

Abstract
Objective
Despite consistent evidence that serotonin functioning affects stress reactivity and vulnerability to aggression, research on serotonin gene-stress interactions (G × E) in the development of aggression remains limited. The present study investigated variation in the promoter region of the serotonin transporter gene (5-HTTLPR) as a moderator of the stress-aggression association at the transition to adulthood.
Methods
Multiple informants and multiple measures were used to assess aggression in a cohort of 381 Australian youth (61% female, 93% Caucasian) interviewed at ages 15 and 20. At age 20, semistructured interviews assessed acute and chronic stressors occurring in the past 12 months.
Results
Structural equation modeling analyses revealed a significant main effect of chronic stress, but not 5-HTTLPR or acute stress, on increases in aggression at age 20. Consistent with G × E hypotheses, 5-HTTLPR short allele carriers demonstrated greater increments in aggression following chronic stress relative to long allele homozygotes. The strength of chronic stress G × E did not vary according to sex.
Conclusions
Variation at 5-HTTLPR appears to contribute to individual differences in aggressive reactions to chronic stress at the transition to adulthood.
Keywords: aggression, serotonin transporter gene, acute stress, chronic stress, gene-environment interaction
Compared to the rapid proliferation of studies investigating the joint contributions of genetic vulnerabilities and environmental stress to the development of internalizing disorders, there has been more limited growth in our understanding of how gene-stress interactions (G × E) affect aggression phenotypes. A landmark molecular genetics study by Caspi et al. (2002) demonstrated an interactive effect between childhood maltreatment and monoamine oxidase A (MAOA) genotype in predicting violent behavior and antisocial traits in young adulthood. Various research groups have since replicated this finding (Kim-Cohen et al., 2006), but the generalizability of serotonergic G × E effects on aggression has rarely been examined across different candidate genes (e.g., tryptophan hydroxylase, serotonin transporter gene), types of stress (e.g., acute life events), and developmental periods. The present study expands on previous G × E research by examining the moderating effect of variation in the serotonin transporter gene on the stress-aggression association for both chronic and acute stressors at the transition to adulthood.
An extensive body of research has established that various forms of early adversity (e.g., economic disadvantage, parental illness) in childhood are concurrently and prospectively related to elevated aggression (e.g., Conger, Ge, Elder, Lorenz, & Simons, 1994; Dodge, Bates, & Petit, 1990). At the same time, a consistent observation in this literature is that a large proportion of youth is resilient to these risk conditions (Cicchetti, Rogosch, Lynch, & Holt, 1993), meaning they do not develop problematic levels of aggression in the face of early adversity. Consequently, researchers have endeavored to identify vulnerability or protective factors that moderate the prospective effect of stress on aggression (Rutter, 1985).
In recent years, genes regulating serotonin neurotransmission have been proposed as key risk factors, in concert with stressful life conditions, for various forms of stress-linked psychopathology. The most extensively studied of these candidate genes is a variable number tandem repeat polymorphism in the promoter region of the serotonin transporter gene (5-HTTLPR) that influences transcription rates of the serotonin transporter protein (5-HTT). Specifically, the lower transcriptional efficiency of the short (s) allele results in reduced levels of 5-HTT expression, relative to the long (l) allele (Lesch et al., 1996). An initial study by Caspi et al. (2003) found that carriers of the s allele were at higher risk for depressive disorders, but only under conditions of high stress. A sizeable body of research has since supported the role of 5-HTTLPR in depressive responses to stress (Karg, Burmeister, Shedden, & Sen, 2011; but see also Risch et al., 2009). There is also evidence that the s allele amplifies the influence of environmental adversity on the development of anxiety disorders (Koenen, Amstadter, & Nugent, 2009; Nugent, Tyrka, Carpenter, & Price, 2011; Stein, Schork, & Gelernter, 2008) and suicidal behavior (Cicchetti, Rogosch, Sturge-Apple, & Toth, 2010; Gibb, McGeary, Beevers, & Miller, 2006).
Although less is known regarding 5-HTTLPR G × E in externalizing problems, there is emerging evidence that interactions between stressful conditions and 5-HTTLPR play a role in the etiology of externalizing pathology, including substance use and impulsivity (e.g., Carver, Johnson, Joormann, Kim, & Nam, 2011; Gunthert et al., 2007). However, only one observational study to date has documented 5-HTTLPR G × E in aggression outcomes. Reif and colleagues (2007) observed higher rates of violent crime among adult males carrying at least one 5-HTTLPR s allele, but only among those exposed to elevated levels of childhood adversity. While this study is suggestive of G × E, childhood adversity was retrospectively reported and psychometric properties for the assessment instrument were not reported, limiting the strength of interpretations that could be made from the findings. Replication studies using psychometrically sound stress assessment instruments are clearly needed to evaluate the robustness of 5-HTTLPR G × E in aggression.
Complementing the findings of Reif et al. (2007), preliminary evidence indicates that 5-HTTLPR may be a predictor of animal and laboratory analogues of aggressive reactions to stressors. A recent study in rhesus macaques (Schwandt et al., 2010) suggested that s allele carriers are predisposed to aggressive behavior following an acute social stressor. Males possessing at least one s allele were more likely to react to the introduction of an unfamiliar macaque with aggression than were males homozygous for the l allele. Similar findings emerged from a study in humans that examined the effect of 5-HTTLPR genotype on administration of electric shock to a fictional confederate following an experimental stressor (Verona, Joiner, Johnson, & Bender, 2006). Male s homozygotes displayed increased aggression, but only under conditions of physical stress (intermittent air blasts). These results suggest a possible interaction between acute stress and 5-HTTLPR genotype in the expression of aggressive behavior.
A growing body of research suggests that 5-HTTLPR may confer risk for aggressive behavior by controlling the intensity of emotion dysregulation precipitated by exposure to stressful conditions. Research in the field of imaging genomics has provided data connecting 5-HTTLPR with activation in neural circuitry mediating affective responses to emotional and stressful cues. Initial studies found that carriers of the s allele displayed exaggerated amygdala excitability following presentation of fearful and angry faces (Hariri et al., 2005). Extensions of this work demonstrated an association of the s allele with attenuated top-down control of the amygdala by regions of the prefrontal cortex (PFC) (Pezawas et al., 2005). Diminished regulatory control of amygdala activity is in turn thought to potentiate emotional and physiological responsivity to stress (Hariri & Holmes, 2006). Therefore, the s allele is theorized to compromise the integrity of neural pathways that provide the executive control necessary to override aggressive tendencies following environmental stress. This prediction is further supported by evidence that the s allele is associated with hypothalamic-pituitary-adrenal axis dysregulation in the presence of acute stressors (Gotlib, Joormann, Minor, & Hallmayer, 2008; Way & Taylor, 2010), slower disengagement of attention from presentations of emotional facial expressions (Beevers, Wells, Ellis, & McGeary, 2009), and exaggerated appraisals of the threat associated with naturally occurring stressful life events (Conway et al., 2011).
