

Abstract
Objective
The most common pathogenesis for familial Alzheimer's disease (FAD) involves misprocessing (or alternative processing) of the amyloid precursor protein (APP) by γ-secretase due to mutations of the presenilin 1 (PS1) gene. This misprocessing/alternative processing leads to an increase in the ratio of the level of a minor γ-secretase reaction product (Aβ42) to that of the major reaction product (Aβ40). Although no PS1 mutations are present, altered Aβ42/40 ratios are also observed in sporadic Alzheimer's disease (SAD), and these altered ratios apparently reflect deposition of Aβ42 as amyloid.
Methods
Using immunoprecipitation-mass spectrometry with quantitative accuracy, we analyzed in the cerebrospinal fluid (CSF) of various clinical populations the peptide products generated by processing of not only APP but also an unrelated protein, alcadein (Alc). Alc undergoes metabolism by the identical APP α-secretases and γ-secretases, yielding a fragment that we have named p3-Alcα because of the parallel genesis of p3-Alcα peptides and the p3 fragment of APP. As with Aβ, both major and minor p3-Alcαs are generated. We studied the alternative processing of p3-Alcα in various clinical populations.
Results
We previously reported that changes in the Aβ42/40 ratio showed covariance in a linear relationship with the levels of p3-Alcα [minor/major] ratio in media conditioned by cells expressing FAD-linked PS1 mutants. Here we studied the speciation of p3-Alcα in the CSF from 3 groups of human subjects (n = 158): elderly nondemented control subjects; mild cognitive impairment (MCI) subjects with a clinical dementia rating (CDR) of 0.5; SAD subjects with CDR of 1.0; and other neurological disease (OND) control subjects. The CSF minor p3-Alcα variant, p3-Alcα38, was elevated (p < 0.05) in MCI subjects or SAD subjects, depending upon whether the data were pooled and analyzed as a single cohort or analyzed individually as 3 separate cohorts.
Interpretation
These results suggest that some SAD may involve alternative processing of multiple γ-secretase substrates, raising the possibility that the molecular pathogenesis of SAD might involve γ-secretase dysfunction.
The most common pathogenesis for familial Alzheimer's disease (FAD) involves misprocessing/alternative processing of the amyloid precursor protein (APP) by γ-secretase (for review, see Gandy1 and Small and Gandy2). This misprocessing/alternative processing leads to a relative increase in the ratio of the level of a minor γ-secretase reaction product, amyloid-β42 (Aβ42), to that of the major reaction product, amyloid-β40 (Aβ40; Borchelt and colleagues3). Until now, little has been known about γ-secretase function in sporadic AD (SAD).
We approached this issue by studying the metabolite peptides, p3-Alcα, that are derived from the processing by α-secretases and γ-secretases of the alcadeinα (Alcα), member of the alcadein (Alc) protein family. Alc proteins colocalize with APP in healthy mouse and SAD human brain,4 but they are entirely distinct from APP in their polypeptide sequence. In neurons, Alc proteins are complexed to APP via X11L adaptor molecules, raising the possibility that Alcs might be sorted and processed together with APP. Experimental evidence supports this reasoning. In the absence of X11L, both Alc and APP proteins are rapidly metabolized by proteolysis.5 Levels of the endogenous APP metabolite, Aβ, are elevated in the brains of X11L-deficient mice.6,7 Thus, taken together with similarities in their structure and cellular distribution, APP and Alc proteins would be predicted to undergo parallel metabolic fates (for APP and X11L, see Gandy1 and Suzuki and Nakaya8; for Alc, see Araki and colleagues,4,5,9).
Alc proteins exist in mammalian neurons as 4 isoforms4: Alcα1, Alcα2, Alcβ, and Alcγ. Alcα, Alcβ, and Alcγ are encoded by independent genes, while Alcα1 and Alcα2 are splice variants derived from the Alcα gene. All 3 members of the Alc family (Alcα, Alcβ, and Alcγ) are cleaved by ADAM 10 and ADAM 17, which have been identified as the α-secretases for APP.10–13 Subsequent cleavage of the remaining Alc C-terminal fragments involves predominantly the presenilin 1-(PS1)-dependent γ-secretase, and this reaction liberates a short peptide that we have designated p3-Alcα into cell-conditioned media and into cerebrospinal fluid (CSF). The amino acid sequences of the various alternatively cleaved p3-Alcα peptides in human CSF are shown with those of APP-p3 and Aβ (Fig 1). The current study is based on the hypothesis that examination of AD-related processing of p3-Alcα might reveal evidence for γ-secretase dysfunction in SAD. In so doing, we seek to confirm and extend the report of Yanagida and colleagues,14 who described similar alternative processing of another γ-secretase substrate, APLP. Taken together with the data from Yanagida and colleagues,14 we suggest that multiple γ-secretase substrates are subjected to altered processing in SAD and that this potentially implicates an “acquired” γ-secretase dysfunction that might contribute the pathogenesis of SAD.
