

Abstract
Synapses are sites of cell-cell contacts that transmit electrical or chemical signals in the brain. Dendritic spines are protrusions on dendritic shaft where excitatory synapses are located. Synapses and dendritic spines are dynamic structures whose plasticity is thought to underlie learning and memory. No wonder neurobiologists are intensively studying mechanisms governing the structural and functional plasticity of synapses and dendritic spines in an effort to understand and eventually treat neurological disorders manifesting learning and memory deficits. One of the best-studied brain disorders that prominently feature synaptic and dendritic spine pathology is Alzheimer's disease (AD). Recent studies have revealed molecular mechanisms underlying the synapse and spine pathology in AD, including a role for mislocalized tau in the postsynaptic compartment. Synaptic and dendritic spine pathology is also observed in other neurodegenerative disease. It is possible that some common pathogenic mechanisms may underlie the synaptic and dendritic spine pathology in neurodegenerative diseases.
1. Introduction
The number of neurons in the human brain approximates the number of stars in the galaxy. Each of these neurons makes an average of 1000 contacts with other neurons. The result is an incredibly complex and sophisticated network made of roughly 100 trillion synapses. Communications between neurons in the brain occur primarily through synapses formed between presynaptic and postsynaptic partners. For fast synaptic transmission, there are two types of synapses: type I synapses use glutamate as the neurotransmitter and are excitatory, whereas type II synapses use gamma-amino butyric acid (GABA) as the major neurotransmitter and are inhibitory. While dendritic shafts are the main location for the inhibitory GABAergic synapses, dendritic spines, which are small membrane protrusions from dendritic shafts that contain glutamate receptors and postsynaptic density components, are the primary locations of excitatory synapses. A functional balance between neuronal excitation and inhibition is established during development for homeostatic control of neuronal excitability and is maintained into adulthood [1–4]. On the other hand, imbalances between neuronal excitation and inhibition have been associated with many neurological disorders including epilepsy [5], schizophrenia [6], fragile X syndrome [7], and autism [8].
Information can be stored in the brain by multiple synaptic mechanisms, including altered structure and chemistry of existing synapses, formation of new synapses, or elimination of old ones. Such synaptic plasticity is thought to be fundamental to learning and memory in the brain [9]. At the electrophysiological level, synaptic plasticity is reflected in processes known as long-term potentiation (LTP) and long-term depression (LTD) [10]. Excitatory synapses contain AMPA and NMDA ionotropic glutamate receptors localized on dendritic spines, with basal synaptic transmission largely mediated by the AMPA receptors. High synaptic activity opens NMDA receptors, leading to long-lasting changes in postsynaptic AMPA receptor number and LTP of synaptic transmission [11]. Alternatively, low levels of synaptic stimulation can activate NMDA receptors to produce LTD [12]. At the morphological level, LTP is generally associated with dendritic spine growth, whereas LTD can induce the removal of postsynaptic AMPA receptors and loss of spines [13–19]. It is thus not surprising that synaptic development, maintenance, and plasticity under normal physiological conditions are frequently associated with changes in the morphology and number of dendritic spines [20].
In many neurodegenerative diseases, particularly those exhibiting cognitive impairments such as Alzheimer's disease (AD) and Parkinson's disease (PD), dendritic spines are altered in number and shape before eventual neuronal death is observed. Changes in dendritic spine number and morphology are also found in other disease conditions such as autism, Down syndrome, drug addiction, fragile X syndrome, and schizophrenia [20–24]. It is worth emphasizing that degeneration of synapses and dendritic spines is one of the earliest features in those neurodegenerative disease conditions, prior to subsequent loss of neurons. Interventions aimed to protect the nervous system from the ravages of these disease would therefore seem more effective when the synaptic and spine pathology are prevented as early as possible.
In this review article, we will summarize recent advances in our understanding of the molecular mechanisms underlying synaptic and dendritic spine pathology in neurodegenerative diseases, particularly in AD and PD. The readers are referred to some excellent previous reviews on the observation of synaptic and dendritic spine pathology in neurological disorders [22–25].
2. Synapse and Dendritic Spine Pathology in AD
AD is the most common neurodegenerative disease and the leading cause of dementia in the elderly. Decades of intensive research have uncovered amyloid plaque and neurofibrillary tangle (NFT) as the pathological hallmarks, and soluble amyloid-β (Aβ) oligomers as the leading candidate for the causative agent of AD [26, 27]. However, the mechanistic link between amyloid plaque and NFT and the mechanism by which Aβ oligomer may cause cognitive impairments remains poorly defined, and there is no effective treatment for this devastating disease. Substantial evidences have accumulated indicating that the memory deficits in AD patients do not correlate well with amyloid plaque burden; instead, the loss of synaptic markers is a better predictor of clinical symptoms and disease progression [28]. Together with studies using animal AD models, these studies have lent support to the hypothesis that AD could be conceptualized as a disease of synaptic failure [28].