Researchers have begun to take interest in the diversity of behavioral manifestations of emotion dysregulation among s allele carriers following exposure to stressful circumstances. Some theorists have proposed that the combination of stress and 5-HTTLPR vulnerability can eventuate in either depressive or aggressive responses, depending on preexisting social and psychological characteristics of the individual (Beauchaine, Klein, Crowell, Derbidge, & Gatzke-Kopp, 2009; Carver, Joormann, & Johnson, 2008). Empirical evidence shows that sex, in particular, may determine the form of emotional reactions to stress among those possessing the s allele. That is, female s allele carriers, as compared to males, appear to be especially susceptible to depressive disorders in the face of stress (Uher & McGuffin, 2008). For instance, a number of studies have reported that 5-HTTLPR G × E in depression is specific to females (Eley et al., 2004; Grabe et al., 2005; Sjöberg et al., 2006). On the other hand, evidence from experimental and naturalistic studies reviewed above suggests that 5-HTTLPR G × E in aggression may be more pronounced among males (e.g., Verona et al., 2006).
With the present study, we sought to make several contributions to the existing literature on G × E in aggression. First, we make use of psychometrically sound interviews to assess recent stress. The one previous study on 5-HTTLPR G × E in aggression was limited by retrospective reports of stress and study-specific assessment instruments. Second, we examine the consistency of G × E across sex, in light of recent theory and research suggesting that males may be especially likely to exhibit 5-HTTLPR G × E for aggression outcomes. A previous study using the present sample found evidence for 5-HTTLPR G × E in depression outcomes for females only (Hammen, Brennan, Keenan-Miller, Hazel, & Najman, 2010). It is possible that male s carriers are also sensitive to the pathogenic effects of stress, but react with more aggressive behavior, as opposed to internalizing distress (Beauchaine et al., 2009).
Third, in the present study we distinguish between acute and chronic forms of stress, which have frequently been confounded in previous 5-HTTLPR G × E research (Monroe & Reid, 2008). Moreover, virtually all studies on the stress-aggression association have examined the effects of chronic stress on aggression (Attar et al., 1994); as a result, little is known about the relationship between acute life stress and aggression outcomes. Interestingly, there have been three prior studies investigating 5-HTTLPR G × E in depression that have differentiated between chronic and acute stress and in each case G × E was stronger for chronic stress (Hammen et al., 2010; Kendler, Kuhn, Vittum, Prescott, & Riley, 2005; Sen et al., 2010). The effects of chronic and acute stress in G × E for aggression will be directly compared in the present study.
Fourth, we examine for the first time the role of recent stressors in 5-HTTLPR G × E research in aggression. The timing of stress exposure may influence the magnitude of 5-HTTLPR G × E, with one recent review highlighting that 5-HTTLPR G × E in depression may vary in strength across developmental periods (Uher & McGuffin, 2008). Consistent with this notion, a recent meta-analysis on 5-HTTLPR G × E in depression demonstrated that G × E results are most robust for childhood stressors, whereas findings were more equivocal among studies assessing recent stressful life events in adulthood (Karg et al., 2011). It remains to be seen whether the timing of stress exposure affects the strength or pattern of 5-HTTLPR G × E in aggression.
The present study examines G × E in a community sample of youth followed from adolescence to young adulthood. Previously validated interview measures are used to assess chronic and acute stress exposure in the past 12 months. Multiple informants and multiple methods are used to measure aggression in both adolescence and young adulthood. In line with prior correlational and experimental evidence, we anticipate a positive association between stress exposure and aggression at the transition to adulthood that will hold even after controlling for aggression levels in mid-adolescence. Further, we hypothesize that the magnitude of this association will be augmented among s allele carriers, relative to l homozygotes. Additionally, the G × E effect is predicted to be stronger for chronic, as compared to acute, stressors. Finally, based on relatively consistent patterns of sex differences in previous 5-HTTLPR G × E research, we expect the strength of G × E to be greater among males.
Method
Participants
A sample of 815 families was drawn from a larger birth cohort study of child health and development between 1981 and 1984 at Mater Misericordiae Mother's Hospital in Brisbane, Queensland, Australia (Keeping et al., 1989). This sample was selected at youth age 15 with the intention of including a range of maternal depression histories, oversampling mothers with a history of depressive symptoms. The sample studied at age 15 was 93% Caucasian and 7% minority (Asian, Pacific Islander, and Aboriginal), and median family income fell in the lower-middle class1. Complete details of the sampling procedures have been published elsewhere (Hammen, Shih, Altman, & Brennan, 2003).
Of the 815 families that participated at age 15, 705 families participated in follow-up assessments at youth age 20. Individuals included in the sample at age 20 did not differ from those not included on age 15 levels of youth-reported aggression, t(701) = 0.04, p = .97, Cohen's d = .01, or maternal-reported aggression, t(692) = 1.15, p = .25, d = .09. Youth were re-contacted between ages 22-25 to request their participation in the genotyping phase of this study. Of the 705 youth participating at age 20, 512 subsequently had blood drawn and transported to the Queensland Institute for Medical Research for genetic analyses. Unavailable participants had either withdrawn from follow-ups, moved, could not be scheduled, had major medical problems, or were deceased. Those participating in the genotyping did not differ from age 20 participants who did not provide DNA in terms of age 20 self-reported aggression, t(701) = 0.56, p = .58, d = .04, youth depression history by age 20, χ2(1, 705) = 0.23, p = .63, φ = .02, or maternal history of depression by age 15, χ2(1, 705) = 0.01, p = .92, φ = .01, but were less likely to be male, χ2(1, 705) = 17.80, p < .01, φ = .16.
The sample for the present analyses consists of 384 youth genotyped for 5-HTTLPR. Of the 512 youth with DNA samples, 384 were randomly selected for 5-HTTLPR genotyping because resources were available for only one 384-well genotyping plating. Due to 3 invalid readings our final sample included 149 males and 232 females. Compared to the 131 youth whose DNA samples were unanalyzed, the 381 with valid readings at 5-HTTLPR did not differ with respect to maternal depression status, χ2(1, 512) = 0.04, p = .51, φ = .01, although males were less likely to have their sample analyzed than females, χ2(1, 512) = 16.49, p < .01, φ = .15. Individuals included in the genotyping analyses did not differ from those not included on age 15 levels of youth-reported aggression, t(701) = 1.63, p = .10, d = .12, or maternal-reported aggression, t(692) = 1.09, p = .28, d = .08.
Procedure
Interviews at age 15 were conducted in the homes of the families by teams of two graduate-level interviewers who were blind to the mother's psychiatric history. Parent and child interviews were conducted separately and final ratings were determined using best-estimate diagnostic procedures. In addition, the mothers and children completed a battery of questionnaires and questionnaire packets were mailed to children's homeroom teachers. In the age 20 follow-up, mothers and children completed similar interviews and questionnaire batteries. Genotyping procedures were conducted at the Genetic Epidemiology Laboratory of the Queensland Institute of Medical Research. Mothers provided written informed consent for themselves and their child at age 15 and the child gave verbal assent. All participants provided written informed consent at the age 20 and genetic follow-ups. All participants were compensated for their time. All procedures were approved by the UCLA Institutional Review Board, Emory University Investigations Committee, and the University of Queensland Ethics Review Committee.