Figure 1.
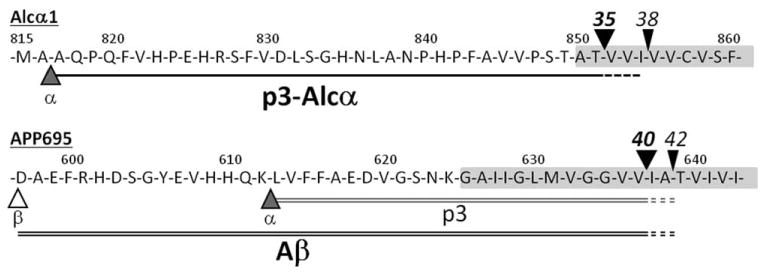
Amino acid sequences and cleavage sites of p3-Alcα and Aβ in human CSF. The amino acid sequences of p3-Alcα (black-underline) along with the sequences of p3 (gray double-underline) and Aβ40 (black double-underline) of APP. The major primary (black arrowheads) and secondary (black arrowheads) cleavage sites of Alcα1 are indicated together with those of APP (α, the cleavage site by α-secretase or ADAM 10/17; β, the cleavage site by β-secretase or BACE). Numbers on amino acids indicate their positions. The shaded area indicates the putative transmembrane region. Alc = alcadein; APP = amyloid β protein precursor; CSF = cerebrospinal fluid.
Materials and Methods
CSF Sample Collection
Standard protocols for CSF collection varied slightly according to site. Complete details of collections protocols are provided in the Supporting Information.
Aggregated characteristics and data for each cohort are shown in the Table. Detailed descriptions of all 158 subjects including clinical backgrounds from the 3 cohorts and the raw values for Aβ42/40 and p3-Alcα38/total p3-Alcα are shown in Supporting Information Table 1. The total p3-Alcα used for the calculation of p3-Alcα38/total p3-Alcα was the sum of all recognized p3-Alcα species; ie, p3-Alcα34, p3-Alcα35, p3Alcα36, p3-Alcα37, and p3-Alcα38. It is worth noting that for US cohort 1, but not for US cohort 2 or the Japanese cohort, CDR 0 subjects were verified as “true” controls by positron emission tomography (PET) scan analysis using [11C]Pittsburgh compound B (PiB).15
Table. Summary of Subject Information.
| CDR 0 | CDR 0.5 | CDR 1 | OND | |
|---|---|---|---|---|
| JP Cohort | ||||
| Age, yr (mean ± SD) | 79.4 ± 0.802 | 75.3 ± 2.79 | 76.9 ± 1.55 | |
| Gender (F%) | 80.0 | 55.6 | 45.5 | |
| MMSE (mean ± SD) | — | 24.7 ± 2.37 | 20.3 ± 3.57 | |
| HDS-R (mean ± SD) | — | 23.5 ± 1.95 | 17.6 ± 2.40 | |
| Duration of disease, yr (mean ± SD) | — | 0.78 ± 0.171 | 1.85 ± 0.700 | |
| US cohort 1 | ||||
| Age, yr (mean ± SD) | 68.8 ± 2.85 | 75.5 ± 1.26 | 76.2 ± 1.73 | 65.6 ± 3.18 |
| Gender (F%) | 50.0 | 55.0 | 53.8 | 18.8 |
| MMSE (mean ± SD) | 28.8 ± 0.244 | 26.4 ± 0.514 | 23.1 ± 1.03 | |
| Duration of disease, yr (mean ± SD) | 2.70 ± 0.334 | 4.25 ± 0.700 | ||
| US cohort 2 | ||||
| Age, yr (mean ± SD) | 71.2 ± 1.13 | 74.5 ± 2.57 | 70.4 ± 2.41 | 73.3 ± 1.18 |
| Gender (F%) | 53.3 | 30.8 | 45.5 | 23.1 |
| MMSE (mean ± SD) | 28.5 ± 0.325 | 27.0 ± 0.361 | 22.4 ± 0.875 |
Details of individual subjects are shown in the Supporting Information Table.
