

Abstract
Identifying which specific neuronal phenotypes are vulnerable to neonatal hypoxia-ischemia, where in the brain they are damaged, and the mechanisms that produce neuronal losses are critical to determine the anatomical substrates responsible for neurological impairments in hypoxic-ischemic brain-injured neonates. Here we describe our current work investigating how the serotonergic network in the brain is disrupted in a rodent model of preterm hypoxia-ischemia. One week after postnatal day 3 hypoxia-ischemia, losses of serotonergic raphé neurons, reductions in serotonin levels in the brain, and reduced serotonin transporter expression are evident. These changes can be prevented using two anti-inflammatory interventions; the postinsult administration of minocycline or ibuprofen. However, each drug has its own limitations and benefits for use in neonates to stem damage to the serotonergic network after hypoxia-ischemia. By understanding the fundamental mechanisms underpinning hypoxia-ischemia-induced serotonergic damage we will hopefully move closer to developing a successful clinical intervention to treat neonatal brain injury.
1. General Characteristics of Neonatal Brain Injury
Approximately 4 in 1000 babies are born each year with brain damage. Being born premature (<37 weeks gestation) and exposure to a hypoxic-ischemic insult (HI; reduced oxygen and blood flow to the brain) are the major risk factors that contribute to this statistic [1, 2]. An HI insult can ensue after many possible factors including placental dysfunction, haemorrhage, hypotension, umbilical cord occlusion, and stroke [1]. A considerable number of these preterm neonates estimate as high as 50% [3], develop neurological and functional impairments such as cerebral palsy, motor deficits, sleep disorders, hyperactivity, anxiety, depression, and cognitive and autonomic disabilities [4–8]. These lifelong disabilities place enormous burdens on the individual as well as family, healthcare, educational, and community resources.
Although significant advances in neonatal care have increased survival rates of preterm infants, particularly those less than 28 weeks gestation, a concomitant decrease in morbidity has not been achieved. In addition, aside from the recent development of early cooling of the neonatal brain [9, 10], there is no therapeutic intervention available to treat neonatal brain injury. Thus the substantial associated life-long burdens are growing and there is an urgent need to identify neuroprotective drugs that target neuronal networks to prevent, slow, or abate the deleterious effects of HI in the neonatal brain.
White matter damage is a hallmark feature of brain injury after HI in the preterm neonate. Enlarged ventricles (ventriculomegaly), loss of vulnerable oligodendrocyte progenitor cells, periventricular leukomalacia (PVL), hypomyelination, thinning of the corpus callosum, astrogliosis, and microgliosis are typical features of white matter damage [11–16]. Characterising white matter injury and searching for the mechanisms contributing to this injury have been major avenues of investigation in the area of preterm HI brain injury. However, neuronal loss is also a critical neuropathological feature of HI and the pattern of brain injury in preterm neonates is described as a combination of white and grey matter damage [11–13]. Moreover, it is plausible that disrupted neuronal function and neural circuit connectivity are a consequence of white matter loss and axonal disruption.
2. Neuronal Damage in the Preterm HI Brain
With the advent of more sophisticated and higher resolution imaging techniques scientists are beginning to discriminate white and gray matter, delineate neural connectivity, and identify biochemical markers so that brain injury in the neonate is increasingly being characterized in much finer detail. It is well established that there are volumetric reductions in certain brain areas of HI-affected preterm infants including the thalamus, basal ganglia, and cerebral cortex and that these effects are manifested in association with PVL and other white matter features [17–21]. Axonal pathology and neuronal injury have been reported in these regions as well as in the brainstem, cerebellum, striatum, hippocampus, and hypothalamus after HI in the human preterm brain [8, 22–24] and animal models [25–28]. Furthermore, long-term changes in neuronal neurotransmitter content and release can also occur after neonatal HI [29–32]. Disruption of neuropeptides and neurotransmitters, critical for the development of synapses and formation of neuronal networks, has been postulated to underlie behavioural deficits and neuroendocrine disorders in the growing child and adult human with a history of preterm HI [33]. It is pertinent that some types of neurons (e.g., dopaminergic, noradrenergic, and cholinergic neurons) may be more vulnerable to perinatal injury than others (e.g., magnocellular neurons in the hypothalamus) [28, 34–36].
Identifying which specific neuronal phenotypes are vulnerable to HI, where in the brain they are damaged, the timing and mechanisms underlying neuronal losses are necessary directions to establish the anatomical substrates underpinning functional impairments in HI-affected neonates. These are important issues to determine because if particular neuronal phenotypes or brain regions are injured at different times or differ in their vulnerability to HI then selective neuroprotective interventions may also be temporally and spatially distinct. One neural network that we have a particular focus on is the serotonergic system in the brain.
3. The Serotonergic System: A Candidate Network Disrupted after Neonatal HI
Virtually all brain regions reportedly injured after neonatal HI receive substantial serotonergic fibre projections from the brainstem. In addition, the rostral brainstem, where serotonergic cell bodies reside, is damaged after neonatal HI [8]. It is well established that interruption of the central serotonergic system can lead to numerous functional deficits and many outcomes are similar to those observed in preterm neonates exposed to HI. These observations prompted us to hypothesise the serotonergic network in the brain is a major system that is disrupted after preterm HI and that this system is a pivotal neural candidate to target with neuroprotective interventions after preterm HI.
Serotonin (5-hydroxytryptamine, 5-HT) is pivotal in fetal and postnatal brain development [37]. The serotonergic network in the brain develops very early during gestation and is one of the first transmitter systems to appear in the developing brain. Indicative of its pervasive innervation of the central nervous system in the postnatal and mature brain, 5-HT is a neurochemical that is involved in a vast array of functions. In addition, dysfunction of serotonergic neurotransmission has been implicated in a host of physiological, metabolic, and behavioural changes in disease states such as epilepsy, depression, movement disorders, autism, anxiety and sudden infant death syndrome (SIDS) [38–43]. In the context of neonatal brain injury, it is pertinent that many of these deficits match those observed in HI-affected neonates [4, 5, 7, 44]. In addition, decreased serotonergic function is a hallmark feature of depression and depressed patients show 31% loss of dorsal raphé neurons [45]. Cerebral palsy is a notable disability in some HI-affected neonates and these patients have been reported to suffer depression [46, 47]. Although, whether altered serotonergic function accounts for certain HI-induced neurological deficits is not known. It is important to first characterise the effects of neonatal HI on major elements of the serotonergic system in the brain and begin to decipher whether these specific nuclei constitute primary candidate networks that underpin neonatal HI-induced neurological deficits.
Utilising a postnatal day 3 (P3) HI model of preterm HI we have recently investigated how P3 HI affects the serotonergic system in the brain. The P3 rat pup is subjected to HI by right common carotid artery ligation followed by 6% oxygen for 30 min. In the rat, the P3 brain development stage is analogous to the preterm human neonate brain at approximately 24–28 weeks gestation in terms of cellular development, number of synapses, neurochemical development, and cortical organization [48]. This preclinical model produces typical behavioural and pathological features including encephalopathy and hypomyelination observed in human preterm neonates affected by HI [4, 28, 48–51].
4. The Synthesis and Release of 5-HT in the Central Nervous System
Serotonin is synthesised in the brain in serotonergic neurons from the amino acid L-tryptophan and its metabolite 5-hydroxytryptophan (5-HTP). Synthesis occurs via tryptophan hydroxylase (TpH), 5-HT's rate-limiting enzyme and a second enzyme amino acid decarboxylase. Two isoforms of TpH are known to exist (TpH1 and TpH2) but only TpH2 is found in the brain [52]. The major regulator of 5-HT levels in the brain is the serotonin transporter (SERT). The transporter consists of 12 transmembrane domains that span the presynaptic membrane of 5-HT-releasing cells [53]. Localised on the presynaptic membrane of serotonergic neurons, SERT terminates serotonergic signalling by the efficient reuptake of extracellular serotonin back into the presynaptic neuron (Figure 1) thereby controlling the duration of action and post-synaptic signalling of 5-HT in the brain. Consequently, SERT is a major target for drugs such as selective serotonin reuptake inhibitors (SSRIs) that can increase 5-HT availability in the brain and are useful drugs in the treatment of depression. Serotonin is also broken down by monoamine oxidase (MAO) enzymes, preferentially MAO-A, into 5-hydroxyindoleacetic acid (5-HIAA); serotonin's major metabolite.
Figure 1.
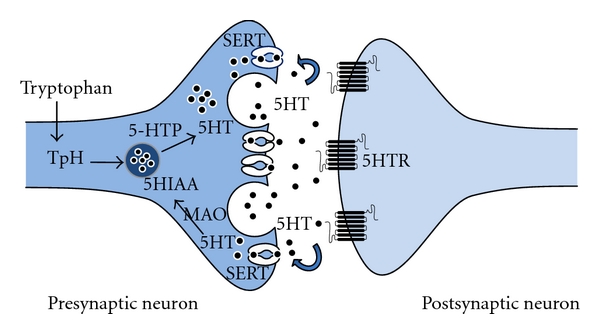
Schematic diagram depicting the major pathways involved in the synthesis, release, re-uptake and metabolism of serotonin in serotonergic neurons. Components of the figure have been modified from Motifolio. TpH: tryptophan hydroxylase; 5-HTP: 5-hydroxy-L-tryptophan; 5-HT: serotonin; SERT: serotonin transporter; MAO: monoamine oxidase; 5-HIAA: 5-hydroxyindoleacetic acid; 5-HTR: serotonergic receptor.