Measures
Youth aggression at age 15
Youth aggression at age 15 was measured using the corresponding scales of the Child Behavior Checklist (CBCL), Youth Self-Report (YSR), and Teacher Report Form (TRF) (Achenbach, 1991). Respondents indicate how true various symptom descriptors are of the target youth, ranging from 0 (not at all true) to 2 (very true or often true). Coefficient alpha was .91 for the maternal-report CBCL, .86 for the YSR, and .91 for the TRF. According to maternal report at age 15, 35 youth (9.18%) displayed clinically significant levels of aggression (i.e., T score greater than 70), with another 38 (9.97%) in the borderline (i.e., T score between 67 and 70) range (Achenbach, 1991).
Youth aggression at age 20
Three questionnaires measured aggression at age 20. Youth completed the Young Adult Self-Report (YASR) and the mothers completed the Young Adult Behavior Checklist (YABC) (Achenbach, 1997). Coefficient alphas for the 12-item YASR and the 17- item YABC Aggression scales were .83 and .91, respectively. Maternal and youth reports on these scales were correlated at .32 (p < .01). Based on clinical cutoffs suggested by Achenbach (1997), 35 youth (9.21%) were in the clinical range by either mother or youth report, and an additional 43 youth (11.31%) were in a borderline clinical range. At age 20 the target youth also completed the 34-item Buss Aggression Questionnaire (Buss & Warren, 2000), which assesses various aggressive behaviors on a 5-point scale ranging from “not at all like me” to “completely like me.” In the current study, coefficient alpha for the total score was .93.
Acute life stress at age 20
Acute stressors were assessed at age 20 using a version of the semistructured UCLA Life Stress Interview (LSI) adapted for use with adolescents and young adults (Hammen, Henry, & Daley, 2000). This interview is based on the contextual threat assessment methods of Brown and Harris (1978), with the goal of finding out what changes have occurred in various aspects of the person's life. Interviewers used a standardized set of probes to obtain the onset date, context, duration, expectations, consequences, and coping resources for interviewer-identified negative life events occurring in the past 12 months. Interviewers then presented a narrative summary of each event based on their interview to a team of independent raters blind to youth psychiatric status and youth reaction to the event. The team scored the objective threat (defined as the impact of the event on the typical person in similar life circumstances) of each event on a 5-point scale ranging from 1 (no negative impact) to 5 (extremely severe negative impact). An objective threat total was created by summing the threat ratings across all reported acute events.
Chronic life stress at age 20
The UCLA LSI queries individuals about how things are going in their lives in different content areas. The goal is to obtain information about level of chronic stress as defined by ongoing, typical conditions in eight domains during the past 12 months: general social life, close friendships, romantic relationships/dating interests, relationships with family members, academic performance, occupational experiences, personal health, and health of close family members. In contrast to acute stressors that typically have a specific onset date, chronic stress refers to typical, continuing circumstances in each major domain of functioning. To minimize the overlap between chronic and acute stress, multiple occurrences of an event or multiple elements of the same enduring situation were considered in the chronic stress ratings and were not included in the acute stress measure. The interviewer probed each domain and then rated each on the level of chronic stress on a 5-point scale, with behaviorally specific anchor points in each domain (with 1 indicating exceptionally good conditions and 5 representing extreme adversity). Previous reports have provided concurrent and predictive validity data for the chronic stress scales (e.g., Hammen et al., 2000). In the present analyses, chronic stress was represented by the sum of ratings across all eight domains.
Depression diagnoses
Maternal diagnoses of major depression prior to offspring age 15, used as a covariate in all analyses, were assessed using the Structured Clinical Interview for DSM-IV (SCID; First, Spitzer, Gibbon, & Williams, 1995) administered at youth age 15. The weighted kappa, computed using blind clinician ratings of 52 taped interviews, for maternal history of depression was .84.
Youth history of major depression was also assessed by the SCID at the age 20 follow-up. Lifetime diagnoses were present among 111 (29.1%) of youth. Based on ratings made by independent judges on recordings of 10% of the interviews, kappa values of .83 and .89 were found for current depressive diagnoses and past five year depressive diagnoses, respectively.
Genotyping
The 43 bp deletion polymorphism was genotyped by agarose gel analysis of PCR products spanning the central portion of the repeats in the 5HTTLPR. PCR was conducted according to methods outlined in Wray et al. (2009). Most samples were subject to triplicate gel analysis. A minimum of two independent results in agreement was required for inclusion which gave a final call rate of 96.4%. To estimate accuracy duplicate samples were genotyped for 764 individuals in a different study using these procedures, with discordance rates of 0.45%. In the present sample the genotype frequencies were ll = 122, sl = 178, and ss = 81, and these frequencies were in Hardy-Weinberg equilibrium. In light of evidence indicating that the minor allele at rs25531 causes the 5-HTTLPR l allele to resemble the s allele with respect to gene transcription (Hu et al., 2004), rs25531 was genotyped (see protocol of Wray et al., 2009) and 31 lG alleles were reclassified as s (ll = 101, sl = 189, ss = 91). The pattern and significance of all G × E results reported below were equivalent in analyses in which rs25531 variation was not taken into account.
Data Analytic Plan
Data were analyzed using structural equation modeling to capitalize on the multiple measures and informants used to assess aggression in the present study. In an initial model, the direct effects of chronic and acute stress and 5-HTTLPR on aggression were examined without testing for interaction effects. Genotype was coded according to the number of s alleles possessed by the individual (i.e., 0, 1, or 2). To ensure that stress at age 20 was predicting changes in aggression levels, as opposed to trait aggression, youth aggression at age 15 was covaried in all analyses. As such, stress effects at age 20 were statistically independent of prior youth aggression levels. Because 5-HTTLPR G × E is linked with liability to depression in the current sample (Hammen et al., 2010), youth lifetime history of depression up to age 20 was modeled as an additional covariate to ensure that covariation between depression and aggression did not produce an artifactual G × E effect for aggression. All exogenous variables were allowed to intercorrelate.
To test G × E hypotheses, a second model was estimated that included product terms representing the interactions between (a) chronic stress and 5-HTTLPR and (b) acute stress and 5-HTTLPR. These variables were entered in the model simultaneously in order to remove any variance shared between the two interaction terms. Finally, in a third model, product terms representing three-way interactions between sex, stress, and 5-HTTLPR were added to test the consistency of 5-HTTLPR G × E across sex. Multiple group structural equation modeling analyses, which estimate the path from stress to aggression for each genotype group separately, were carried out to examine simple effects in the event of a significant interaction (Muthén & Muthén, 1998-2007).