CDR = clinical dementia rating; F = female; HDS-R = Hasegawa's dementia scale; JP = Japanese; MMSE = Mini-Mental State Examination; OND = other neuronal and neurodegenerative diseases except for AD; SD = standard deviation; US = United States.
Matrix-Assisted Laser Desorption/Ionization–Time of Flight/Mass Spectrometry and Matrix-Assisted Laser Desorption/Ionization–Tandem Mass Spectrometry Analysis of p3-Alcα Secreted into Human CSF
In the initial pilot study (Fig 2), pooled CSF (300μl) from 5 individuals (70–90 years old) was subjected to immunoprecipitation with anti-Alcα UT135 and anti-Aβ 82E1 (Immuno-Biological Laboratories/IBL, Fujloka, Japan) antibodies. In the extended studies (Fig 3A–D), aliquots (300μl) of CSF from well-characterized individual subjects were derived from 2 U.S. cohorts (designated US cohort 1 and US cohort 2) and 1 Japanese cohort (see Table). Samples were subjected to immunoprecipitation individually with anti-Alcα UT135 antibody and Protein G Sepharose.10
Figure 2.
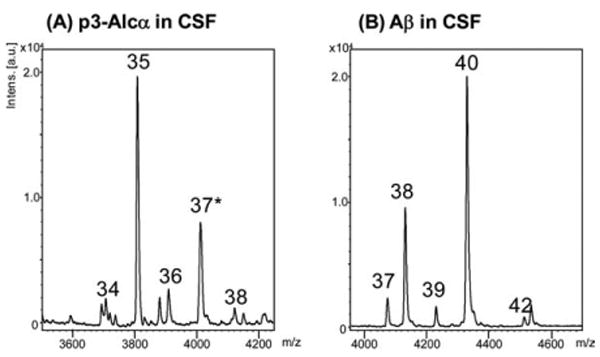
Representative MS spectra of p3-Alcα peptides and Aβ peptides in human CSF. (A) p3-Alcα in CSF and (B) Aβ in CSF. The CSF (300μl) were subjected to immunoprecipitation with (A) UT135 or (B) 82E1 antibodies, respectively. The precipitates were analyzed for molecular mass with MALDI-TOF/MS. (A) “34”, p3-Alcα34; “35”, p3-Alcα35; “36”, p3-Alcα36; “37*”, a mixture of p3-Alcα37 and p3-Alcα2N+35 (see Hata and colleagues10 for p3-Alcα2N+35); “38”, p3Alcα38. (B) “37”, Aβ37; “38”, Aβ38; “39”, Aβ39; “40”, Aβ40; “42”, Aβ42. Alc = alcadein; CSF = cerebrospinal fluid; MALDI-TOF/MS = matrix-assisted laser desorption ionization–time-of-flight/mass spectrometry.
Figure 3.