Nine groups of 5-HT-containing cell bodies represented in raphé subdivisions in the pons and upper brainstem were first described using histochemical techniques and designated B1−9 [54]. The bilateral raphé subdivisions are predominantly populated with serotonergic neuronal cell bodies and provide an extensive serotonergic network throughout the central nervous system. Based on their cytoarchitecture, neurochemistry, and neural projections, nomenclature for the clusters of 5-HT neurons describes their location in the dorsal, lateral, midline, or caudal portion of the pons and medulla oblongata [55, 56].
5. Serotonergic Damage in the Immature Brain after HI
In human neonates with HI encephalopathy tryptophan hydroxylase, the 5-HT rate-limiting enzyme, is reduced in the brainstem [41, 57]. Damage to human dorsal brainstem nuclei, where serotonergic cell bodies are located, is also apparent [8]. However, until our initial study in 2010 in a rodent P3 HI model [58], information about the effects of HI on specific raphé nuclei was scarce. We found an overall significant loss of 5-HT-positive raphé neurons after P3 HI, consistent with previous animal studies [59, 60] and reports from human neonates [8]. However it is interesting that certain serotonergic raphé nuclei appear to be more vulnerable to P3 HI-induced injury than others. One week after P3 HI, 5-HT-positive neuronal losses occur in the dorsal raphé caudal, dorsal raphé ventrolateral, and dorsal raphé dorsal nuclei. In contrast, the dorsal raphé interfascicular and the raphé magnus nuclei showed no reduction in number of 5-HT-positive neurons on P10 and P45. Six weeks after P3 HI, on P45, only the dorsal raphé ventrolateral and the dorsal raphé dorsal demonstrated a maintained and significant decrease in numbers of 5-HT-positive neurons [58].
The rostrocaudal distribution of the raphé serotonergic neurons may determine their vulnerability to HI injury. It is evident that the anterior raphé subdivisions are more affected by P3 HI than the more posterior and caudally located raphé nuclei such as the raphé magnus and the dorsal raphé interfascicular nuclei [58]. The topographical clustering of different raphé subdivisions in the midbrain and brainstem also represents differential connectivity patterns in the brain. As such the dorsal raphé caudal, dorsal raphé ventrolateral, and dorsal raphé dorsal nuclei primarily innervate the cerebral cortex, basal ganglia, thalamus, hypothalamus, hippocampus, and amygdala [55, 61, 62]. In contrast, the more caudal nuclei predominantly send neural projections to the spinal cord and other parts of the brainstem [63]. The afferent and efferent connections of each raphé subdivision are integral to producing characteristic serotonergic-dependent functions. Thus selective losses of serotonergic raphé nuclei may underpin particular deficits reported in HI-affected preterm infants. On the other hand, it appears that the more caudal raphé magnus and dorsal raphé interfascicular nuclei are not susceptible to P3 HI injury and therefore the serotonergic innervation of the spinal cord remains relatively intact and functional after P3 HI [58]. Indeed previous reports suggest that spinal cord injury only occurs after severe neonatal HI insults [64, 65].
Functional disruption of the serotonergic system after neonatal HI is clearly reflected in the reduction in 5-HT levels in the brain. We and others have demonstrated reduced 5-HT levels in cortical, thalamic, and brainstem regions after HI produced in the immature rodent brain [66, 67]. The losses of brainstem dorsal raphé neurons and their neural projections after HI are most likely responsible for the reduced 5-HT levels in the forebrain. Although regional differences are apparent, the association between direct serotonergic neural inputs to forebrain regions from specific raphé nuclei in the brainstem is not known. Thus determining whether specific ascending and descending neural connections are disrupted after HI injury may predict raphé nuclei vulnerability to P3 HI injury and the effects they have on brain regions innervated by serotonergic afferents and efferents.
In concert with the loss of raphé neurons and reductions in 5-HT levels in the brain, SERT expression is significantly reduced in the brain [58, 67, 68]. We have characterised SERT losses after P3 HI using both Western blot and immunolabelling techniques. The distribution of SERT in fibres, dendrites, cell bodies, and axon terminals [69] makes it an excellent marker of the serotonergic network in the brain [70, 71]. As such the distribution of SERT in the brain closely reflects that of serotonergic neuronal cell bodies and innervating fibres [72, 73]. Serotonergic fibre losses and damage are observed after P3 HI in several key forebrain regions such as the motor and somatosensory cortex, lateral hypothalamus, ventrolateral thalamus, and horizontal limb of the diagonal band [67]. The parallel and concomitant reductions in 5-HT levels and SERT indicate that there was reduced availability of 5-HT for release as well as limited reuptake of 5-HT. This is analogous to findings after ischemia in P7 rat pups whereby there is concurrent attenuation of 5-HT and its major metabolite, 5-hydroxyindoleacetic acid (5-HIAA), suggesting that injury to the serotonergic neuronal network ensues rather than direct modulation of SERT itself or of serotonergic metabolism [66]. We therefore speculate that P3 HI induces disruption to the serotonergic system as a result of loss or damage to serotonergic neurons.
The efficient reuptake of 5-HT is primarily dependent on the localisation of SERT on cell bodies, dendrites, and fibres of serotonergic neurons in the central nervous system [72]. However there is evidence that the reuptake of 5-HT can occur by glial cells whereby SERT may be also localised on astrocytes [74–77] and/or microglial cells [78]. Thus glial SERT could potentially assist in the clearance of 5-HT from the serotonergic synapse [74]. However of the few studies that have specifically examined this possibility, most have only reported localisation of SERT in glial cell lines and primary cultures. Through our own in vivo investigations, we have found no evidence of SERT localisation on microglia or astrocytes in the normal or P3 HI-injured neonatal rodent brain (unpublished).
From our studies it is interesting to note that, in general, proportionately greater serotonergic changes occur in the forebrain regions compared to the brainstem raphé nuclei [58, 67, 68]. This observation has led us to speculate that damage to the serotonergic fibres in the forebrain core/penumbral areas of the HI-injured brain may occur before injury to the brainstem raphé nuclei. In our P3 HI model in the rodent, ligation of the common carotid artery affects a vascular field encompassing primarily forebrain regions, whereas the brainstem lies outside this vascular field and is seemingly spared of immediate hypoxic and ischemic conditions. It has been shown that blood flow to the brainstem tends to increase during HI [79]. In addition we have consistently found that, unlike the forebrain, there is no change in brainstem hemisphere area after P3 HI [28, 58, 67, 68]. The dorsal raphé nuclei can be considered remote from the damaged forebrain sites and therefore serotonergic neuronal injury in the brainstem might develop as a result of secondary mechanisms. One such secondary injury mechanism that we have had a particular focus on is P3 HI-induced neuroinflammation.
6. Role of Neuroinflammation in Producing Neuronal Injury
Two phases of injury can be defined after a neonatal HI insult; an early primary phase within 24–48 h causing mainly irreversible injury in the brain and a later secondary injury phase then ensues. Early neuronal injury after HI is thought to evolve primarily via necrosis resulting from excitotoxic damage produced by excessive release of glutamate from presynaptic nerve terminals and astrocytes, causing calcium overload and cell death [80]. Brain injury during the primary phase can also result from high levels of free radicals including reactive nitrogen species and reactive oxygen species accumulating in the brain tissue [81, 82]. Both caspase-dependent and caspase-independent mediators of cell death are also initiated after neonatal HI [83, 84].
The subsequent secondary phase can continue for weeks, months, or longer after HI. A vast array of mechanisms may contribute to neuronal injury during this phase and the majority of these have been identified as features of neuroinflammation. Key features of this phase include increased numbers of activated microglia, astrogliosis, increased levels of proinflammatory cytokines (e.g., interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), decreased levels of anti-inflammatory cytokines, increased cyclooxygenase (COX-1 and COX-2) expression, prostaglandins (PGE2 and PGI2), nuclear factor kappa-light-chain-enhancer of activated B cells, increased expression of chemokines and chemokine receptors, cell adhesion molecule expression, and matrix metalloproteinases [85–89]. Proinflammatory cytokines, particularly IL-1β and TNF-α, are synthesized and released by activated microglia although IL-1β is also expressed by astrocytes and developing oligodendrocytes [90–92]. Astrocytes are important sources of lactate, neurotrophic factors, and pyruvate for neurons and contribute to maintaining neurotransmitter and metabolic homeostasis in the brain [93, 94]. However, although inhibition of astrogliosis after neonatal HI improves the survival of newborn neurons it does not alter infarct volume [95]. The infiltration of peripheral cells such as lymphocytes, neutrophils, and mast cells can also ensue if there is sufficient breakdown and leakage across the blood-brain barrier [89, 96, 97]. The hallmark feature of neuroinflammation in the HI-affected brain that we have focused on, in terms of a potential mechanism underpinning serotonergic neurodegeneration, is the elevated number of activated microglia.