Although prior neural, cognitive, and behavioral research has supported the functional equivalence of the ss and sl genotypes with regard to stress reactivity phenotypes (e.g., Beevers et al., 2009; Hariri & Holmes, 2006; Reif et al., 2007), the role of heterozygosity in 5-HTTLPR G × E in aggression remains unclear. As such, simple effects analyses examined each genotype group separately, instead of combining all s allele carriers. Wald tests were used to directly compare the strength of the stress-aggression association across genotype groups (Chou & Bentler, 2002). Wald tests evaluate whether model fit deteriorates when the path from stress to aggression is constrained to be equal for two distinct genotype groups (e.g., ss versus ll). Significant Wald values would suggest that the stress-aggression association is significantly different across these two groups.
All models were estimated using Mplus (Muthén & Muthén, 1998-2007), which employs full information maximum likelihood (FIML) procedures for missing data. FIML estimates yield unbiased parameter estimates and standard errors when data are missing at random (MAR). These estimates are generally considered superior to those obtained using listwise deletion and other imputation strategies even when the MAR assumption is not fully satisfied (Schafer & Graham, 2002). Robust maximum likelihood estimation was used to calculate parameter standard errors to account for univariate and multivariate non-normality in the data.
Results
Stratified by genotype group, descriptive statistics for all study variables are presented in Table 1. Table 2 presents correlations among all variables. Analyses of variance revealed no significant differences in exposure to chronic, F(2, 378) = 0.88, p = .58, η2 = .005, or acute, F(2, 378) = 0.26, p = .77, η2 = .001, stress by genotype. This indicated that gene-environment correlations were unlikely to confound analysis and interpretation of G × E effects.
Table 1. Descriptive Statistics for Main Study Variables.
| 5-HTTLPR Genotype | ||||
|---|---|---|---|---|
| Variable | LL | SL | SS | Total |
| N | 101 | 189 | 91 | 381 |
| No. (%) maternal depression | 40 (39%) | 88 (47%) | 36 (22%) | 164 (43%) |
| Mean (SD) age 15 TRF | 3.35 (6.59) | 2.47 (5.34) | 1.70 (3.38) | 2.58 (5.45) |
| Mean (SD) age 15 CBCL | 5.63 (5.76) | 6.12 (6.20) | 4.90 (4.62) | 5.69 (5.76) |
| Mean (SD) age 15 YSR | 7.70 (5.17) | 7.61 (5.19) | 6.56 (4.61) | 7.41 (5.07) |
| Mean (SD) age 20 YABC | 4.65 (5.11) | 4.76 (5.28) | 4.47 (4.42) | 4.67 (5.04) |
| Mean (SD) age 20 YASR | 3.43 (3.09) | 3.60 (3.90) | 3.91 (3.16) | 3.55 (3.49) |
| Mean (SD) age 20 Buss | 62.29 (17.77) | 62.49 (19.72) | 61.42 (16.28) | 62.10 (18.35) |
| No. (%) youth lifetime MDD | 40 (30%) | 63 (37%) | 23 (29%) | 126 (33%) |
| Mean (SD) chronic stress | 13.62 (2.03) | 13.79 (2.11) | 13.39 (2.15) | 13.85 (2.23) |
| Mean (SD) acute stress | 2.30 (0.48) | 2.26 (0.46) | 2.19 (0.40) | 2.25 (0.45) |
TRF, Teacher Report Form; CBCL, Child Behavior Checklist; YSR, Youth Self-Report; YABC, Young Adult Behavior Checklist; YASR, Young Adult Self-Report; Buss, Buss Aggression Scale; MDD, major depressive disorder.
Table 2. Bivariate Correlations for All Variablesa.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. Chronic Stress | — | ||||||||||
| 2. Acute Stress | .33 | — | |||||||||
| 3. Age 15 TRF | .18 | .01 | — | ||||||||
| 4. Age 15 CBCL | .30 | .10 | .41 | — | |||||||
| 5. Age 15 YSR | .31 | .15 | .23 | .46 | — | ||||||
| 6. Age 20 YASR | .41 | .20 | .24 | .42 | .51 | — | |||||
| 7. Age 20 YABC | .40 | .15 | .25 | .58 | .43 | .54 | — | ||||
| 8. Age 20 Buss | .42 | .20 | .22 | .41 | .50 | .79 | .52 | — | |||
| 9. Maternal Depression | .17 | .09 | .16 | .18 | .06 | .15 | .22 | .15 | — | ||
| 10. Youth Depression | .30 | .28 | .03 | .15 | .23 | .32 | .22 | .31 | .16 | — | |
| 11. Sex | −.07 | −.07 | .07 | .10 | −.02 | −.04 | −.02 | .08 | .01 | −.20 | — |
All rs > ∣.10∣ are significant at an alpha level of .05; all rs > ∣.17∣ are significant at alpha = .01
Main Effects of Acute Stress, Chronic Stress, and 5-HTTLPR
A schematic of the baseline (main effects) model is displayed in Figure 1. This model provided an adequate fit to the data as indicated by a comparative fit index of 0.95 (CFI; Hu & Bentler, 1999), a root mean square error of approximation of 0.08 with a 90% confidence interval of .05-.10 (RMSEA; Browne & Cudeck, 1993), and a standardized root mean square residual of .05 (Muthén & Muthén, 1998-2007). As expected, aggression levels at age 15 were strongly correlated with aggression levels at age 20, b = 0.51, SE = 0.15, p < .001, β = 0.58. Even after controlling for this continuity in aggression, chronic stress from the past 12 months exerted a significant unique effect on aggression at age 20, b = 0.14, SE = 0.07, p < .05, β = 0.20. In contrast, acute stress was not significantly associated with individual differences in aggression, b = 0.04, SE = 0.04, p = .32, β = 0.06. Additionally, neither sex, b = −0.13, SE = 0.41, p = .76, β = −0.02, nor history of depression, b = 0.67, SE = 0.52, p = . 19, β = 0.09, had significant effects on age 20 aggression. Finally, although mean levels of aggression were somewhat higher among those with one or two s alleles at age 20, there was no significant direct effect of 5-HTTLPR on aggression, b = 0.21, SE = 0.22, p = .30, β = 0.06.
Figure 1.