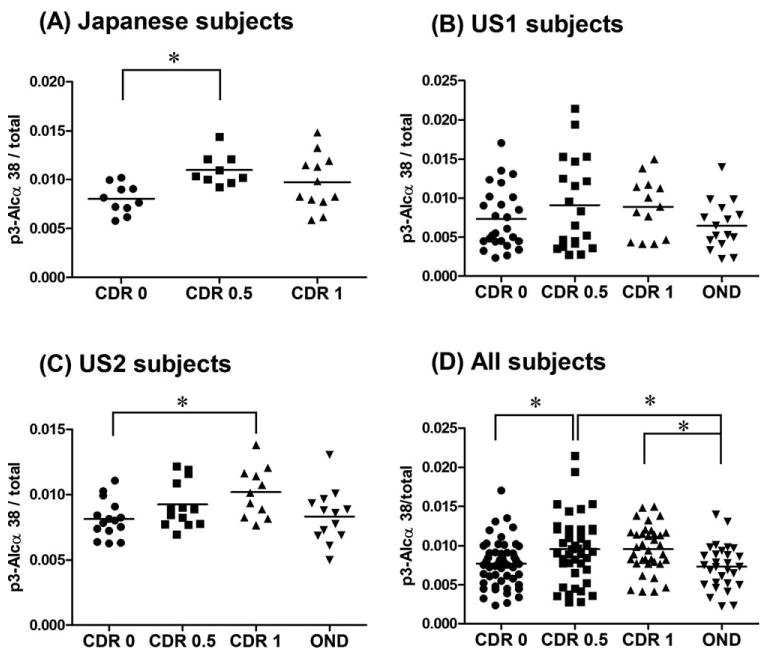
Comparison of the distribution of the ratio of p3-Alcα38/total p3-Alcα in CSF of elderly nondemented subjects, AD subjects and other neurological disease subjects according to cohort. (A) Japanese cohort; nondemented CDR 0 (n = 10), AD CDR 0.5 (n = 9), and AD CDR 1 (n = 12). (B) US1 cohort; nondemented CDR 0 (n = 26), AD CDR 0.5 (n = 20), AD CDR 1 (n = 13), and OND (n = 16). (C) US2 cohort; nondemented CDR 0 (n = 15), AD CDR 0.5 (n = 13), AD CDR 1 (n = 11), and OND (n = 13). (D) Combined subjects of 3 cohorts; nondemented CDR 0 (n = 51), AD CDR 0.5 (n = 42), AD CDR 1 (n = 36), and OND (n = 29). See the Table for raw data of the ratio and subject information. Statistical analysis was performed by a one-way analysis of variance followed by the Tukey Kramer's test (*p < 0.05). AD = Alzheimer's disease; Alc = alcadein; CDR = clinical dementia rating; CSF = cerebrospinal fluid; OND = other neuronal and neurodegenerative diseases except for AD.
After washing the beads, samples were eluted with trifluoroacetic acid/acetonitrile/water (1:20:20) saturated with sinapinic acid. The dissolved samples were dried on a target plate, and matrix-assisted laser desorption/ionization–time of flight/mass spectrometry (MALDI-TOF/MS) analysis was performed using an Ultraflex II TOF/TOF (Bruker Daltonics, Bremen, Germany). In all immunoprecipitation studies prior to MALDI-TOF/MS analysis, protease inhibitor mixture (5μg/ml chymostatin, 5μg/ml leupeptin, and 5μg/ml pepstatin) was added in samples to prevent nonspecific proteolysis. Molecular masses were calibrated using the peptide calibration standard (Bruker Daltonics). The quantitative accuracy of mass spectrometry analysis with immunoprecipitation was confirmed by studies with a mixture of synthetic p3-Alcα peptides as described previously.10 Aβ40 and Aβ42 levels were quantified with sELISA systems (Innotest; Innogenetics, Ghent, Belgium and IBL, Fujioka, Japan). All analyses were performed with operators blinded to diagnosis until data tables were generated. Diagnoses and data tables were exchanged at the time of unblinding.
Results
The recent production of an anti-Alcα antibody raised against the Alcα extracellular juxtamembrane sequences enabled us to recover p3-Alcα secreted into conditioned media and CSF.10 The amino acid sequences of the major and minor p3-Alcα were determined by matrix-assisted laser desorption/ionization–tandem mass spectrometry (MALDI-MS/MS) analysis and shown in Figure 1. The p3-Alcα species with Thr851 as the C-terminal residue is the major species in human CSF (p3-Alcα35), as described in detail elsewhere.10
Secondary cleavage sites, determined by MALDI-MS/MS analysis,10 are also shown in Figure 1 (black arrowheads), together with the major secondary γ-secretase-dependent cleavage sites of APP that generate Aβ40 and Aβ42. We have shown that HEK293 cells expressing FAD-linked PS1 mutants generated qualitatively altered p3-Alcα, and the ratios of p3-Alcα 38/35 [minor/major] were strongly correlated with the ratio of Aβ42/40 [minor/major] (R2 > 0.5).10 This suggested that altered processing of Alcs can reflect intrinsic (ie, genetic) γ-secretase dysfunction. Thus, we undertook a study of CSF from various clinical populations including sporadic AD.