Numbers of activated microglia peak within the first week after HI although can remain elevated for weeks or months after the initial ischemic episode as observed in human and preclinical studies [68, 84, 89, 91, 98–100]. Microglia are the resident immune cells of the CNS that, in the normal brain, survey the extracellular environment and scavenge and clear the brain of debris and dying cells [101–103]. However microglia can also respond quickly to changes induced by HI in the brain and within 48 h can switch from a resting to an active state, multiply and migrate to sites of ischemic injury [104, 105]. Activated microglia produce and release excessive levels of IL-1β and TNF-α [50, 84, 89, 100, 106], that are toxic to neurons, can cause neurodegeneration, and negatively affect the neurodevelopment of neonates [107, 108]. We have demonstrated that numbers of activated microglia and levels of TNF-α and IL-1β in the brain are elevated over the critical first week after P3 HI, particularly in the cortex, thalamic nuclei, and white matter, and closely parallel injury to the serotonergic system [67, 109]. Thus an association between neuroinflammation and serotonergic injury is evident and the period after the P3 HI insult is a critical window of opportunity for interventions that target neuroinflammation. Two anti-inflammatory drugs are proving to be potential interventions to ameliorate HI-induced damage to the serotonergic system. These are minocycline and ibuprofen.
(a) Effects of Minocycline on the Serotonergic System after Neonatal HI —
The role of activated microglia and raised levels of proinflammatory cytokines in contributing to serotonergic neuronal disruption can be addressed by blocking microglial function with anti-inflammatory drugs. Minocycline is a broad-spectrum antibiotic that also has anti-inflammatory properties in the brain primarily because it is a potent inhibitor of activated microglia [110, 111]. Minocycline does not appear to affect astrocytes after neonatal HI [84, 111, 112]. Minocycline readily crosses the blood-brain barrier after systemic delivery [113, 114] and is an effective neuroprotective intervention when delivered after-insult [50, 67, 83, 115, 116]. The opportunity to alter HI-induced brain injury after the insult is an important prospect because clinical diagnosis of HI in the preterm neonate is often not made until 3 days after birth, well into the secondary injury phase. Furthermore it is difficult to predict if an HI insult is imminent and therefore prophylactic treatments during pregnancy or labour are difficult to administer.
Recent studies in the adult rat demonstrate that minocycline reverses 3-nitropropionic acid neurotoxicity-induced changes in 5-HT levels [117] and reduces the 3,4-methylenedioxymethamphetamine-induced reduction in SERT expression [118]. We have now shown in our neonatal model that minocycline, initiated 2 h after P3 HI and administered daily for 1 week, inhibits P3 HI-induced microglial activation and TNF-α and IL-1β levels and also results in fewer raphé neurons being lost, maintenance of normal 5-HT levels, and increases SERT expression [67]. However not all effects of minocycline on the serotonergic system damage are completely prevented. Furthermore, using the same 1-week-long minocycline regimen, HI-induced neuroinflammation is still inhibited 6 weeks later [68] but minocycline's long-term neuroprotection of the serotonergic system is less effective than at P10. At 6 weeks after-HI SERT expression and serotonergic fibre content appear to be close to control levels but 5-HT levels remain reduced [68]. Nonetheless minocycline, a robust inhibitor of P3 HI-induced neuroinflammatory mediators, significantly improves serotonergic outcomes; however, HI induced damage to the serotonergic network.
Although minocycline treatment could be a novel therapy to minimise serotonergic changes after neonatal HI and preserve the integrity of 5-HT neurocircuitry in the brain, the use of minocycline in neonates is controversial. Minocycline is an excellent tool to block microglial activation and has considerable neuroprotective effects, not only in neonatal HI animal models. Moreover minocycline has proven to be highly beneficial in numerous adult human trials to treat a variety of neurodegenerative conditions [119–122]. Nevertheless its use in human neonates must be undertaken with caution because of the adverse effects associated with chronic tetracycline that historically have tarnished their administration to neonates [123]. Minocycline can produce bone stunting, staining, and pitting of teeth [123–125]. Tetracyclines may also prevent the binding of bilirubin to albumin and possibly lead to bilirubin-induced brain damage in neonates. In contrast, recent studies have demonstrated that minocycline does not produce some of the side effects historically associated with tetracycline use in neonates [126–129]. The development of new derivatives of minocycline, with fewer adverse side effects, could be promising interventions to develop for clinical translation. Alternatively, given the potential of anti-inflammatory interventions to prevent serotonergic injury, testing other anti-inflammatory drugs that may be more clinically acceptable for use in neonates is a rational approach.
(b) Effects of Ibuprofen on the Serotonergic System after Neonatal HI —
Nonsteroidal anti-inflammatory drugs (NSAIDs) constitute an alternative anti-inflammatory treatment to stem brain injury after neonatal HI. In this class, drugs such as ibuprofen and indomethacin are commonly used to treat patent ductus arteriosus in preterm neonates [130, 131]. Ibuprofen is a lipophilic compound and after systemic delivery easily crosses the blood-brain barrier [132]. A canonical mechanism of action of NSAIDs is to inhibit cyclooxygenase 1 and 2 enzymes (COX-1, COX-2) and the conversion and synthesis of arachidonic acid to downstream inflammatory effectors such as cytokines and prostaglandins.
Systemic delivery of ibuprofen can inhibit central neuroinflammation and elicit neuroprotective effects although these outcomes have primarily been demonstrated in adult models of cerebral ischemia [133–136]. In human preterm neonates (<28 weeks gestation) indomethacin reduces white matter loss [137] although ibuprofen combined with ascorbic acid treatment in neonates reportedly has little effect on brain injury after severe HI [138]. Consistent with previous studies [86, 139–142], we have shown that COX-2 is elevated in the brain after P3 HI and that ibuprofen significantly prevents this effect as well as P3 HI-induced increases in numbers of activated microglia, IL-1β, and TNF-α levels [143]. In association with these anti-inflammatory effects ibuprofen ameliorated reductions in cerebral hemisphere size, O4-positive pre-myelinating, O1-positive immature oligodendrocyte cell counts, and myelin content [143].
The potential of ibuprofen to be a neuroprotective agent in neonates to stem HI brain injury is further supported by findings that systemic indomethacin or COX-2 inhibitors (NS398) attenuate inflammatory changes as well as functional impairments after neonatal HI in the rodent [142, 144, 145]. In contrast, the effects of NSAIDs on serotonergic neuronal injury after HI are not known. Preliminary evidence in our preclinical HI model indicates that ibuprofen prevents reductions in SERT expression, 5-HT levels (in the frontal cortex and thalamic nuclei), and serotonergic raphé neuronal counts (unpublished). Our findings suggest that ibuprofen is as effective at preventing serotonergic injury however, like minocycline, it does not appear to completely ameliorate damage to this neuronal network. Thus it is plausible that other mechanisms of injury also contribute to HI-induced serotonergic damage in the neonatal brain.
7. Lack of P3 HI-Induced Neuroinflammatory Mediators in the Brainstem
From our studies, it is interesting to note that a pattern of neuroinflammation is beginning to emerge. The brainstem dorsal raphé and frontal cortex, for example, represent two areas where neuroinflammatory mediator profiles differ markedly. In the frontal cortex substantial and significant increases in activated microglia and proinflammatory cytokines occur after P3 HI. In contrast, in the brainstem, we have observed that the brainstem does not elicit any major signs of neuroinflammation after P3 HI. Numbers of activated microglia are relatively small, there are no apparent changes in proinflammatory cytokines [67] and more recent data from our laboratory indicates there are no changes in COX-2 expression in raphé serotonergic subdivisions (unpublished). It therefore appears that serotonergic raphé cell bodies are not lost because of local neuroinflammation in the brainstem. Thus, even though inhibition of neuroinflammation has a significant beneficial effect on P3 HI-induced losses of raphé neurons [67], we speculate that the effects of anti-inflammatory drugs such as minocycline are not directly effective at the level of the raphé nuclei. We postulate that the losses of 5-HT-positive neurons in the brainstem after P3 HI, and the neuroprotective effects of minocycline are therefore likely to occur via other, indirect secondary mechanisms.
As stated earlier, at least in our model, the brainstem is located outside the vascular field of the common carotid artery and should not be directly affected by immediate changes in perfusion after HI. Instead neuroinflammation could contribute to brainstem injury via remote actions originating from primary injury sites. Inflammatory mediators may damage afferent and efferent fibres of dorsal raphé nuclei in the forebrain and subsequently compromise the survival of brainstem nuclei by retrograde degeneration and/or target deprivation. The thalamus, substantia nigra, hippocampus, and amygdala have substantial neural connections with primary injury sites (e.g., the cerebral cortex) and can undergo prolonged periods of apoptosis and degeneration in the neonatal brain after HI [25–27, 146]. It has been shown that after ischemic conditions, disrupted somatosensory transmission in the thalamus is associated with increased numbers of thalamic neurons degenerating in the secondary phase [26, 147, 148]. Progressive loss of serotonergic neural connections with damaged areas could lead to the disruption and loss of raphé serotonergic neurons in the brainstem. Indeed the regional differences in vulnerability of 5-HT-positive neurons in the dorsal raphé nuclei after P3 HI [58] might be attributed to the serotonergic innervation pattern to damaged and undamaged forebrain regions.