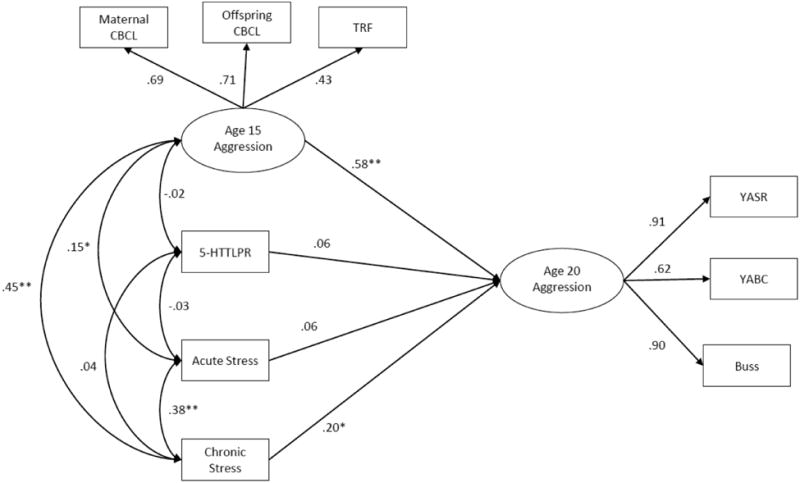
Schematic of the structural equation model testing the main effects of stress and 5-HTTLPR on aggression at age 20. For ease of presentation, model predictors with nonsignificant effects on the age 20 latent aggression variable are omitted. Standardized path coefficients are presented. All factor loadings are significant at the .01 level. * p < .05, ** p < .01.
Gene-Environment Interaction
When the two G × E terms were added to the baseline model, the interaction between chronic stress and 5-HTTLPR was found to be significant, b = 0.15, SE = 0.05, p < .01, β = 0.21, whereas the interaction between acute stress and 5-HTTLPR was not, b = 0.07, SE = 0.05, p = . 13, β = 0.06. Simple effects analyses indicated that among l homozygotes the structural path linking chronic stress and age 20 aggression did not significantly differ from 0, b = 0.07, SE = 0.07, p = .29, β = 0.10. On the other hand, both the sl, b = 0.37, SE = 0.09, p < .001, β = 0.27, and ss, b = 0.39, SE = 0.11, p < .001, β = 0.29, groups displayed a significant positive relation between chronic stress and aggression. Wald tests indicated that the regression of aggression on chronic stress did not significantly differ for the ss and sl groups, Δχ2(1, N = 381) = 0.04, p = .83. In contrast, the magnitude of this association in the ll group was significantly smaller than that of the sl, Δχ2(1, N = 381) = 7.44, p < .01, and ss, Δχ2(1, N = 381) = 6.09, p < .05, groups2.
Analyses revealed that G × E was not moderated by sex for either chronic, b = −0.01, SE = 0.01, p = .51, β = −0.04, or acute, b = 0.04, SE = 0.04, p =.30, β = 0.06, stress3. The form of the interaction between chronic stress and 5-HTTLPR in predicting aggression levels is presented in Figure 2 for both sexes, revealing nearly identical patterns of interaction for males and females.
Figure 2.
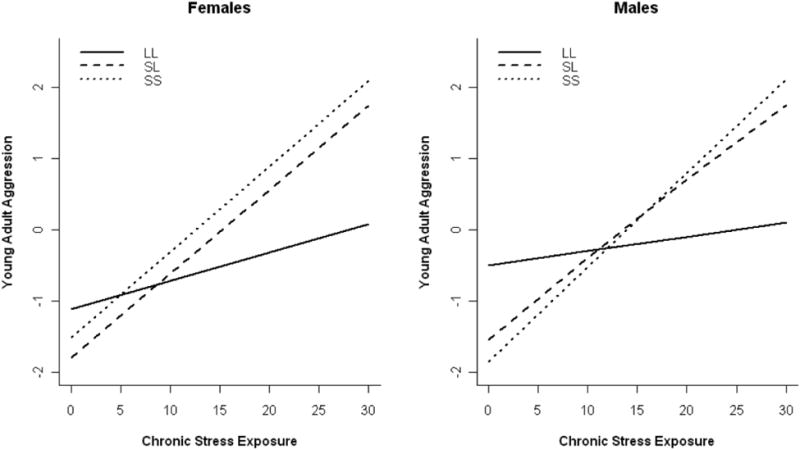
Association between age 20 aggression levels and chronic stress in the past 12 months as a function of 5-HTTLPR genotype. On the y-axis, standing on the latent aggression variable is presented in standardized units.
Discussion
Our aim in conducting the present study was to advance our understanding of how life stress and 5-HTTLPR genotype contribute jointly to the development of aggression. Prior developmental psychopathology work has suggested that stress exposure portends risk for aggression, particularly for genetically vulnerable youth (Caspi et al., 2002). Yet, nearly all existing research has concentrated on G × E among children; it is unclear to what extent stressors encountered in adolescence and adulthood predispose to aggression both directly and in conjunction with 5-HTTLPR (Moffitt, 2005).
Regarding main effects, the present findings suggest that chronic stressors occurring in the past 12 months have a direct, positive association with aggression levels in young adulthood. Supporting the robustness of the chronic stress-aggression link, a significant main effect was observed for chronic stress even after statistically controlling for youths' propensity to aggression in adolescence. This indicates that past year chronic stressful conditions predicted changes in, and not simply trait levels of, aggression. In contrast, acute stressful life events in the past 12 months did not significantly increase aggression levels after adjusting for covariation between acute and chronic stress. As expected, 5-HTTLPR did not have a significant main effect on the latent aggression variable at age 20.
Generally consistent with one prior 5-HTTLPR G × E study in aggression (Reif et al., 2007), the present findings suggest that 5-HTTLPR genotype may confer susceptibility to aggressive reactions to proximal stressors. Interestingly, G × E was observed for chronic, but not acute, stress. Specifically, increasing chronic stress had no influence on aggression levels for l homozygotes, whereas the chronic stress-aggression association was significant and positive among s carriers. This distinction between chronic and acute stress with regard to interaction with 5-HTTLPR has been established previously in the G × E literature. To date, three studies have compared the effects of chronic and acute stress in potentiating 5-HTTLPR vulnerability to depression and in each case G × E was stronger for chronic stress (Hammen et al., 2010; Kendler et al., 2005; Sen et al., 2010). Moreover, a recent meta-analysis documented that the interaction of acute stressful life events and 5-HTTLPR only inconsistently predicted risk for depression, whereas interaction effects were more robust for childhood adversity and chronic medical conditions (Karg et al., 2011). Results from the present study suggest that a similar pattern may characterize 5-HTTLPR G × E in aggression.
The present findings are partially consistent with recent work in animal and experimental models of 5-HTTLPR G × E demonstrating greater increments in aggressive behavior following physical and social stressors among s allele carriers (Schwandt et al., 2010; Verona et al., 2006). It is important to note, however, that these previous were designed specifically to assess the G × E effects of acute stress. Our results suggest that, in naturalistic settings, a more robust G × E effect is likely to be observed for chronic, as compared to acute, stress. Further longitudinal research is clearly needed to clarify the roles of different types of stress in models of 5-HTTLPR G × E in aggression.