Early structural studies of postmortem tissues showed that when compared with age-matched control brains, AD brains had reduced synapse density and number of dendritic spines in the cortex and hippocampus, principal brain areas affected by the disease, and that greater loss of dendritic spines was associated with lower mental status [29, 30]. These findings suggested that progressive loss of dendritic spines is directly related to the pathogenesis of AD and represents a good indicator of disease progression. Studies of transgenic mouse models of AD have shown that, in the vicinity of amyloid plaques, there were dramatic spine loss and neurite dystrophy, structural changes that could lead to altered neuronal circuits and brain functions [31–33]. Further studies showed that the accumulation of soluble Aβ might be the culprit that leads to dendritic spines loss [34]. Aβ is the proteolytic product of a large protein called amyloid precursor protein (APP), which is cleaved by beta- and gamma-secretases to produce Aβ and other fragments of the precursor protein [35]. Interestingly, the formation and secretion of Aβ peptides are positively regulated by neuronal activity, and excess Aβ peptide can in turn depress excitatory synaptic transmission onto neurons that produce Aβ as well as nearby neurons that do not produce Aβ [36]. Thus, activity-dependent modulation of Aβ production may normally participate in a negative feedback regulatory loop to restrain neuronal hyperactivity, the impairment of which could contribute to AD pathogenesis [36]. Under normal conditions, Aβ monomers could be cleared by proteolytic enzymes like neprilysin, chaperone molecule ApoE, or the lysosomal and proteasomal pathways. However, under pathological conditions, soluble Aβ levels are increased, leading to the buildup of Aβ oligomers, which can be further sequestered into protofibrils and fibrils as seen in plaques [27].
Several lines of evidence support that Aβ is the primary causative agent of AD. First, genetic studies of familial forms of AD have identified rare genetic mutations that cause AD by altering the production or metabolism of Aβ peptides, leading to their aberrant accumulation [27, 37]. Soluble Aβ levels have been found to better correlate with disease progression and severity than amyloid plaques or NFTs [34]. Second, Aβ oligomers formed in vitro from synthetic peptides, purified from cultured cells expressing APP, or from cortex of AD patient brains can induce synaptic dysfunction and neuritic degeneration [38–41]. Third, the reduction of soluble Aβ levels using an immunization method in mouse AD models rescued the cognitive deficits [42]. However, despite the overwhelming supporting evidences, the Aβ hypothesis of AD as described above still faces challenge, since several highly publicized clinical trials targeting Aβ had failed.
3. Mechanisms Underlying the Synapse and Dendritic Spine Pathology in AD
The molecular mechanisms through which Aβ might cause synaptic loss and neuronal death remain uncertain. Aβ has been found to form pore-like structures with calcium channel activity, which could interfere with calcium signaling [43, 44]. Aβ can also affect LTP and LTD by modulating glutamate receptor-dependent signaling pathways [45–47] and trigger aberrant patterns of neural network activity [48]. Aβ may also cause mitochondrial dysfunction [49] and lysosomal failure [50].
One of the earliest clues about the mechanisms of Aβ-induced synaptic dysfunction came from studies of cultured neurons derived from Tg2576 mutant APP transgenic mice [51]. Among the synaptic changes observed were fewer and smaller postsynaptic compartments and fewer and enlarged active presynaptic compartments. Notably, the earliest observable change in synaptic components was the reduction of PSD-95, which is a master regulator of the assembly and anchoring of postsynaptic density components such as glutamate receptor subunits [52]. Aβ was shown to be the toxic agent causing these synaptic changes since the effects were blocked by gamma-secretase inhibitor treatment and recapitulated by application of synthetic Aβ to wild-type neurons [51]. Similar PSD-95-related synaptic defects were also observed in human AD brain samples [53]. The molecular mechanisms through which Aβ influences PSD-95 remain to be determined. Studies in Drosophila models showed that PAR-1 kinase, the fly homologue of mammalian microtubule affinity regulating kinases (MARKs), can directly phosphorylate the fly PSD-95 homologue Dlg, and this phosphorylation event caused the delocalization of Dlg from the postsynaptic membrane [54]. PAR-1/MARK kinases are known to be activated by APP or Aβ in Drosophila or mammalian neurons [55, 56]. It would be interesting to test whether MARKs are critical mediators of Aβ toxicity on mammalian synapses and dendritic spines.
