, Bye N. Glucocorticoid regulation of glial responses during hippocampal neurodegeneration and regeneration. Brain Res Brain Res Rev. 2005;48(2):287–301. doi: 10.1016/j.brainresrev.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Nybo T, Sainio M, Muller K. Middle age cognition and vocational outcome of childhood brain injury. Acta Neurol Scand. 2005;112(5):338–342. doi: 10.1111/j.1600-0404.2005.00489.x. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17(10):3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard-Riera N, Nait-Oumesmar B, Baron-Van Evercooren A. Endogenous adult neural stem cells: limits and potential to repair the injured central nervous system. J Neurosci Res. 2004;76(2):223–231. doi: 10.1002/jnr.20040. [DOI] [PubMed] [Google Scholar]
- Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, Manvelyan HM, Noble-Haeusslein LJ. Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. NeuroRx. 2006;3(2):143–153. doi: 10.1016/j.nurx.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullela R, Raber J, Pfankuch T, Ferriero DM, Claus CP, Koh SE, Yamauchi T, Rola R, Fike JR, Noble-Haeusslein LJ. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev Neurosci. 2006;28(4-5):396–409. doi: 10.1159/000094166. [DOI] [PubMed] [Google Scholar]
- Rice AC, Khaldi A, Harvey HB, Salman NJ, White F, Fillmore H, Bullock MR. Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp Neurol. 2003;183(2):406–417. doi: 10.1016/s0014-4886(03)00241-3. [DOI] [PubMed] [Google Scholar]
- Sato M, Chang E, Igarashi T, Noble LJ. Neuronal injury and loss after traumatic brain injury: time course and regional variability. Brain Res. 2001;917(1):45–54. doi: 10.1016/s0006-8993(01)02905-5. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Jiang X, Francisco C, Christen S, Vexler ZS, Tauber MG, Ferriero DM. Manipulation of antioxidant pathways in neonatal murine brain. Pediatr Res. 2004;56(4):656–662. doi: 10.1203/01.PDR.0000139413.27864.50. [DOI] [PubMed] [Google Scholar]
- Sun D, McGinn MJ, Zhou Z, Harvey HB, Bullock MR, Colello RJ. Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp Neurol. 2007;204(1):264–272. doi: 10.1016/j.expneurol.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Tong W, Igarashi T, Ferriero DM, Noble LJ. Traumatic brain injury in the immature mouse brain: characterization of regional vulnerability. Exp Neurol. 2002;176(1):105–116. doi: 10.1006/exnr.2002.7941. [DOI] [PubMed] [Google Scholar]
- Tsuro-Aoyagi K, Caus CP, Potts MB, Manvelyan HM, Koh SE, Noble-Haeusslein LJ. Society for Neuroscience. 2006. The kinetics of leukocyte infiltration differ in the immature relative to the adult brain after traumatic brain injury. abstract 682.15. [Google Scholar]
- Yager JY, Thornhill JA. The effect of age on susceptibility to hypoxic-ischemic brain damage. Neurosci Biobehav Rev. 1997;21(2):167–174. doi: 10.1016/s0149-7634(96)00006-1. [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Teramoto T, Whalen MJ, Irizarry MC, Takagi Y, Qiu J, Harada J, Waeber C, Breakefield XO, Moskowitz MA. FGF-2 regulates neurogenesis and degeneration in the dentate gyrus after traumatic brain injury in mice. J Clin Invest. 2003;112(8):1202–1210. doi: 10.1172/JCI16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Li X, Cui X, Zuo P. D-galactose injured neurogenesis in the hippocampus of adult mice. Neurol Res. 2005;27(5):552–556. doi: 10.1179/016164105X25126. [DOI] [PubMed] [Google Scholar]