It is also noteworthy that prior studies in this area were either conducted among entirely male samples or found that G × E was stronger for males. These data, in combination with a number of studies—including one conducted with the present sample (Hammen et al., 2010)—finding female-specific 5-HTTLPR G × E in the development of depression (see Uher & McGuffin, 2008), have led some theorists to suggest that female s allele carriers react to increasing stress with internalizing distress, whereas male s allele carriers respond with externalizing behavior (Beauchaine et al., 2009; Carver et al., 2011). Contrary to this hypothesis, our results showed that the strength and pattern of 5-HTTLPR G × E were nearly identical across sex. We speculate that this discrepancy may be due in large part to the operationalization of aggression in the present study. The latent aggression variable at age 20 reflected shared variation among a set of measures that assessed affective and cognitive components of aggression in addition to aggressive behavior (Achenbach, 1997; Buss & Perry, 1992). In contrast, prior studies showing male-specific G × E specifically assessed physical aggression outcomes, which are consistently higher among males (Martin, Watson, & Wan, 2000).
Deficits in emotion regulation capabilities associated with the 5-HTTLPR s allele represent a possible mechanism for the observed 5-HTTLPR G × E effect. Research in the field of imaging genomics has linked the s allele to attenuated prefrontal cortex control over amygdala reactivity following presentations of stressful stimuli (Pezawas et al., 2005). Diminished inhibition of amygdala excitability is in turn associated with weaker executive control over prepotent aggressive tendencies arising in response to provocation and stress (Siever, 2008). Thus, the influence of 5-HTTLPR on neural pathways mediating executive control or affect regulation may predispose s allele carriers to more intense and persistent affective, cognitive, and behavioral manifestations of aggression in the face of stress (Carver et al., 2008).
Several limitations of the present study may be useful in guiding future research. First, the sample size for the present analyses was modest in comparison to molecular genetic studies in epidemiological samples. Given the potential for false positive findings in molecular G × E analyses, some caution is warranted in the interpretation of these results until replication studies in large samples are available. Second, it was impossible to establish that stress exposure always preceded changes in aggression levels in young adulthood. However, as mentioned previously, controlling for the influence of aggression in mid-adolescence allowed a test of stress exposure that was independent of trait levels of aggression. Third, the present sample was predominantly Caucasian. Future research is needed to assess the generalizability of the main effects of chronic and acute stress, as well as their interactions with 5-HTTLPR, on aggression among more ethnically diverse samples. Fourth, although 5-HTTLPR genotype was not associated with exposure to chronic or acute stress in the present sample, it is highly unlikely that stress occurrence is entirely independent of genetic contributions. Thus, it is possible that some portion of the observed G × E effect may be due to unmeasured gene-gene interactions. One further limitation is that mediators of G × E were not assessed in the present study, and it is unclear to what extent the mechanisms of 5-HTTLPR G × E in aggression differ from those operative for G × E in internalizing problems. Although our analyses controlled for lifetime history of depressive disorders, indicating that G × E effects were not artifacts of comorbidity between aggression and depression, this does not imply that mechanisms of G × E for internalizing and externalizing problems do not overlap. Future work would do well to examine the specificity of mediators of 5-HTTLPR G × E across different forms of stress-linked psychopathology.
In spite of these limitations, this study offers novel findings on the link between stressful experiences and the development of aggression at the transition to adulthood. Results suggest that exposure to chronic stressful conditions in the past 12 months is associated with elevated aggression, and also highlight one potential protective factor—5-HTTLPR l allele homozygosity—that may underlie variability in aggressive responses to stress. Future research may benefit from assessing possible mechanisms of G × E and clarifying common versus unique pathways by which 5-HTTLPR affects risk for both aggression and depression in the face of stress.
Acknowledgments
The authors greatly appreciate the assistance of Robyne LeBrocque, Cheri Dalton Comber, and Sascha Hardwicke (project coordinators) and their interview staff. We also thank staff of the Genetic Epidemiological Laboratory of the Queensland Institute of Medical Research: Professor Nick Martin (Head) for cooperation and access, Michael James and Leanne Ryan for 5-HTTLPR and rs25531 genotyping, and Megan Campbell and Dixie Statham who coordinated genetic data collection and analysis. The 5-HTTLPR assays were carried out by Leanne Ryan and Troy Dumenil at Queensland Institute of Medical Research. Thanks also to the original MUSP principals, William Bor, MD, Michael O'Callaghan, MD, and Professor Gail Williams. This study was supported by NIMH R01 MH52239 to Brennan, Hammen, and Najman.
Footnotes
Financial Disclosures: The authors report no biomedical financial interests or potential conflicts of interest.
To confirm that results were not confounded by population stratification, analyses were conducted among Caucasians only. The significance and overall pattern of results were unaltered.
To clarify the nature of the significant interaction between chronic stress and 5-HTTLPR in predicting propensity to aggression, exploratory analyses examined G × E separately for each indicator of the aggression latent variable. Significant G × E was found for the Buss Aggression Questionnaire, b = 0.81, SE = 0.25, p < .01, and YASR, b = 0.16, SE = 0.04, p < .001, but not maternal report of aggression on the YABC, b = 0.04, SE = 0.08, p =.67. Regarding acute stress G × E models, interaction effects were nonsignificant for all three aggression indicators, zs < 1.70, ps > .10. Complete results are available from the authors upon request.
The significance of all main effect and interaction results was not changed when offspring depression history and/or aggression levels in adolescence were removed from the model. Similarly, results were unaltered when maternal depression history was included in the model as an additional covariate.