The two mechanisms of HI-induced neuroinflammation and neural disruption may not be mutually exclusive. Activated microglia have also been shown to be present in brain regions as a consequence of a loss of connectivity with a target region or axonal interruption [149–151]. It remains to be investigated whether forebrain neuroinflammation after neonatal HI initiates subsequent serotonergic neuronal damage in the remote brainstem via retrograde degeneration and/or target deprivation mechanisms.
8. Conclusions and Future Directions
We have identified the serotonergic system as a pervasive network that is disrupted after neonatal HI in a rodent model. The concomitant reductions in SERT, 5-HT levels and 5-HT-positive raphé neurons suggest that serotonergic network injury is a consequence of degenerating serotonergic neurons that project to the HI-damaged forebrain. A change in the levels of 5-HT in the brain gives a “readout” of the functional integrity of the serotonergic system. However determining how the synthesis of 5-HT is affected, the storage, release mechanisms, postsynaptic signaling and the breakdown of 5-HT would further our understanding of how HI-injury affects the serotonergic network and possibly reveal new targets for selective interventions. Moreover key components of the serotonergic system have been a critical focus of our recent work, but whether serotonergic changes manifest as specific impairments of neurological performance is not known. It is plausible that disruption of the serotonergic system may underpin impairments such as hyperactivity, cardiorespiratory, cognitive, and attention deficits observed in preterm children who have experienced neonatal HI [4–7, 152]. Also, current theories implicate a disrupted 5-HT neurocircuitry in the brainstem raphé nuclei as the putative underlying mechanism of cardiorespiratory dysfunction in neonates and increased susceptibility to SIDS [41, 153–155]. Being born preterm is a significant risk factor for SIDS [156] and exposure to a HI insult may be sufficient to alter raphé serotonergic function and increase a neonate's susceptibility to later cardiorespiratory complications and possibly SIDS [41, 155].
The serotonergic system does show some degree of recovery weeks after the initial P3 HI insult [58, 68]. Greater density of serotonergic innervation, increased arborization and axonal length, and higher expression of the SERT occur in the postnatal brain; indicating plasticity and temporal differences depending on the region examined [157–159]. It is also remarkable that serotonergic neurons have an ability to sprout and potentially reinnervate after injury [160–162]. In the HI-injured neonatal brain this avenue of investigation remains to be explored, and possibly exploited, to test new therapeutic strategies.
To date, evidence suggests that both minocycline and ibuprofen are successful postinsult interventions to ameliorate neuroinflammation and reducing neuronal loss. Both of these potential anti-inflammatory treatments could be beneficial for HI-induced injury to other neurons in the brain [163–165]. However neither intervention appears to be sufficient to completely reverse the HI-induced decrease in brain 5-HT levels. The dose, timing, and specificity of anti-inflammatory interventions are likely to be key parameters that dictate their success. Alternatively, selectively targeting the serotonergic system to improve its function, in concert with changes produced by anti-inflammatory drugs, could be an ideal combination treatment to achieve long-term improvement of the serotonergic system after neonatal HI. By understanding the fundamental mechanisms of serotonergic damage after neonatal HI we will hopefully move closer to providing a clinical intervention.
References
- 1.Volpe JJ. Brain injury in the premature infant: neuropathology, clinical aspects, and pathogenesis. Mental Retardation and Developmental Disabilities Research Reviews. 1997;3(1):3–12. [Google Scholar]
- 2.Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatric Research. 2001;50(5):553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Larroque B, Ancel PY, Marret S, et al. Neurodevelopmental disabilities and special care of 5-year-old children born before 33 weeks of gestation (the EPIPAGE study): a longitudinal cohort study. The Lancet. 2008;371(9615):813–820. doi: 10.1016/S0140-6736(08)60380-3. [DOI] [PubMed] [Google Scholar]
- 4.Peterson BS, Anderson AW, Ehrenkranz R, et al. Regional brain volumes and their later neurodevelopmental correlates in term and preterm infants. Pediatrics. 2003;111(5):939–948. doi: 10.1542/peds.111.5.939. [DOI] [PubMed] [Google Scholar]
- 5.Anderson P, Doyle LW. Neurobehavioral outcomes of school-age children born extremely low birth weight or very preterm in the 1990s. Journal of the American Medical Association. 2003;289(24):3264–3272. doi: 10.1001/jama.289.24.3264. [DOI] [PubMed] [Google Scholar]
- 6.O’Brien F, Roth S, Stewart A, Rifkin L, Rushe T, Wyatt J. The neurodevelopmental progress of infants less than 33 weeks into adolescence. Archives of Disease in Childhood. 2004;89(3):207–211. doi: 10.1136/adc.2002.006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hack M, Youngstrom EA, Cartar L, et al. Behavioral outcomes and evidence of psychopathology among very low birth weight infants at age 20 years. Pediatrics. 2004;114(4):932–940. doi: 10.1542/peds.2003-1017-L. [DOI] [PubMed] [Google Scholar]
- 8.Logitharajah P, Rutherford MA, Cowan FM. Hypoxic-ischemic encephalopathy in preterm infants: antecedent factors, brain imaging, and outcome. Pediatric Research. 2009;66(2):222–229. doi: 10.1203/PDR.0b013e3181a9ef34. [DOI] [PubMed] [Google Scholar]
- 9.Selway LD. State of the science: hypoxic ischemic encephalopathy and hypothermic intervention for neonates. Advances in Neonatal Care. 2010;10(2):60–66. doi: 10.1097/ANC.0b013e3181d54b30. [DOI] [PubMed] [Google Scholar]
- 10.Pfister RH, Soll RF. Hypothermia for the treatment of infants with hypoxic-ischemic encephalopathy. Journal of Perinatology. 2010;30(1):S82–S87. doi: 10.1038/jp.2010.91. [DOI] [PubMed] [Google Scholar]
- 11.Pierson CR, Folkerth RD, Billiards SS, et al. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathologica. 2007;114(6):619–631. doi: 10.1007/s00401-007-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. The Lancet Neurology. 2009;8(1):110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leviton A, Gressens P. Neuronal damage accompanies perinatal white-matter damage. Trends in Neurosciences. 2007;30(9):473–478. doi: 10.1016/j.tins.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 14.Billiards SS, Haynes RL, Folkerth RD, et al. Development of microglia in the cerebral white matter of the human fetus and infant. Journal of Comparative Neurology. 2006;497(2):199–208. doi: 10.1002/cne.20991. [DOI] [PubMed] [Google Scholar]
- 15.Haynes RL, Folkerth RD, Keefe RJ, et al. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. Journal of Neuropathology and Experimental Neurology. 2003;62(5):441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 16.Nosarti C, Rushe TM, Woodruff PWR, Stewart AL, Rifkin L, Murray RM. Corpus callosum size and very preterm birth: relationship to neuropsychological outcome. Brain. 2004;127(9):2080–2089. doi: 10.1093/brain/awh230. [DOI] [PubMed] [Google Scholar]
- 17.Inder TE, Huppi PS, Warfield S, et al. Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Annals of Neurology. 1999;46(5):755–760. doi: 10.1002/1531-8249(199911)46:5<755::aid-ana11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 18.Ricci D, Anker S, Cowan F, et al. Thalamic atrophy in infants with PVL and cerebral visual impairment. Early Human Development. 2006;82(9):591–595. doi: 10.1016/j.earlhumdev.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Peterson BS, Vohr B, Staib LH, et al. Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. Journal of the American Medical Association. 2000;284(15):1939–1947. doi: 10.1001/jama.284.15.1939. [DOI] [PubMed] [Google Scholar]
- 20.Kesler SR, Ment LR, Vohr B, et al. Volumetric analysis of regional cerebral development in preterm children. Pediatric Neurology. 2004;31(5):318–325. doi: 10.1016/j.pediatrneurol.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zubiaurre-Elorza L, Soria-Pastor S, Junque C, et al. Gray matter volume decrements in preterm children with periventricular leukomalacia. Pediatric Research. 2011;69(6):554–560. doi: 10.1203/PDR.0b013e3182182366. [DOI] [PubMed] [Google Scholar]