A significant recent advance in our understanding of the mechanisms of the synaptic toxicity of Aβ has been the finding that Aβ uses LTD-related signaling mechanisms to affect synaptic function and dendritic spine morphology [45]. One of the principle mechanisms of LTD induction is the removal of AMPA receptors from the postsynaptic membrane through endocytosis. Significant parallels were found between Aβ-induced synaptic changes and LTD. Overexpression of Aβ resulted in decreased spine density and postsynaptic AMPA receptor number, through signaling molecules implicated in LTD, such as p38 MAP kinase and calcineurin. Importantly, expression of a mutant form of AMPA receptor that resists LTD-driven endocytosis blocked the morphological effects and synaptic depression induced by Aβ [45]. This study implicated the endocytosis of AMPA receptors as a major mechanism through which Aβ causes synaptic dysfunction and subsequent degeneration, but the detailed molecular mechanisms remain unclear.
Recent studies using transgenic mouse models of AD have implicated the microtubule-binding protein tau as a major mediator of the toxicity of Aβ at the postsynaptic compartment and dendritic spines. Although tau abnormality has long been observed in AD, as exemplified by the formation of NFTs by tau that accompany plaque pathology, and tau abnormality can cause neurodegeneration in the absence of plaque pathology as in frontotemporal dementia cases [57, 58], the direct involvement of tau in Aβ-induced synaptic and dendritic spine pathology may initially appear surprising, since tau is generally considered a presynaptic protein that is primarily localized to axons. In fact, the relationship between NFTs and amyloid plaques in disease pathogenesis has long been a source of considerable debate [37, 59, 60]. Studies in mice suggested that the two lesions might be causally linked. In transgenic mouse models, intracranial injection of synthetic Aβ, or crossing of APP transgenic mice with tau transgenic mice, promoted NFT pathology [61–63], and immunization of APP/Psn/tau triple transgenic mice with antibodies against Aβ reduced the levels of hyperphosphorylated tau [64]. This was consistent with earlier studies showing that the removal of tau could relieve Aβ-induced neurotoxicity in cultured neurons [65]. Together, these studies support the notion that the initiating event in AD is the accumulation of the toxic Aβ peptides, and that tau abnormality is a major downstream molecular event that contributes to disease pathogenesis [37].
How tau abnormality arises in AD is not well understood. Current efforts have focused on the role of aberrant phosphorylation of tau [27]. Previous studies have shown that Aβ could lead to abnormal activation of a number of kinases, including cyclin-dependent kinase-5 (CDK5) [66, 67], Fyn kinase [68, 69], glycogen synthase kinase-3beta (GSK3β) [70], and MARK [71–73], all of which promote tau hyperphosphorylation and could potentially affect synaptic structure and function. However, very few in vivo studies have been done to assess the roles of tau kinases or phosphatases in conferring tau toxicity and in causing AD-related memory deficit. Identification of the relevant kinases or phosphatases will provide attractive therapeutic targets for AD.
Recently studies have shown that removing endogenous tau can prevent Aβ-induced behavioral deficits in a mouse AD model expressing human APP, and block excitotoxin-induced neuronal dysfunction in both transgenic and nontransgenic mice [74]. Since current data support postsynaptic toxicity as a primary mechanism of Aβ action in causing learning and memory deficits in AD, this study raised the possibility that tau may also act in the postsynaptic compartment. Indeed, under both physiological and pathological conditions, tau was found in dendrites [75, 76], albeit the level of dendritic tau was much higher under disease conditions. Tau was known to interact with microtubules through its microtubule-binding domain to stabilize microtubule and regulate axonal transport. It has many putative phosphorylation sites and becomes hyperphosphorylated in AD patients and transgenic animal models [57, 58]. Apart from the notion that phosphorylation can lead to the dissociation of tau from the microtubules, other pathophysiological effects of this molecular event are unknown.