References
- Achenbach TM. Manual for the Child Behavior Checklist/ 4-18 and 1991 Profile. Burlington, VT: University of Vermont Department of Psychiatry; 1991. [Google Scholar]
- Achenbach TM. Manual for the Young Adult Self-Report and Young Adult Behavior Checklist. Burlington, VT: University of Vermont Department of Psychiatry; 1997. [Google Scholar]
- Attar BK, Guerra NG, Tolan PH. Neighborhood disadvantage, stressful life events, and adjustment in urban elementary-school children. Journal of Clinical Child Psychology. 1994;23:391–400. [Google Scholar]
- Barling J, Rosenbaum A. Work Stressors and wife abuse. Journal of Applied Psychology. 1986;71:346–348. [PubMed] [Google Scholar]
- Beauchaine TP, Klein DN, Crowell SE, Debridge C, Gatzke-Kopp L. Multifinality in the development of personality disorders: A biology × sex × environment interaction model of antisocial and borderline traits. Development and Psychopathology. 2009;21:735–770. doi: 10.1017/S0954579409000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beevers CG, Wells TT, Ellis AJ, McGeary JE. Association of the serotonin transporter gene promoter region (5-HTTLPR) polymorphism with biased attention for emotional stimuli. Journal of Abnormal Psychology. 2009;118:670–681. doi: 10.1037/a0016198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GW, Harris TO. Social Origins of Depression: A Study of Psychiatric Disorder in Women. New York: Free Press; 1978. [Google Scholar]
- Browne MW, Cudeck R. Alternative ways of assessing model fit. In: Bollen KA, Long JS, editors. Testing structural models. Newbury Park, CA: Sage; 1993. pp. 136–162. [Google Scholar]
- Buss AH, Warren WL. Aggression Questionnaire. Los Angeles, CA: Psychological Services; 2000. [Google Scholar]
- Carver CS, Johnson SL, Joorman J, Kim Y, Nam JY. Serotonin transporter polymorphism interacts with childhood adversity to predict aspects of impulsivity. Psychological Science. 2011 doi: 10.1177/0956797611404085. [DOI] [PubMed] [Google Scholar]
- Carver CS, Johnson SL, Joormann J. Serotonergic function, two-mode models of self-regulation, and vulnerability to depression: What depression has in common with impulsive aggression. Psychological Bulletin. 2008;134:912–943. doi: 10.1037/a0013740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi AC, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: The case of the serotonin transporter gene and its implications for studying complex diseases and traits. American Journal of Psychiatry. 2010;167:509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Sudgen K, Moffitt TE, Taylor A, Craig IW, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Chou CP, Bentler PM. Model modification in structural equation modeling by imposing constraints. Computational Statistics & Data Analysis. 2002;41(2):271–287. [Google Scholar]
- Cicchetti D, Rogosch FA, Lynch M, Holt K. Resilience in maltreated children: Processes leading to adaptive outcomes. Development and Psychopathology. 1993;9:799–817. [Google Scholar]
- Cicchetti D, Rogosch FA, Sturge-Apple M, Toth SL. Interaction of child maltreatment and 5-HTT polymorphisms: Suicidal ideation among children from low-SES backgrounds. Journal of Pediatric Psychology. 2010;35:536–546. doi: 10.1093/jpepsy/jsp078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conger RD, Ge X, Elder GH, Lorenz FO, Simons RL. Economic stress, coercive family process, and developmental problems of adolescents. Child Development. 1994;65:541–561. [PubMed] [Google Scholar]
- Conway CC, Hammen C, Espejo EP, Wray NR, Najman JM, Brennan PA. Appraisals of stressful life events as a genetically-linked mechanism in the stress-depression relationship. Cognitive Therapy and Research. 2011 doi: 10.1007/s10608-011-9368-9. [DOI] [Google Scholar]
- Craig IW, Halton KE. Genetics of human aggressive behavior. Human Genetics. 2009;126:101–113. doi: 10.1007/s00439-009-0695-9. [DOI] [PubMed] [Google Scholar]
- Dodge KA, Bates JE, Petit GS. Mechanisms in the cycle of violence. Science. 1990;250:1678–1683. doi: 10.1126/science.2270481. [DOI] [PubMed] [Google Scholar]
- Eley TC, Sugden K, Corsico A, Gregory AM, Sham P, McGuffin P, et al. Gene– environment interaction analysis of serotonin system markers with adolescent depression. Molecular Psychiatry. 2004;9:908–915. doi: 10.1038/sj.mp.4001546. [DOI] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW. Structured clinical Interview for DSM-IV Axis I disorders. Washington, D.C: American Psychiatric Press; 1995. [Google Scholar]
- Frankle WG, Lombardo I, New AS, Goodman M, Talbot PS, Huang Y, et al. Brain serotonin transporter distribution in subjects with impulsive aggressivity: A positron emission study with [11C]McN 5652. American Journal of Psychiatry. 2005;162:915–923. doi: 10.1176/appi.ajp.162.5.915. [DOI] [PubMed] [Google Scholar]
- Gibb BE, McGeary JE, Beevers CG, Miller IW. Serotonin transporter (5-HTTLPR) genotype, child abuse, and suicide attempts in adult psychiatric inpatients. Suicide and Life-Threatening Behavior. 2006;36:687–693. doi: 10.1521/suli.2006.36.6.687. [DOI] [PubMed] [Google Scholar]
- Gotlib IH, Joormann J, Minor KL, Hallmayer J. HPA axis reactivity: a mechanism underlying the associations among 5-HTTLPR, stress, and depression. Biological Psychiatry. 2008;63(9):847–851. doi: 10.1016/j.biopsych.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabe HJ, Lange M, Wolff B, Völzke H, Lucht M, Freyberger HJ, et al. Mental and physical distress is modulated by a polymorphism in the 5-HT transporter gene interacting with social stressors and chronic disease burden. Molecular Psychiatry. 2005;10:220–224. doi: 10.1038/sj.mp.4001555. [DOI] [PubMed] [Google Scholar]
- Gunthert KC, Conner TS, Armeli S, Tennen H, Covault J, Kranzler HR. Serotonin transporter gene polymorphism (5-HTTLPR) and anxiety reactivity in daily life: A daily process approach to gene-environment interaction. Psychosomatic Medicine. 2007;69(8):762–768. doi: 10.1097/PSY.0b013e318157ad42. [DOI] [PubMed] [Google Scholar]
- Hammen C, Brennan PA, Keenan-Miller D, Hazel NA, Najman JM. Chronic and acute stress, gender, and serotonin transporter gene-environment interactions predicting depression symptoms in youth. Journal of Child Psychology and Psychiatry. 2010;51:180–187. doi: 10.1111/j.1469-7610.2009.02177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammen C, Henry R, Daley S. Depression and sensitization to stressors among young women as a function of childhood adversity. Journal of Consulting and Clinical Psychology. 2000;68:782–787. [PubMed] [Google Scholar]
- Hammen CL, Shih JH, Altman T, Brennan PA. Interpersonal impairment and the prediction of depressive symptoms in adolescent children of depressed and nondepressed mothers. Journal of the American Academy of Child and Adolescent Psychiatry. 2003;42:571– 577. doi: 10.1097/01.CHI.0000046829.95464.E5. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Drabant EM, Munoz KE, Kolachana BS, Mattay VS, Egan MF, et al. A susceptibility gene for affective disorders and the response of the human amygdala. Archives of General Psychiatry. 2005;62:146–152. doi: 10.1001/archpsyc.62.2.146. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Holmes A. Genetics of emotional regulation: The role of the serotonin transporter in neural function. Trends in Cognitive Sciences. 2006;10:182–191. doi: 10.1016/j.tics.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Hu L, Bentler PM. Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives. Structural Equation Modeling. 1999;6:1–55. [Google Scholar]