- 22.Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatric Research. 2008;63(6):656–661. doi: 10.1203/PDR.0b013e31816c825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larouche A, Roy M, Kadhim H, Tsanaclis AM, Fortin D, Sébire G. Neuronal injuries induced by perinatal hypoxic-ischemic insults are potentiated by prenatal exposure to lipopolysaccharide: animal model for perinatally acquired encephalopathy. Developmental Neuroscience. 2005;27(2–4):134–142. doi: 10.1159/000085985. [DOI] [PubMed] [Google Scholar]
- 24.Marín-Padilla M. Developmental neuropathology and impact of perinatal brain damage. III: gray matter lesions of the neocortex. Journal of Neuropathology and Experimental Neurology. 1999;58(5):407–429. doi: 10.1097/00005072-199905000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Nakajima W, Ishida A, Lange MS, et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. Journal of Neuroscience. 2000;20(21):7994–8004. doi: 10.1523/JNEUROSCI.20-21-07994.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Northington FJ, Ferriero DM, Flock DL, Martin LJ. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. Journal of Neuroscience. 2001;21(6):1931–1938. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oo TF, Henchcliffe C, Burke RE. Apoptosis in substantia nigra following developmental hypoxic-ischemic injury. Neuroscience. 1995;69(3):893–901. doi: 10.1016/0306-4522(95)00282-n. [DOI] [PubMed] [Google Scholar]
- 28.Buller KM, Wixey JA, Pathipati P, et al. Selective losses of brainstem catecholamine neurons after hypoxia-ischemia in the immature rat pup. Pediatric Research. 2008;63(4):364–369. doi: 10.1203/PDR.0b013e3181659774. [DOI] [PubMed] [Google Scholar]
- 29.Loidl CF, Herrera-Marschitz M, Andersson K, et al. Long-term effects of perinatal asphyxia on basal ganglia neurotransmitter systems studied with microdialysis in rat. Neuroscience Letters. 1994;175(1-2):9–12. doi: 10.1016/0304-3940(94)91065-0. [DOI] [PubMed] [Google Scholar]
- 30.Johnston MV, Hudson C. Effects of postnatal hypoxia-ischemia on cholinergic neurons in the developing rat forebrain: choline acetyltransferase immunocytochemistry. Brain Research. 1987;431(1):41–50. doi: 10.1016/0165-3806(87)90193-3. [DOI] [PubMed] [Google Scholar]
- 31.Allen GV, Cheung RTF, Cechetto DF. Neurochemical changes following occlusion of the middle cerebral artery in rats. Neuroscience. 1995;68(4):1037–1050. doi: 10.1016/0306-4522(95)00198-r. [DOI] [PubMed] [Google Scholar]
- 32.Kohlhauser C, Kaehler S, Mosgoeller W, et al. Histological changes and neurotransmitter levels three months following perinatal asphyxia in the rat. Life Sciences. 1999;64(23):2109–2124. doi: 10.1016/s0024-3205(99)00160-5. [DOI] [PubMed] [Google Scholar]
- 33.Herlenius E, Lagercrantz H. Development of neurotransmitter systems during critical periods. Experimental Neurology. 2004;190(1):S8–S21. doi: 10.1016/j.expneurol.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 34.Currás-Collazo MC, Patel UB, Hussein MO. Reduced susceptibility of magnocellular neuroendocrine nuclei of the rat hypothalamus to transient focal ischemia produced by middle cerebral artery occlusion. Experimental Neurology. 2002;178(2):268–279. doi: 10.1006/exnr.2002.8032. [DOI] [PubMed] [Google Scholar]
- 35.Gross J, Müller I, Chen Y, et al. Perinatal asphyxia induces region-specific long-term changes in mRNA levels of tyrosine hydroxylase and dopamine D1 and D2 receptors in rat brain. Molecular Brain Research. 2000;79(1-2):110–117. doi: 10.1016/s0169-328x(00)00106-6. [DOI] [PubMed] [Google Scholar]
- 36.Cheung RTF, Cechetto DF. Colchicine affects cortical and amygdalar neurochemical changes differentially after middle cerebral artery occlusion in rats. Journal of Comparative Neurology. 1997;387(1):27–41. [PubMed] [Google Scholar]
- 37.Gaspar P, Cases O, Maroteaux L. The developmental role of serotonin: news from mouse molecular genetics. Nature Reviews Neuroscience. 2003;4(12):1002–1012. doi: 10.1038/nrn1256. [DOI] [PubMed] [Google Scholar]
- 38.Gartside SE, Johnson DA, Leitch MM, Troakes C, Ingram CD. Early life adversity programs changes in central 5-HT neuronal function in adulthood. European Journal of Neuroscience. 2003;17(11):2401–2408. doi: 10.1046/j.1460-9568.2003.02668.x. [DOI] [PubMed] [Google Scholar]
- 39.Gardner KL, Thrivikraman KV, Lightman SL, Plotsky PM, Lowry CA. Early life experience alters behavior during social defeat: focus on serotonergic systems. Neuroscience. 2005;136(1):181–191. doi: 10.1016/j.neuroscience.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 40.Lowry CA, Johnson PL, Hay-Schmidt A, Mikkelsen J, Shekhar A. Modulation of anxiety circuits by serotonergic systems. Stress. 2005;8(4):233–246. doi: 10.1080/10253890500492787. [DOI] [PubMed] [Google Scholar]
- 41.Duncan JR, Paterson DS, Hoffman JM, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. Journal of the American Medical Association. 2010;303(5):430–437. doi: 10.1001/jama.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nemeroff CB, Owens MJ. The role of serotonin in the pathophysiology of depression: as important as ever. Clinical Chemistry. 2009;55(8):1578–1579. doi: 10.1373/clinchem.2009.123752. [DOI] [PubMed] [Google Scholar]
- 43.Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathology. 2007;17(4):434–447. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ferriero DM. Neonatal brain injury. The New England Journal of Medicine. 2004;351:1985–1995. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- 45.Baumann B, Bielau H, Krell D, et al. Circumscribed numerical deficit of dorsal raphe neurons in mood disorders. Psychological Medicine. 2002;32(1):93–103. doi: 10.1017/s0033291701004822. [DOI] [PubMed] [Google Scholar]
- 46.Rone LA, Ferrando SJ. Serotonin reuptake inhibitor-related extrapyramidal side effects in two patients with cerebral palsy. Psychosomatics. 1996;37(2):165–166. doi: 10.1016/S0033-3182(96)71586-4. [DOI] [PubMed] [Google Scholar]
- 47.Krakovsky G, Huth MM, Lin L, Levin RS. Functional changes in children, adolescents, and young adults with cerebral palsy. Research in Developmental Disabilities. 2007;28(4):331–340. doi: 10.1016/j.ridd.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 48.Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-Ischemic, and excitotoxic insults. Mental Retardation and Developmental Disabilities Research Reviews. 2002;8(1):30–38. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- 49.Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. Journal of Neuroscience. 2001;21(4):1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carty ML, Wixey JA, Colditz PB, Buller KM. Post-insult minocycline treatment attenuates hypoxia-ischemia-induced neuroinflammation and white matter injury in the neonatal rat: a comparison of two different dose regimens. International Journal of Developmental Neuroscience. 2008;26(5):477–485. doi: 10.1016/j.ijdevneu.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 51.Sizonenko SV, Kiss JZ, Inder T, Gluckman PD, Williams CE. Distinctive neuropathologic alterations in the deep layers of the parietal cortex after moderate ischemic-hypoxic injury in the P3 immature rat brain. Pediatric Research. 2005;57(6):865–872. doi: 10.1203/01.PDR.0000157673.36848.67. [DOI] [PubMed] [Google Scholar]
- 52.Walther DJ, Peter JU, Bashammakh S, et al. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299(5603):p. 76. doi: 10.1126/science.1078197. [DOI] [PubMed] [Google Scholar]
- 53.Blakely RD, Berson HE, Fremeau RT, et al. Cloning and expression of a functional serotonin transporter from rat brain. Nature. 1991;353(6348):66–70. doi: 10.1038/354066a0. [DOI] [PubMed] [Google Scholar]
- 54.Dahlström A, Fuxe K. Localization of monoamines in the lower brain stem. Experientia. 1964;20(7):398–399. doi: 10.1007/BF02147990. [DOI] [PubMed] [Google Scholar]
- 55.Hornung JP. The human raphe nuclei and the serotonergic system. Journal of Chemical Neuroanatomy. 2003;26(4):331–343. doi: 10.1016/j.jchemneu.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 56.Kinney HC, Broadbelt KG, Haynes RL, Rognum IJ, Paterson DS. The serotonergic anatomy of the developing human medulla oblongata: implications for pediatric disorders of homeostasis. Journal of Chemical Neuroanatomy. 2011;41(4):182–199. doi: 10.1016/j.jchemneu.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayashi M, Itoh M, Araki S, et al. Immunohistochemical analysis of brainstem lesions in infantile spasms. Neuropathology. 2000;20(4):297–303. doi: 10.1046/j.1440-1789.2000.00353.x. [DOI] [PubMed] [Google Scholar]