A recent study has indicated that phosphorylated tau could accumulate in dendritic spines, where it may affect the synaptic trafficking and/or anchoring of glutamate receptors, thereby influencing postsynaptic function [76]. Interestingly, this effect of tau on synaptic function occurred without causing the loss of synapses or dendritic spines. This study thus revealed a critical role for tau phosphorylation in causing tau mislocalization and subsequent synaptic impairment, and it established dendritic spines as pathogenic targets of tau action. Another study provided further mechanistic insights into the dendritic function of tau [75]. Tau interacts with fyn [77], a protein tyrosine kinase that can phosphorylate tau and whose activity is increased in AD brain [78]. Ittner et al. showed that the interaction of tau with fyn leads to the targeting of fyn to dendritic spines, where fyn can phosphorylate NMDA receptor subunit 2 (GluR2), resulting in stabilization of the interaction between GluR2 and PSD-95 and enhanced excitotoxicity. Tau also shows strong interaction with PSD-95, providing further support for a dendritic role of tau besides its known axonal function. Importantly, the toxic effects of APP/Aβ were attenuated by interfering with GluR2/PSD-95 interaction with a cell-permeable peptide [75], supporting that dendritic tau-mediated fyn recruitment and GluR2/PSD-95 interaction confer Aβ toxicity at the postsynapse.
Thus, a “tau hypothesis” has been put forward based on these recent results; Aβ triggers the phosphorylation of tau, causing tau to dissociate from the microtubules and accumulate at the dendritic compartments. Phosphorylated tau exhibits stronger interaction with Fyn and thus facilitates the targeting of fyn to dendritic spines. The targeting of fyn to postsynaptic density sensitizes the NMDA receptors and renders neurons more vulnerable to the toxicity of Aβ in the postsynaptic compartment [79]. It remains to be determined whether tau becomes hyperphosphorylated in situ in the dendritic spines as a result of altered kinase/phosphatase activities there, or that it becomes hyperphosphorylated elsewhere and is then transported to the dendritic spines. Nevertheless, targeting the tau-dependent pathway, for example, by reducing tau protein level, inhibiting tau kinase activities, or increasing phosphatase activities, would represent suitable new ways of treating AD.
In summary, we can consider the toxic effect of Aβ on neuronal synapses and dendritic spines as a normal physiological process gone awry, instead of some pathological process unique to the disease process. Aβ is continuously produced in the brain, and its production can be stimulated by neuronal activity. Aβ can then feedback on the hyperactive neuron using a LTD-related mechanism to tune down neuronal activity, for example, by promoting AMPAR removal. This process normally acts as a homeostatic mechanism to restrain neuronal hyperactivation. In the disease process, however, the buildup of Aβ tips the balance of this process toward excessive synaptic depression and AMPAR removal, resulting in synapse and spine loss (Figure 1). The molecular mechanisms involved in Aβ toxicity on synapses and dendritic spines are just beginning to be elucidated. We propose that a signaling cascade from Aβ to tau and PSD-95, involving tau kinases such as PAR-1/MARK and its activating kinase LKB1, might be involved (Figure 1).
Figure 1.
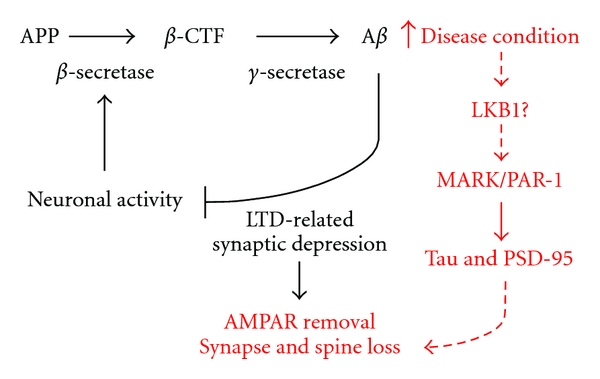
A diagram depicting the physiological and pathological roles of Aβ. The pathway in black represents the normal function of Aβ in restraining neuronal hyperactivation. In response to neuronal activation, there is upregulation of BACE, leading to overproduction of Aβ, which then acts through LTD-related mechanism involving AMPAR removal to tune down neuronal activity. In disease condition (depicted in red), however, the excessive accumulation of Aβ leads to excessive synaptic depression and AMPAR removal, which eventually results in synapse and spine loss. Based on our unpublished work (Yu et al., manuscript submitted), we propose that Aβ can act through the LKB1→MARK→tau/PSD-95 signaling cascade to cause synapse and spine loss.
4. Possible Nonneuronal Contribution to Synapse and Spine Pathology in AD
Although much of the research on the mechanisms of Aβ toxicity to synapses and spines has taken a “neuron-centric” approach, it is worth noting that other nonneuronal cell types in the brain play critical roles in the formation and maturation of synapses during development, and similar mechanisms may operate in the adult brain to mediate the effects of Aβ on neuronal synapses and dendritic spines.