In initial experiments, p3-Alcα species were recovered from pooled human CSF samples by immunoprecipitation with anti-p3-Alcα and anti-Aβ antibodies. The major and minor species of p3-Alcα and Aβ were compared by analyses with MALDI-TOF/MS (see Fig 2). This preliminary study demonstrated that the levels of p3-Alcα35 [major] and p3-Alcα38 [minor] are correlated with those of Aβ40 [major] and Aβ42 [minor], supporting the usefulness of our immunoprecipitation-MS analysis in estimating the relative amounts of p3-Alcα species and obtaining p3-Alcα38/total p3-Alcα ratios with quantitative accuracy.10
We first studied CSF p3-Alcα38 in a small Japanese (JP) population (see Fig 3A). CSF p3-Alcα38/total p3-Alcα tended to be elevated in both CDR 0.5 and CDR 1 but only CSF p3-Alcα38/total p3-Alcα in the CDR 1 group reached statistical significance when compared against age-matched nondemented elderly (p < 0.05). In the US1 cohort (see Fig 3B), a nonsignificant trend toward elevation of CSF p3-Alcα38/total p3-Alcα was observed. In the US2 cohort (see Fig 3C) as in the Japanese cohort (see Fig 3A), CSF p3-Alcα38/total p3-Alcα tended to be elevated in both CDR 0.5 and CDR 1 but only CSF p3-Alcα38/total p3-Alcα in the CDR 1 group reached statistical significance (p < 0.05). Thus, elevated CSF p3-Alcα38/total p3-Alcα was associated with CDR 1 SAD in 2 of the 3 cohorts tested. Whenever all 3 cohorts were pooled and analyzed as a single group (see Fig 3D), both CDR 0.5 and CDR 1 were elevated and reached statistical significance, depending upon whether CDR 0 control or OND control was used as the reference group.
We also sought to determine whether correlations existed between the ratios of p3-Alcα38/total p3-Alcα and Mini-Mental State Examination (MMSE) scores (N.B., for some Japanese subjects, revised Hasegawa's dementia scale [HDS-R] scores were also examined). The results are shown in Supporting Information Figure 1. The ratios of p3-Alcα38/total p3-Alcα and MMSE scores were negatively correlated in all 3 cohorts. We also sought to determine whether there existed any correlation between the ratio of p3-Alcα38/total p3-Alcα and disease duration in US cohort 1. A weakly positive correlation was detected. Disease duration was not available for other cohorts. There was no correlation between age at onset and ratios of p3-Alcα38/total p3-Alcα.
Discussion
Our preferred interpretation of the foregoing data is that a non-APP γ-secretase substrate undergoes alternative γ-secretase processing in SAD. Because CSF Aβ42 levels fall during MCI and SAD, presumably due to deposition as amyloid,15 we also considered the possibility that the altered levels of p3-Alcα38 were caused not by γ-secretase dysfunction but by aggregation of Aβ and/or p3-Alcα peptides. Although we cannot completely exclude this possibility, we would note that Alc immunoreactivity is apparently confined to intraneuronal vesicles and dystrophic neurites in AD brains,4 mimicking the distribution of APP. Neither non-Aβ holoAPP epitopes nor p3-Alcα epitopes are detectable in amyloid deposits.4 Further, in unpublished experiments, we have determined that synthetic p3-Alcα peptides undergo little or no detectable spontaneous aggregation. For this reason, too, we do not favor the idea that the altered CSF p3-Alcα38 levels are generated by differential deposition.
We have demonstrated that C-terminal speciation of p3-Alcα enables an analysis that distinguishes SAD CSF from CSF taken from other clinical populations. Yanagida and colleagues14 have recently proposed that levels of another non-APP γ-secretase reaction product, derived from APLP1 and designated APL1β28, may serve as a surrogate marker for Aβ42. Likewise, our data imply that APP is not the only γ-secretase substrate that undergoes variant processing in association with the AD clinical phenotype. Since alternative γ-secretase processing was not observed in the OND subjects, the most parsimonious explanation is that environmental or otherwise acquired γ-secretase modulators may occur in nature, and, conceivably, that these compounds contribute specifically to the risk for SAD. Indeed, examples of such compounds (eg, fenofibrate16) have been described, and a major challenge will be to determine whether this or some other compound with similar allosteric action on γ-secretase17 can be associated with an increase in risk for SAD.