- 58.Reinebrant HE, Wixey JA, Gobe GC, Colditz PB, Buller KM. Differential effects of neonatal hypoxic-ischemic brain injury on brainstem serotonergic raphe nuclei. Brain Research. 2010;1322:124–133. doi: 10.1016/j.brainres.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 59.Takeuchi Y, Fujiwara K, Ishimura K, et al. Effects of neonatal asphyxlia on the serotonin neuron system in the developing brain studied by immunohistochemistry. Developmental Neuroscience. 1992;14(5-6):394–402. doi: 10.1159/000111688. [DOI] [PubMed] [Google Scholar]
- 60.Shiraishi M, Takizawa Y, Ide S, Obonai T, Goto YI, Itoh M. Brainstem monoamine pathology of neonatal hypoxic-ischemic brain damage: a model of acute stage of neonatal asphyxia. Brain Research. 2008;1213:120–126. doi: 10.1016/j.brainres.2008.03.068. [DOI] [PubMed] [Google Scholar]
- 61.Kosofsky BE, Molliver ME. The serotoninergic innervation of cerebral cortex: different classes of axon terminals arise from dorsal and median raphe nuclei. Synapse. 1987;1(2):153–168. doi: 10.1002/syn.890010204. [DOI] [PubMed] [Google Scholar]
- 62.Michelsen KA, Schmitz C, Steinbusch HWM. The dorsal raphe nucleus-from silver stainings to a role in depression. Brain Research Reviews. 2007;55(2):329–342. doi: 10.1016/j.brainresrev.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Bowker RM, Westlund KN, Coulter JD. Origins of serotonergic projections to the spinal cord in rat: an immunocytochemical-retrograde transport study. Brain Research. 1981;226(1-2):187–199. doi: 10.1016/0006-8993(81)91092-1. [DOI] [PubMed] [Google Scholar]
- 64.Clancy RR, Stadky JT, Rorke LB. Hypoxic-ischemic spinal cord injury following perinatal asphyxia. Annals of Neurology. 1989;25(2):185–189. doi: 10.1002/ana.410250213. [DOI] [PubMed] [Google Scholar]
- 65.Groenendaal F, Vles J, Lammers H, De Vente J, Smit D, Nikkels PGJ. Nitrotyrosine in human neonatal spinal cord after perinatal asphyxia. Neonatology. 2008;93(1):1–6. doi: 10.1159/000106432. [DOI] [PubMed] [Google Scholar]
- 66.Hadjiconstantinou M, Yates AJ, Neff NH. Hypoxia-induced neurotransmitter deficits in neonatal rats are partially corrected by exogenous GM1 ganglioside. Journal of Neurochemistry. 1990;55(3):864–869. doi: 10.1111/j.1471-4159.1990.tb04571.x. [DOI] [PubMed] [Google Scholar]
- 67.Wixey JA, Reinebrant HE, Buller KM. Inhibition of neuroinflammation prevents injury to the serotonergic network after hypoxia-ischemia in the immature rat brain. Journal of Neuropathology and Experimental Neurology. 2011;70(1):23–35. doi: 10.1097/NEN.0b013e3182020b7b. [DOI] [PubMed] [Google Scholar]
- 68.Wixey JA, Reinebrant HE, Spencer SJ, Buller KM. Efficacy of post-insult minocycline administration to alter long-term hypoxia-ischemia-induced damage to the serotonergic system in the immature rat brain. Neuroscience. 2011;182:184–192. doi: 10.1016/j.neuroscience.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 69.Zhou FC, Tao-Cheng JH, Segu L, Patel T, Wang Y. Serotonin transporters are located on the axons beyond the synaptic junctions: anatomical and functional evidence. Brain Research. 1998;805(1-2):241–254. doi: 10.1016/s0006-8993(98)00691-x. [DOI] [PubMed] [Google Scholar]
- 70.Smith HR, Porrino LJ. The comparative distributions of the monoamine transporters in the rodent, monkey, and human amygdala. Brain Structure and Function. 2008;213(1-2):73–91. doi: 10.1007/s00429-008-0176-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto H, Fujimiya M, Shirai Y, Nakashita M, Oyasu M, Saito N. Immunohistochemical localization of serotonin transporter in normal and colchicine treated rat brain. Neuroscience Research. 1998;32(4):305–312. doi: 10.1016/s0168-0102(98)00097-2. [DOI] [PubMed] [Google Scholar]
- 72.Qian Y, Melikian HE, Rye DB, Levey AI, Blakely RD. Identification and characterization of antidepressant-sensitive serotonin transporter proteins using site-specific antibodies. Journal of Neuroscience. 1995;15(2):1261–1274. doi: 10.1523/JNEUROSCI.15-02-01261.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Murphy DL, Fox MA, Timpano KR, et al. How the serotonin story is being rewritten by new gene-based discoveries principally related to SLC6A4, the serotonin transporter gene, which functions to influence all cellular serotonin systems. Neuropharmacology. 2008;55(6):932–960. doi: 10.1016/j.neuropharm.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dave V, Mullaney KJ, Goderie S, Kimelberg HK, Aschner M. Astrocytes as mediators of methylmercury neurotoxicity: effects on D-aspartate and serotonin uptake. Developmental Neuroscience. 1994;16(3-4):222–231. doi: 10.1159/000112110. [DOI] [PubMed] [Google Scholar]
- 75.Bel N, Figueras G, Vilaró MT, Suñol C, Artigas F. Antidepressant drugs inhibit a glial 5-hydroxytryptamine transporter in rat brain. European Journal of Neuroscience. 1997;9(8):1728–1738. doi: 10.1111/j.1460-9568.1997.tb01530.x. [DOI] [PubMed] [Google Scholar]
- 76.Kubota N, Kiuchi Y, Nemoto M, et al. Regulation of serotonin transporter gene expression in human glial cells by growth factors. European Journal of Pharmacology. 2001;417(1-2):69–76. doi: 10.1016/s0014-2999(01)00906-2. [DOI] [PubMed] [Google Scholar]
- 77.Inazu M, Takeda H, Ikoshi H, Sugisawa M, Uchida Y, Matsumiya T. Pharmacological characterization and visualization of the glial serotonin transporter. Neurochemistry International. 2001;39(1):39–49. doi: 10.1016/s0197-0186(01)00010-9. [DOI] [PubMed] [Google Scholar]
- 78.Horikawa H, Kato TA, Mizoguchi Y, et al. Inhibitory effects of SSRIs on IFN-γ induced microglial activation through the regulation of intracellular calcium. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2010;34(7):1306–1316. doi: 10.1016/j.pnpbp.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 79.Vannucci RC, Lyons DT, Vasta F. Regional cerebral blood flow during hypoxia-ischemia in immature rats. Stroke. 1988;19(2):245–250. doi: 10.1161/01.str.19.2.245. [DOI] [PubMed] [Google Scholar]
- 80.Choi DW. Excitotoxicity, apoptosis, and ischemic stroke. Journal of Biochemistry and Molecular Biology. 2001;34(1):8–14. [Google Scholar]
- 81.Wayenberg JL, Ransy V, Vermeylen D, Damis E, Bottari SP. Nitrated plasma albumin as a marker of nitrative stress and neonatal encephalopathy in perinatal asphyxia. Free Radical Biology and Medicine. 2009;47(7):975–982. doi: 10.1016/j.freeradbiomed.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 82.Buonocore G, Groenendaal F. Anti-oxidant strategies. Seminars in Fetal and Neonatal Medicine. 2007;12(4):287–295. doi: 10.1016/j.siny.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 83.Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Annals of Neurology. 2002;52(1):54–61. doi: 10.1002/ana.10242. [DOI] [PubMed] [Google Scholar]
- 84.Cai Z, Lin S, Fan LW, Pang Y, Rhodes PG. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137(2):425–435. doi: 10.1016/j.neuroscience.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 85.Kremlev SG, Roberts RL, Palmer C. Minocycline modulates chemokine receptors but not interleukin-10 mRNA expression in hypoxic-ischemic neonatal rat brain. Journal of Neuroscience Research. 2007;85(11):2450–2459. doi: 10.1002/jnr.21380. [DOI] [PubMed] [Google Scholar]
- 86.Toti P, De Felice C, Schürfeld K, et al. Cyclooxygenase-2 immunoreactivity in the ischemic neonatal human brain: an autopsy study. Journal of Submicroscopic Cytology and Pathology. 2001;33(3):245–249. [PubMed] [Google Scholar]
- 87.Yin W, Signore AP, Iwai M, et al. Preconditioning suppresses inflammation in neonatal hypoxic ischemia via Akt activation. Stroke. 2007;38(3):1017–1024. doi: 10.1161/01.STR.0000258102.18836.ca. [DOI] [PubMed] [Google Scholar]
- 88.Šumanović-Glamuzina D, Čulo F, Čulo M-I, et al. Vasodilatory prostaglandins in perinatal hypoxic brain damage. Collegium Antropologicum. 2008;32:183–187. [PubMed] [Google Scholar]
- 89.Bona E, Andersson AL, Blomgren K, et al. Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatric Research. 1999;45(4):500–509. doi: 10.1203/00006450-199904010-00008. [DOI] [PubMed] [Google Scholar]
- 90.Girard S, Sébire G, Kadhim H. Proinflammatory orientation of the interleukin 1 system and downstream induction of matrix metalloproteinase 9 in the pathophysiology of human perinatal white matter damage. Journal of Neuropathology and Experimental Neurology. 2010;69(11):1116–1129. doi: 10.1097/NEN.0b013e3181f971e4. [DOI] [PubMed] [Google Scholar]