Besides providing trophic factors for neurons, glial cells have been shown to play key roles in regulating neuronal migration, axon guidance, and synapse formation [80]. In one of the better-characterized cases, astrocytes were shown to secret signals that induce synapse formation by retinal ganglion cells (RGCs). A family of extracellular matrix proteins called thrombospondins (TSPs) was identified as the synaptogenic signals coming from astrocytes [81]. The TSP receptor from the neuronal side involved in synaptogenesis was found to be the calcium channel subunit α2δ-1 [82]. Interestingly, the synapses formed by TSPs are postsynaptically silent due to the lack of surface AMPA receptors, whereas those formed by astrocyte conditioned medium are postsynaptically active, suggesting that additional factors are secreted by the astrocytes to control synaptic strength and plasticity [83]. The identity of these additional factors is currently unknown. Also, synapses are made in excess during development, and the extra synapses or weak synapses are eliminated by a process involving signals from astrocytes that induce the classical complement pathway protein C1q in neurons [84]. In addition to secreted factors, astrocytes can regulate synapse formation using contact-mediated mechanisms. Astrocytes also regulate dendritic spine morphology through a contact-mediated mechanism involving bidirectional ephrin/EphA signaling. In the hippocampus, for example, astrocytes express ephrin A3, whereas neurons express the ephrin receptor EphA4. Perturbing ephrin/EphA signaling results in defects in spine formation and maturation [85]. One can imagine that disruption of astrocyte-neuron interaction by Aβ could affect synapse and spine morphology through the above-mentioned mechanisms. In this respect, it is interesting to note that a recent study has shown that lentiviral-mediated delivery of EphB2 expression constructs in the dentate gyrus of hAPP transgenic mice reversed deficits in NMDA receptor-dependent LTP and memory impairments [86]. Whether there is glial involvement in this experimental setting has not been examined.
The other abundant glial cells in the brain are microglia. Unlike the astrocytes, these cells are of mesodermal origin. The roles of microglia in disease pathogenesis in AD and other neurodegenerative diseases are very complex and controversial [87, 88]. This probably has to do with the diverse activities of these cells in the brain. Relevant to AD pathogenesis, microglia can promote Aβ clearance, release anti-inflammatory cytokines and neurotrophic factors on one hand, and they can also affect the activation of complement systems and elimination of synapses and spines on the other hand [88]. Thus microglia can exert neuroprotective as well as neurodegenerative effects, depending on the strength, timing, and duration of their activation. Imaging studies showed that activated microglia were found in patients with MCI [89], suggesting that neuroinflammation is an early event in the disease process. Consistent with this finding, microglial activation was observed early in a tauopathy mouse model, preceding NFT formation and roughly concurrent with synapse loss and impairment of synaptic function [90]. Interestingly, supplement of immunosuppressant FK506 to young mice attenuated tau pathology and increased lifespan, suggesting that microglia activation may contribute to disease. In another AD mouse model expressing the E693Δ mutation that causes AD by enhanced Aβ oligomerization without fibrillization, it was found that the mice displayed age-dependent accumulation of intraneuronal Aβ oligomers at around 8 months, when abnormal tau phosphorylation, and impairments of hippocampal synaptic plasticity and memory were observed. However, microglial activation was observed from 12 months, astrocyte activation from 18 months, and neuronal loss at 24 months [91]. It is not known in this case whether microglial and astrocyte activation plays a neurodegenerative role or as part of a neuroprotective, compensatory response.
Despite the large amount of literature documenting a detrimental role for microglia and astrocyte activation in the disease process, these cells are important for neuronal heath during development and later in adult life. For example, microglia are proposed to play a surveillance role by constantly monitoring and sensing synaptic health [92], and, in addition to the critical roles, astrocytes play in synapse formation as mentioned earlier, and these cells can also control extracellular glutamate levels, remove excess extracellular K+, release gliotransmitters, store glucose and transform it into lactate as energy source of neurons, and scavenge ROS to protect against oxidative damages [88]. Given these essential roles of glia to neuronal function and health, it is possible that damaging of glial cells by Aβ may have equally harmful effect on the neurons eventually. In fact, there is evidence that glial cells can release ROS upon Aβ exposure [93], and glial-released cytokines may even trigger a signaling process that promotes tau hyperphosphorylation [94]. Thus, a possible role of dysfunction glial cells in AD pathogenesis should be considered, especially in the early stages of the disease process (Figure 2).
Figure 2.