- Hu X, Oroszi G, Chun J, Smith TL, Goldman D, Schuckit MA. An expanded evaluation of the relationship of four alleles to the level of response to alcohol and the alcoholism risk. Alcoholism Clinical and Experimental Research. 2005;29:8–16. doi: 10.1097/01.alc.0000150008.68473.62. [DOI] [PubMed] [Google Scholar]
- Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: Evidence of genetic moderation. Archives of General Psychiatry. 2011;68:444–454. doi: 10.1001/archgenpsychiatry.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeping JD, Najman JM, Morrison J, Western JS, Andersen MJ, Williams GM. A prospective longitudinal study of social, psychological, and obstetrical factors in pregnancy: Response rates and demographic characteristics of the 8,556 respondents. British Journal of Obstetrics and Gynaecology. 1989;96:289–297. doi: 10.1111/j.1471-0528.1989.tb02388.x. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Kuhn JW, Vittum J, Prescott CA, Riley B. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression. Archives of General Psychiatry. 2005;62:529–535. doi: 10.1001/archpsyc.62.5.529. [DOI] [PubMed] [Google Scholar]
- Kim-Cohen J, Caspi A, Tayor A, Williams B, Newcombe R, Craig IW, et al. MAOA, maltreatment, and gene-environment interaction predicting children's mental health: New evidence and a meta-analysis. Molecular Psychiatry. 2006;11:903–913. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- Koenen KC, Amstadter AB, Nugent NR. Gene-environment interaction in posttraumatic stress disorder: An update. Journal of Traumatic Stress. 2009;22:416–426. doi: 10.1002/jts.20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- Lyons-Ruth K, Holmes BM, Sasvari-Szekely M, Ronai Z, Nemoda Z, Pauls D. Serotonin transporter polymorphism and borderline or antisocial traits among low-income young adults. Psychiatric Genetics. 2007;17:339–343. doi: 10.1097/YPG.0b013e3281ac237e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R, Watson D, Wan CK. A three-factor model of trait anger: Dimensions of affect, behavior, and cognition. Journal of Personality. 2000;68:869–897. doi: 10.1111/1467-6494.00119. [DOI] [PubMed] [Google Scholar]
- Moffitt TE. The new look of behavior genetics in developmental psychopathology: Gene-Environment interplay in antisocial behaviors. Psychological Bulletin. 2005;131:533–554. doi: 10.1037/0033-2909.131.4.533. [DOI] [PubMed] [Google Scholar]
- Monroe SM, Reid MW. Gene–environment interactions in depression research. Genetic polymorphisms and life-stress polyprocedures. Psychological Science. 2008;19:947–956. doi: 10.1111/j.1467-9280.2008.02181.x. [DOI] [PubMed] [Google Scholar]
- Muthén LK, Muthén BO. Mplus User's Guide. 5th. Muthén & Muthén; Los Angeles: 1998-2007. [Google Scholar]
- Nugent NR, Tyrka AR, Carpenter LL, Price LH. Gene-environment interactions: Early life stress and risk for depressive and anxiety disorders. Psychopharmacology. 2011;214:175–196. doi: 10.1007/s00213-010-2151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS, et al. 5-HTTLPR polymorphism impacts human cingulateamygdala interactions: A genetic susceptibility mechanism for depression. Nature Neuroscience. 2005;8:828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- Reif A, Rosler M, Freitag CM, Schneider M, Eujen A, Kissling C, et al. Nature and nurture predispose to violent behavior: Serotonergic genes and adverse childhood environment. Neuropsychopharmacology. 2007;32:2375–2383. doi: 10.1038/sj.npp.1301359. [DOI] [PubMed] [Google Scholar]
- Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression. Journal of the American Medical Association. 2009;301:2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KD, Troop-Gordon W, Granger DA. Peer victimization and aggression: Moderation by individual differences in salivary cortisol and alpha-amylase. Journal of Abnormal Child Psychology. 2010;38:843–856. doi: 10.1007/s10802-010-9412-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter M. Resilience in the face of adversity: Protective factors and resistance to psychiatric disorder. British Journal of Psychiatry. 1985;147:598–661. doi: 10.1192/bjp.147.6.598. [DOI] [PubMed] [Google Scholar]
- Schafer JL, Graham JW. Missing data: Our view of the state of the art. Psychological Methods. 2002;7:147–177. [PubMed] [Google Scholar]
- Schwandt ML, Lindell SG, Sjoberg RL, Chisholm KL, Higley JD, Suomi SJ, et al. Gene-environment interactions and response to social intrusion in male and female rhesus macaques. Biological Psychiatry. 2010;67:323–330. doi: 10.1016/j.biopsych.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Burmeister M, Ghosh D. Meta-analysis of the association between a serotonin transporter promoter polymorphism (5-HTTLPR) and anxiety-related personality traits. American Journal of Medical Genetics Part B (Neuropsychiatric Genetics) 2004;127B:85–89. doi: 10.1002/ajmg.b.20158. [DOI] [PubMed] [Google Scholar]
- Sen S, Kranzler HR, Krystal JH, Speller H, Chan G, Gelernter J, Guille C. A prospective cohort study investigating factors associated with depression during medical internship. Archives of General Psychiatry. 2010;67:557–565. doi: 10.1001/archgenpsychiatry.2010.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siever LJ. Neurobiology of aggression and violence. American Journal of Psychiatry. 2008;165:429–442. doi: 10.1176/appi.ajp.2008.07111774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöberg RL, Nilsson KW, Nordquist N, O hrvik J, Leppert J, Lindström L, et al. Development of depression: Sex and the interaction between environment and a promoter polymorphism of the serotonin transporter gene. International Journal of Neuropsychopharmacology. 2006;9:443–449. doi: 10.1017/S1461145705005936. [DOI] [PubMed] [Google Scholar]
- Sontag LM, Graber JA. Coping With Perceived Peer Stress: Gender-Specific and Common Pathways to Symptoms of Psychopathology. Developmental Psychology. 2010;46:1605–1620. doi: 10.1037/a0020617. [DOI] [PubMed] [Google Scholar]
- Stein MB, Schork NJ, Gelernter J. Gene-by-environment (serotonin transporter and childhood maltreatment) interaction for anxiety sensitivity, an intermediate phenotype for anxiety disorders. Neuropsychopharmacology. 2008;33:312–319. doi: 10.1038/sj.npp.1301422. [DOI] [PubMed] [Google Scholar]
- Uher R, McGuffin P. The moderation by the serotonin transporter gene of environmental adversity in the etiology of mental illness: Review and methodological analysis. Molecular Psychiatry. 2008;13:131–146. doi: 10.1038/sj.mp.4002067. [DOI] [PubMed] [Google Scholar]
- Verona E, Joiner TE, Johnson F, Bender TW. Gender specific gene-environment interactions on laboratory-assessed aggression. Biological Psychology. 2006;71:33–41. doi: 10.1016/j.biopsycho.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Way BM, Taylor SE. The serotonin transporter promoter polymorphism is associated with cortisol response to psychosocial stress. Biological Psychiatry. 2010;67(5):487–492. doi: 10.1016/j.biopsych.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland JR, Lesch KP, Newman TK, Timme A, Gachot-Neveau H, Thierry B, et al. Differential functional variability of serotonin transporter and monoamine oxidase A genes in macaque species displaying contrasting levels of aggression-related behavior. Behavior Genetics. 2006;36:163–172. doi: 10.1007/s10519-005-9017-8. [DOI] [PubMed] [Google Scholar]
- Wray NR, James MR, Gordon SD, Dumenil T, Ryan L, Coventry WL. Accurate, large-scale genotyping of 5HTTLPR and flanking single nucleotide polymorphisms in an association study of depression, anxiety, and personality measures. Biological Psychiatry. 2009;66:468–476. doi: 10.1016/j.biopsych.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]