- 91.Kadhim H, Tabarki B, Verellen G, De Prez C, Rona AM, Sébire G. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology. 2001;56(10):1278–1284. doi: 10.1212/wnl.56.10.1278. [DOI] [PubMed] [Google Scholar]
- 92.Vela JM, Molina-Holgado E, Arévalo-Martín Á, Almazán G, Guaza C. Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Molecular and Cellular Neuroscience. 2002;20(3):489–502. doi: 10.1006/mcne.2002.1127. [DOI] [PubMed] [Google Scholar]
- 93.Dringen R, Gebhardt R, Hamprecht B. Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Research. 1993;623(2):208–214. doi: 10.1016/0006-8993(93)91429-v. [DOI] [PubMed] [Google Scholar]
- 94.Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Canadian Journal of Physiology and Pharmacology. 1997;75(10-11):1149–1163. [PubMed] [Google Scholar]
- 95.Järlestedt K, Rousset CI, Faiz M, et al. Attenuation of reactive gliosis does not affect infarct volume in neonatal hypoxic-ischemic brain injury in mice. PLoS ONE. 2010;5(4) doi: 10.1371/journal.pone.0010397. Article ID e10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jin Y, Silverman AJ, Vannucci SJ. Mast cells are early responders after hypoxia-ischemia in immature rat brain. Stroke. 2009;40(9):3107–3112. doi: 10.1161/STROKEAHA.109.549691. [DOI] [PubMed] [Google Scholar]
- 97.Brochu M-E, Girard S, Lavoie K, Sébire G. Developmental regulation of the neuroinflammatory responses to LPS and/or hypoxia-ischemia between preterm and term neonates: an experimental study. Journal of Neuroinflammation. 2011;20:55–69. doi: 10.1186/1742-2094-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huh Y, Jung JW, Park C, et al. Microglial activation and tyrosine hydroxylase immunoreactivity in the substantia nigral region following transient focal ischemia in rats. Neuroscience Letters. 2003;349(1):63–67. doi: 10.1016/s0304-3940(03)00743-2. [DOI] [PubMed] [Google Scholar]
- 99.Bhat RV, DiRocco R, Marcy VR, et al. Increased expression of IL-1β converting enzyme in hippocampus after ischemia: selective localization in microglia. Journal of Neuroscience. 1996;16(13):4146–4154. doi: 10.1523/JNEUROSCI.16-13-04146.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Silveira RC, Procianoy RS. Interleukin-6 and tumour necrosis factor-a levels in plasma and cerebral spinal fluid of term newborn infants with hypoxic-ischemic encephalopathy. Journal of Pediatrics. 2003;143:625–629. doi: 10.1067/S0022-3476(03)00531-6. [DOI] [PubMed] [Google Scholar]
- 101.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 102.Raivich G. Like cops on the beat: the active role of resting microglia. Trends in Neurosciences. 2005;28(11):571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 103.Kettenmann H. Neuroscience: the brain’s garbage men. Nature. 2007;446(7139):987–989. doi: 10.1038/nature05713. [DOI] [PubMed] [Google Scholar]
- 104.Ivacko JA, Sun R, Silverstein FS. Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatric Research. 1996;39(1):39–47. doi: 10.1203/00006450-199601000-00006. [DOI] [PubMed] [Google Scholar]
- 105.McRae A, Gilland E, Bona E, Hagberg H. Microglia activation after neonatal hypoxic-ischemia. Developmental Brain Research. 1995;84(2):245–252. doi: 10.1016/0165-3806(94)00177-2. [DOI] [PubMed] [Google Scholar]
- 106.Biran V, Joly L-M, Héron A, et al. Glial activation in white matter following ischemia in the neonatal P7 rat brain. Experimental Neurology. 2006;199(1):103–112. doi: 10.1016/j.expneurol.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 107.Basu A, Lazovic J, Krady JK, et al. Interleukin-1 and the interleukin-1 type 1 receptor are essential for the progressive neurodegeneration that ensues subsequent to a mild hypoxic/ischemic injury. Journal of Cerebral Blood Flow and Metabolism. 2005;25(1):17–29. doi: 10.1038/sj.jcbfm.9600002. [DOI] [PubMed] [Google Scholar]
- 108.Bartha AI, Miller SP, Foster-Barber A, et al. Cytokines are associated with cerebral metabolism and outcome in term neonatal encephalopathy. Developmental Neuroscience. 2005;27, article 270 [Google Scholar]
- 109.Wixey JA, Reinebrant HE, Carty ML, Buller KM. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. Journal of Neuroimmunology. 2009;212(1-2):35–43. doi: 10.1016/j.jneuroim.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 110.Amin AR, Attur MG, Thakker GD, et al. A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):14014–14019. doi: 10.1073/pnas.93.24.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brogden RN, Speight TM, Avery GS. Minocycline: a review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs. 1975;9(4):251–291. doi: 10.2165/00003495-197509040-00005. [DOI] [PubMed] [Google Scholar]
- 114.Macdonald H, Kelly RG, Allen ES. Pharmacokinetic studies on minocycline in man. Clinical Pharmacology and Therapeutics. 1973;14(5):852–861. doi: 10.1002/cpt1973145852. [DOI] [PubMed] [Google Scholar]
- 115.Leonardo CC, Eakin AK, Ajmo JM, et al. Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia-ischemia in the neonatal rat. Journal of Neuroinflammation. 2008;5:34–45. doi: 10.1186/1742-2094-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lechpammer M, Manning SM, Samonte F, et al. Minocycline treatment following hypoxic/ischaemic injury attenuates white matter injury in a rodent model of periventricular leucomalacia. Neuropathology and Applied Neurobiology. 2008;34(4):379–393. doi: 10.1111/j.1365-2990.2007.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ahuja M, Bishnoi M, Chopra K. Protective effect of minocycline, a semi-synthetic second-generation tetracycline against 3-nitropropionic acid (3-NP)-induced neurotoxicity. Toxicology. 2008;244(2-3):111–122. doi: 10.1016/j.tox.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 118.Zhang L, Shirayama Y, Shimizu E, Iyo M, Hashimoto K. Protective effects of minocycline on 3,4-methylenedioxymethamphetamine-induced neurotoxicity in serotonergic and dopaminergic neurons of mouse brain. European Journal of Pharmacology. 2006;544(1–3):1–9. doi: 10.1016/j.ejphar.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 119.Zabad RK, Metz LM, Todoruk TR, et al. The clinical response to minocycline in multiple sclerosis is accompanied by beneficial immune changes: a pilot study. Multiple Sclerosis. 2007;13(4):517–526. doi: 10.1177/1352458506070319. [DOI] [PubMed] [Google Scholar]
- 120.Lampl Y, Boaz M, Gilad R, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69(14):1404–1410. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 121.Griffin MO, Fricovsky E, Ceballos G, Villarreal F. Tetracyclines: a pleitropic family of compounds with promising therapeutic properties. Review of the literature. American Journal of Physiology. 2010;299(3):C539–C548. doi: 10.1152/ajpcell.00047.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Plane JM, Shen Y, Pleasure DE, Deng W. Prospects for minocycline neuroprotection. Archives of Neurology. 2010;67(12):1442–1448. doi: 10.1001/archneurol.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Buller KM, Carty ML, Reinebrant HE, Wixey JA. Minocycline: a neuroprotective agent for hypoxic-ischemic brain injury in the neonate? Journal of Neuroscience Research. 2009;87(3):599–608. doi: 10.1002/jnr.21890. [DOI] [PubMed] [Google Scholar]
- 124.Laurence DR, Bennett PN, editors. Clinical Pharmacology. 6th edition. New York, NY, USA: Churchill Livingstone; 1987. [Google Scholar]
- 125.Hardman JG, Limbird LE, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 9th edition. New York, NY, USA: The Mcgraw-Hill Companies; 1996. [Google Scholar]
- 126.Cascio A, Di Liberto C, D'Angelo M, et al. No findings of dental defects in children treated with minocycline. Antimicrobial Agents and Chemotherapy. 2004;48(7):2739–2741. doi: 10.1128/AAC.48.7.2739-2741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Geiger AS, Rice AC, Shapiro SM. Minocycline blocks acute bilirubin-induced neurological dysfunction in jaundiced Gunn rats. Neonatology. 2007;92(4):219–226. doi: 10.1159/000103740. [DOI] [PubMed] [Google Scholar]
- 128.Lin S, Wei X, Bales KR, et al. Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. European Journal of Neuroscience. 2005;22(1):21–27. doi: 10.1111/j.1460-9568.2005.04182.x. [DOI] [PubMed] [Google Scholar]