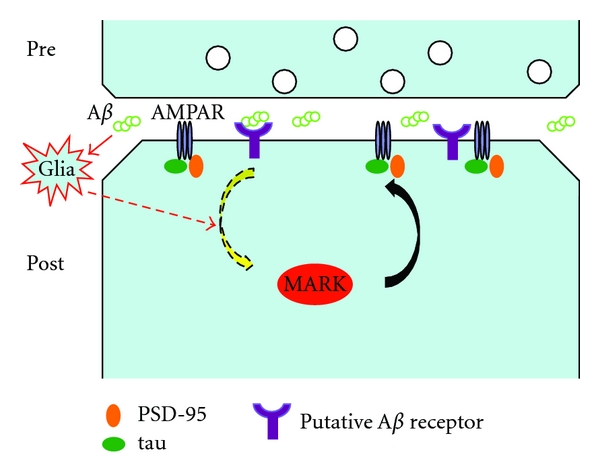
A diagram depicting a potential role of glia in mediating the synaptic toxicity of Aβ. Aβ oligomers presumably secreted from the presynaptic neuron could bind to its putative receptor on the postsynaptic cell, and this could then initiate a signaling cascade leading to activation kinases such as MARK, which then acts on tau, PSD-95, and possibly other synaptic substrates to affect AMPAR removal from the synaptic surface, leading to synapse and spine loss. Alternatively, Aβ could act on glial cells near neuronal synapses, which then release factors such as cytokines to activate signaling molecules such as MARK and cause synapse and spine loss. These two mechanisms are not mutually exclusive and could in fact occur simultaneously to mediate Aβ toxicity.
5. Conclusions and Future Directions
Synapse and dendritic spine pathology have been observed in the early stages of neurodegenerative diseases before neuronal death is evident, suggesting that these cellular locations represent pathogenic sites of action by the disease-causing agents early in the disease process. At least in the case of AD, there is compelling evidence supporting a pathogenic role for the synaptic and dendritic spine abnormalities. An intriguing possibility is that, as in AD, defects in the morphology and function of synapses and dendritic spines may play a critical role in the pathogenesis of PD. In fact, alterations in synaptic plasticity as represented by LTP and LTD are observed in PD, and some familial PD-associated genes have been shown to affect synapse and dendritic spine morphology and function [95, 96]. It would thus be interesting to examine whether LTP- and LTD-related signaling mechanisms are involved in PINK1/Parkin-induced synapse and dendritic spine changes. In this respect, it would also be interesting to test the potential role of dendritic tau in mediating the synaptic effects of the FPD genes. This is particularly relevant, given the identification of tau as a susceptibility factor for PD [97]. Future studies along these directions could lead to the identification of common molecular mechanisms underlying the pathogenesis of AD, PD, and possibly other neurological disorders and offer new therapeutic strategies.
Acknowledgments
This research is supported by Dean's Postdoctoral Fellowship, Stanford University School of Medicine (W. Yu), Brain Disorders Award from the McKnight Endowment Fund for Neurosciences (B. Lu), and National Institute of Health Grants no. R01MH080378 and R01AR054926 (B. Lu).
References
- 1.Eichler SA, Meier JC. E-I balance and human diseases—from molecules to networking. Frontiers in Molecular Neuroscience. 2008;1, article 2 doi: 10.3389/neuro.02.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keith D, El-Husseini A. Excitation control: balancing PSD-95 function at the synapse. Frontiers in Molecular Neuroscience. 2008;1, article 4 doi: 10.3389/neuro.02.004.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nature Reviews Neuroscience. 2004;5(2):97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- 4.Yu W, Blas ALD. Gephyrin expression and clustering affects the size of glutamatergic synaptic contacts. Journal of Neurochemistry. 2008;104(3):830–845. doi: 10.1111/j.1471-4159.2007.05014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stief F, Zuschratter W, Hartmann K, Schmitz D, Draguhn A. Enhanced synaptic excitation-inhibition ratio in hippocampal interneurons of rats with temporal lobe epilepsy. European Journal of Neuroscience. 2007;25(2):519–528. doi: 10.1111/j.1460-9568.2006.05296.x. [DOI] [PubMed] [Google Scholar]
- 6.Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annual Review of Neuroscience. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- 7.Tsiouris JA, Brown WT. Neuropsychiatric symptoms of fragile X syndrome: pathophysiology and pharmacotherapy. CNS Drugs. 2004;18(11):687–703. doi: 10.2165/00023210-200418110-00001. [DOI] [PubMed] [Google Scholar]
- 8.Deonna T, Roulet E. Autistic spectrum disorder: evaluating a possible contributing or causal role of epilepsy. Epilepsia. 2006;47(supplement 2):79–82. doi: 10.1111/j.1528-1167.2006.00697.x. [DOI] [PubMed] [Google Scholar]
- 9.Kandel ER, Schwartz JH. Molecular biology of learning: modulation of transmitter release. Science. 1982;218(4571):433–443. doi: 10.1126/science.6289442. [DOI] [PubMed] [Google Scholar]
- 10.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 11.Bliss TVP, Lomo T. Long lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. The Journal of Physiology. 1973;232(2):331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch GS, Dunwiddie T, Gribkoff V. Heterosynaptic depression: a postsynaptic correlate of long term potentiation. Nature. 1977;266(5604):737–739. doi: 10.1038/266737a0. [DOI] [PubMed] [Google Scholar]
- 13.Lüscher C, Xia H, Beattie EC, et al. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24(3):649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- 14.Man HY, Lin JW, Ju WH, et al. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25(3):649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 15.Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nature Neuroscience. 2001;4(11):1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- 16.Xiao MY, Zhou Q, Nicoll RA. Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology. 2001;41(6):664–671. doi: 10.1016/s0028-3908(01)00134-4. [DOI] [PubMed] [Google Scholar]