Supplementary Material
Acknowledgments
This research was supported by a grant from the Ministry of Education, Science, Culture, Sports, and Technology, Japan (Grants-in-Aid for Scientific Research on Priority Areas 20023001 to T.S.).
We thank Patrick Hof, Brandon Wustman, Rosalie Crouch, and Yuhki Saito for helpful discussions. Some CSF samples from aged nondemented control subjects and from subjects suffering from various neurological diseases were provided by the Washington University Biomarkers Core, which is supported by the Washington University Alzheimer's Disease Research Center grant P50 AG05681 and P01AG03991. Some CSF samples were provided by the Washington University Resource Bank, which is supported by P50 AG05681 and P01 AG03991, and the University of Washington Resource Bank, which is supported by the University of Washington P50 Alzheimer's Disease Research Center and the US Department of Veterans Affairs. Some samples were also contributed by the Tottori University School of Health Sciences.
T.S. and T.N. are supported by Kurozumi Medical Foundation and Suzuken Memorial Foundation. S.G. is supported by National Institute on Aging grants P01 AG10491 and P50 AG005138. Authors S.H., Y.A., and M.A. are the recipients of research fellowships from the Japan Society for the Promotion of Science (JSPS) for young scientists.
Footnotes
Additional Supporting Information can be found in the online version of this article.
Potential Conflicts of Interest: The other authors report no conflicts of interest.
References
- 1.Gandy S. The role of cerebral amyloid b accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 4.Araki Y, Tomita S, Yamaguchi H, et al. Novel cadherin-related membrane proteins, alcadeins, enhance the X11-like protein-mediated stabilization of amyloid β-protein precursor metabolism. J Biol Chem. 2003;278:49448–49458. doi: 10.1074/jbc.M306024200. [DOI] [PubMed] [Google Scholar]
- 5.Araki Y, Miyagi N, Kato N, et al. Coordinated metabolism of Alcadein and amyloid β-protein precursor regulates FE65-dependent gene transactivation. J Biol Chem. 2004;279:24343–24354. doi: 10.1074/jbc.M401925200. [DOI] [PubMed] [Google Scholar]
- 6.Sano Y, Syuzo-Takabatake A, Nakaya T, et al. Enhanced amyloidogenic metabolism of the amyloid β-protein precursor in the X11L-deficient mouse brain. J Biol Chem. 2006;281:37853–37860. doi: 10.1074/jbc.M609312200. [DOI] [PubMed] [Google Scholar]
- 7.Saito Y, Sano Y, Vassar R, et al. X11 proteins regulate the translocation of APP into detergent resistant membrane and suppress the amyloidogenic cleavage of APP by BACE in brain. J Biol Chem. 2008;283:35763–35771. doi: 10.1074/jbc.M801353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki T, Nakaya T. Regulation of APP by phosphorylation and protein interaction. J Biol Chem. 2008;283:29633–29637. doi: 10.1074/jbc.R800003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Araki Y, Kawano T, Taru H, et al. The novel cargo receptor Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 2007;26:1475–1486. doi: 10.1038/sj.emboj.7601609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hata S, Fujishige S, Araki Y, et al. Alcadein cleavages by APP α-and γ-secretases generate small peptides p3-Alcs indicating Alzheimer disease-related γ-secretase dysfunction. J Biol Chem. 2009;284:36024–36033. doi: 10.1074/jbc.M109.057497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buxbaum JD, Liu KN, Luo Y, et al. Evidence that tumor necrosis factor a converting enzyme is involved in regulated a-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 12.Lammich S, Kojro E, Postina R, et al. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allinson TMJ, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein α-secretases. J Neurosci Res. 2003;74:342–352. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- 14.Yanagida K, Okochi M, Tagami S, et al. The 28-amino acid form of an APLP1derived Aβ-like peptide is a surrogate marker for Aβ42 production in the central nervous system. EMBO Mol Med. 2009;1:1–13. doi: 10.1002/emmm.200900026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 16.Kukar T, Murphy MP, Eriksen JL, et al. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- 17.Uemura K, Lill CM, Li X, et al. Allosteric modulation of PS1/ gamma-secretase conformation correlates with amyloid beta(42/ 40) ratio. PLoS One. 2009;4:e7893. doi: 10.1371/journal.pone.0007893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.