- 17.Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36(4):661–674. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- 18.Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44(5):759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44(5):749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annual Review of Neuroscience. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Research Reviews. 2002;39(1):29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- 22.Schulz-Schaeffer WJ. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathologica. 2010;120(2):131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Spronsen M, Hoogenraad CC. Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports. 2010;10(3):207–214. doi: 10.1007/s11910-010-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Penzes P, Cahill ME, Jones KA, Vanleeuwen J-E, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature Neuroscience. 2011;14(3):285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathologica. 2010;119(5):523–541. doi: 10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annual Review of Pharmacology and Toxicology. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 27.Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Science Translational Medicine. 2011;3(77, article 77sr1) doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 29.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Annals of Neurology. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 30.Selkoe DJ. Amyloid β protein precursor and the pathogenesis of Alzheimer’s disease. Cell. 1989;58(4):611–612. doi: 10.1016/0092-8674(89)90093-7. [DOI] [PubMed] [Google Scholar]
- 31.Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nature Neuroscience. 2004;7(11):1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 32.Spires TL, Meyer-Luehmann M, Stern EA, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. Journal of Neuroscience. 2005;25(31):7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spires-Jones TL, Meyer-Luehmann M, Osetek JD, et al. Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. The American Journal of Pathology. 2007;171(4):1304–1311. doi: 10.2353/ajpath.2007.070055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behavioural Brain Research. 2008;192(1):106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sisodia SS, Price DL. Role of the β-amyloid protein in Alzheimer’s disease. The FASEB Journal. 1995;9(5):366–370. doi: 10.1096/fasebj.9.5.7896005. [DOI] [PubMed] [Google Scholar]
- 36.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37(6):925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 37.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 38.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotixicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245(4916):417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 39.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 40.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45(5):675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 43.Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(2):567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418(6895, article 291):p. 291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 45.Hsieh H, Boehm J, Sato C, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52(5):831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. Journal of Neuroscience. 2007;27(11):2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55(5):697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin MT, Beal MF. Alzheimer’s APP mangles mitochondria. Nature Medicine. 2006;12(11):1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 50.Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2006;9(supplement 3):277–289. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- 51.Almeida CG, Tampellini D, Takahashi RH, et al. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiology of Disease. 2005;20(2):187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 52.Kim E, Sheng M. PDZ domain proteins of synapses. Nature Reviews Neuroscience. 2004;5(10):771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 53.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Snaptic changes in alzheimer’s disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. The American Journal of Pathology. 2004;165(5):1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Guo H, Kwan H, Wang JW, Kosek J, Lu B. PAR-1 kinase phosphorylates Dlg and regulates its postsynaptic targeting at the Drosophila neuromuscular junction. Neuron. 2007;53(2):201–215. doi: 10.1016/j.neuron.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. The Journal of Neuroscience. 2007;27(3):574–581. doi: 10.1523/JNEUROSCI.5094-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. The Journal of Neuroscience. 2010;30(36):11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annual Review of Neuroscience. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 58.Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends in Neurosciences. 2009;32(3):150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 59.Davies P. A very incomplete comprehensive theory of Alzheimer’s disease. Annals of the New York Academy of Sciences. 2000;924:8–16. doi: 10.1111/j.1749-6632.2000.tb05553.x. [DOI] [PubMed] [Google Scholar]
- 60.Lee VMY. Biomedicine: tauists and ßaptists united—well almost! Science. 2001;293(5534):1446–1447. doi: 10.1126/science.1064684. [DOI] [PubMed] [Google Scholar]
- 61.Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 62.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 63.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 64.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(9):6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patrick GN, Zukerberg L, Nikolic M, De La Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 67.Cruz JC, Kim D, Moy LY, et al. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid β in vivo. The Journal of Neuroscience. 2006;26(41):10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang C, Qiu HE, Krafft GA, Klein WL. Aβ peptide enhances focal adhesion kinase/Fyn association in a rat CNS nerve cell line. Neuroscience Letters. 1996;211(3):187–190. doi: 10.1016/0304-3940(96)12761-0. [DOI] [PubMed] [Google Scholar]
- 69.Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. The Journal of Neuroscience. 2004;24(19):4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baum L, Hansen L, Masliah E, Saitoh T. Glycogen synthase kinase 3 alteration in Alzheimer disease is related to neurofibrillary tangle formation. Molecular and Chemical Neuropathology. 1996;29(2-3):253–261. doi: 10.1007/BF02815006. [DOI] [PubMed] [Google Scholar]
- 71.Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule- associated proteins and trigger microtubule disruption. Cell. 1997;89(2):297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 72.Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116(5):671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- 73.Chin JY, Knowles RB, Schneider A, Drewes G, Mandelkow EM, Hyman BT. Microtubule-affinity regulating kinase (MARK) is tightly associated with neurofibrillary tangles in Alzheimer brain: a fluorescence resonance energy transfer study. Journal of Neuropathology and Experimental Neurology. 2000;59(11):966–971. doi: 10.1093/jnen/59.11.966. [DOI] [PubMed] [Google Scholar]
- 74.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 75.Ittner LM, Ke YD, Delerue F, et al. Dendritic function of tau mediates amyloid-β toxicity in alzheimer’s disease mouse models. Cell. 2010;142(3):387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 76.Hoover BR, Reed MN, Su J, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68(6):1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee G, Todd Newman S, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. Journal of Cell Science. 1998;111(21):3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- 78.Shirazi SK, Wood JG. The protein tyrosine kinase, fyn, in Alzheimer’s disease pathology. NeuroReport. 1993;4(4):435–437. doi: 10.1097/00001756-199304000-00024. [DOI] [PubMed] [Google Scholar]
- 79.Ittner LM, Götz J. Amyloid-β and tau—a toxic pas de deux in Alzheimer’s disease. Nature Reviews Neuroscience. 2011;12(2):65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 80.Bolton MM, Eroglu C. Look who is weaving the neural web: glial control of synapse formation. Current Opinion in Neurobiology. 2009;19(5):491–497. doi: 10.1016/j.conb.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 81.Christopherson KS, Ullian EM, Stokes CCA, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120(3):421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 82.Eroglu Ç, Allen NJ, Susman MW, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139(2):380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60(3):430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 84.Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 85.Carmona MA, Murai KK, Wang L, Roberts AJ, Pasqualea EB. Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(30):12524–12529. doi: 10.1073/pnas.0903328106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cissé M, Halabisky B, Harris J, et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469(7328):47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nature Medicine. 2006;12(9):1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 88.Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Current Pharmaceutical Design. 2010;16(25):2766–2778. doi: 10.2174/138161210793176572. [DOI] [PubMed] [Google Scholar]
- 89.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. The Lancet. 2001;358(9280):461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 90.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 91.Tomiyama T, Matsuyama S, Iso H, et al. A mouse model of amyloid β oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. The Journal of Neuroscience. 2010;30(14):4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathologica. 2010;119(1):89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- 93.Matos M, Augusto E, Oliveira CR, Agostinho P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience. 2008;156(4):898–910. doi: 10.1016/j.neuroscience.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 94.Arnaud L, Robakis NK, Figueiredo-Pereira ME. It may take inflammation, phosphorylation and ubiquitination to “tangle” in Alzheimer’s disease. Neurodegenerative Diseases. 2007;3(6):313–319. doi: 10.1159/000095638. [DOI] [PubMed] [Google Scholar]
- 95.Helton TD, Otsuka T, Lee MC, Mu Y, Ehlers MD. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(49):19492–19497. doi: 10.1073/pnas.0802280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu W, Sun Y, Guo S, Lu B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Human Molecular Genetics. 2011;20(16):3227–3240. doi: 10.1093/hmg/ddr235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Skipper L, Wilkes K, Toft M, et al. Linkage disequilibrium and association of MAPT H1 in Parkinson disease. American Journal of Human Genetics. 2004;75(4):669–677. doi: 10.1086/424492. [DOI] [PMC free article] [PubMed] [Google Scholar]