- 129.Shapiro SM, Bhutani VK, Johnson L. Hyperbilirubinemia and kernicterus. Clinics in Perinatology. 2006;33(2):387–410. doi: 10.1016/j.clp.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 130.Aranda JV, Thomas R. Systematic review: intravenous ibuprofen in preterm newborns. Seminars in Perinatology. 2006;30(3):114–120. doi: 10.1053/j.semperi.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 131.Varvarigou A, Bardin CL, Beharry K, Chemtob S, Papageorgiou A, Aranda JV. Early ibuprofen administration to prevent patent ductus arteriosus in premature newborn infants. Journal of the American Medical Association. 1996;275(7):539–544. [PubMed] [Google Scholar]
- 132.Parepally JMR, Mandula H, Smith QR. Brain uptake of nonsteroidal anti-inflammatory drugs: ibuprofen, flurbiprofen, and indomethacin. Pharmaceutical Research. 2006;23(5):873–881. doi: 10.1007/s11095-006-9905-5. [DOI] [PubMed] [Google Scholar]
- 133.Park EM, Cho BP, Volpe BT, Cruz MO, Joh TH, Cho S. Ibuprofen protects ischemia-induced neuronal injury via up-regulating interleukin-1 receptor antagonist expression. Neuroscience. 2005;132(3):625–631. doi: 10.1016/j.neuroscience.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 134.Antezana DF, Clatterbuck RE, Alkayed NJ, et al. High-dose ibuprofen for reduction of striatal infarcts during middle cerebral artery occlusion in rats. Journal of Neurosurgery. 2003;98(4):860–866. doi: 10.3171/jns.2003.98.4.0860. [DOI] [PubMed] [Google Scholar]
- 135.Patel PM, Drummon JC, Sano T, Cole DJ, Kalkman CJ, Yaksh TL. Effect of ibuprofen on regional eicosanoid production and neuronal injury after forebrain ischemia in rats. Brain Research. 1993;614(1-2):315–324. doi: 10.1016/0006-8993(93)91050-3. [DOI] [PubMed] [Google Scholar]
- 136.Cole DJ, Patel PM, Reynolds L, Drummond JC, Marcantonio S. Temporary focal cerebral ischemia in spontaneously hypertensive rats: the effect of ibuprofen on infarct volume. Journal of Pharmacology and Experimental Therapeutics. 1993;266(3):1713–1717. [PubMed] [Google Scholar]
- 137.Miller SP, Mayer EE, Clyman RI, Glidden DV, Hamrick SEG, Barkovich AJ. Prolonged indomethacin exposure is associated with decreased white matter injury detected with magnetic resonance imaging in premature newborns at 24 to 28 weeks’ gestation at birth. Pediatrics. 2006;117(5):1626–1631. doi: 10.1542/peds.2005-1767. [DOI] [PubMed] [Google Scholar]
- 138.Aly H, Abd-Rabboh L, El-Dib M, et al. Ascorbic acid combined with ibuprofen in hypoxic ischemic encephalopathy: a randomized controlled trial. Journal of Perinatology. 2009;29(6):438–443. doi: 10.1038/jp.2009.1. [DOI] [PubMed] [Google Scholar]
- 139.Lim GP, Yang F, Chu T, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for alzheimer’s disease. Journal of Neuroscience. 2000;20(15):5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yokota C, Kaji T, Kuge Y, Inoue H, Tamaki N, Minematsu K. Temporal and topographic profiles of cyclooxygenase-2 expression during 24 h of focal brain ischemia in rats. Neuroscience Letters. 2004;357(3):219–222. doi: 10.1016/j.neulet.2003.12.109. [DOI] [PubMed] [Google Scholar]
- 141.Candelario-Jalil E, Fiebich BL. Cyclooxygenase inhibition in ischemic brain injury. Current Pharmaceutical Design. 2008;14(14):1401–1418. doi: 10.2174/138161208784480216. [DOI] [PubMed] [Google Scholar]
- 142.Fathali N, Ostrowski RP, Lekic T, et al. Cyclooxygenase-2 inhibition provides lasting protection against neonatal hypoxic-ischemic brain injury. Critical Care Medicine. 2010;38(2):572–578. doi: 10.1097/CCM.0b013e3181cb1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Carty ML, Wixey JA, Reinebrant HE, Gobe G, Colditz PB, Buller KM. Ibuprofen inhibits neuroinflammation and attenuates white matter damage following hypoxia-ischemia in the immature rodent brain. Brain Research. 2011;1402:9–19. doi: 10.1016/j.brainres.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 144.Taskin E, Ozcan K, Canacankatan N, Satar M, Yapıcıoğlu H, Erdoğan Ş. The effects of indomethacin on caspases, glutathione level and lipid peroxidation in the newborn rats with hypoxic-ischemic cerebral injury. Brain Research. 2009;1289:118–123. doi: 10.1016/j.brainres.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 145.Tutak E, Satar M, Zorludemir S, Erdoğan Ş, Yapıcıoğlu H, Narlı N. Neuroprotective effects of indomethacin and aminoguanidine in the newborn rats with hypoxic-ischemic cerebral injury. Neurochemical Research. 2005;30(8):937–942. doi: 10.1007/s11064-005-5978-5. [DOI] [PubMed] [Google Scholar]
- 146.Cheng Y, Deshmukh M, D’Costa A, et al. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. Journal of Clinical Investigation. 1998;101(9):1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dihné M, Grommes C, Lutzenburg M, Witte OW, Block F. Different mechanisms of secondary neuronal damage in thalamic nuclei after focal cerebral ischemia in rats. Stroke. 2002;33(12):3006–3011. doi: 10.1161/01.str.0000039406.64644.cb. [DOI] [PubMed] [Google Scholar]
- 148.Tokuno T, Kataoka K, Asai T, et al. Functional changes in thalamic relay neurons after focal cerebral infarct: a study of unit recordings from VPL neurons after MCA occlusion in rats. Journal of Cerebral Blood Flow and Metabolism. 1992;12(6):954–961. doi: 10.1038/jcbfm.1992.132. [DOI] [PubMed] [Google Scholar]
- 149.Banati RB, Cagnin A, Brooks DJ, et al. Long-term trans-synaptic glial responses in the human thalamus after peripheral nerve injury. Neuroreport. 2001;12(16):3439–3442. doi: 10.1097/00001756-200111160-00012. [DOI] [PubMed] [Google Scholar]
- 150.Cho BP, Song DY, Sugama S, et al. Pathological dynamics of activated microglia following medial forebrain bundle transection. GLIA. 2006;53(1):92–102. doi: 10.1002/glia.20265. [DOI] [PubMed] [Google Scholar]
- 151.Block F, Dihné M, Loos M. Inflammation in areas of remote changes following focal brain lesion. Progress in Neurobiology. 2005;75(5):342–365. doi: 10.1016/j.pneurobio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 152.Marlow N, Rose AS, Rands CE, Draper ES. Neuropyschological and educational problems at school age associated with neonatal encephalopathy. Archives of Disease in Childhood. 2005;90:380–387. doi: 10.1136/adc.2004.067520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Paterson DS, Belliveau RA, Trachtenberg F, Kinney HC. Differential development of 5-HT receptor and the serotonin transporter binding in the human infant medulla. Journal of Comparative Neurology. 2004;472(2):221–231. doi: 10.1002/cne.20105. [DOI] [PubMed] [Google Scholar]
- 154.Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. Journal of the American Medical Association. 2006;296(17):2124–2132. doi: 10.1001/jama.296.17.2124. [DOI] [PubMed] [Google Scholar]
- 155.Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annual Review of Pathology: Mechanisms of Disease. 2009;4:517–550. doi: 10.1146/annurev.pathol.4.110807.092322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Malloy MH, Hoffman HJ. Prematurity, sudden infant death syndrome, and age of death. Pediatrics. 1995;96(3):464–471. [PubMed] [Google Scholar]
- 157.Tarazi FI, Tomasini EC, Baldessarini RJ. Postnatal development of dopamine and serotonin transporters in rat caudate-putamen and nucleus accumbens septi. Neuroscience Letters. 1998;254(1):21–24. doi: 10.1016/s0304-3940(98)00644-2. [DOI] [PubMed] [Google Scholar]
- 158.Moll GH, Mehnert C, Wicker M, et al. Age-associated changes in the densities of presynaptic monoamine transporters in different regions of the rat brain from early juvenile life to late adulthood. Developmental Brain Research. 2000;119(2):251–257. doi: 10.1016/s0165-3806(99)00182-0. [DOI] [PubMed] [Google Scholar]
- 159.Galineau L, Kodas E, Guilloteau D, Vilar MP, Chalon S. Ontogeny of the dopamine and serotonin transporters in the rat brain: an autoradiographic study. Neuroscience Letters. 2004;363(3):266–271. doi: 10.1016/j.neulet.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 160.Zhou FC, Azmitia EC, Bledsoe S. Rapid serotonergic fiber sprouting in response to ibotenic acid lesion in the striatum and hippocampus. Developmental Brain Research. 1995;84(1):89–98. doi: 10.1016/0165-3806(94)00158-v. [DOI] [PubMed] [Google Scholar]
- 161.Inman DM, Steward O. Ascending sensory, but not other long-tract axons, regenerate into the connective tissue matrix that forms at the site of a spinal cord injury in mice. Journal of Comparative Neurology. 2003;462(4):431–449. doi: 10.1002/cne.10768. [DOI] [PubMed] [Google Scholar]
- 162.Hawthorne AL, Hu H, Kundu B, et al. The unusual response of serotonergic neurons after CNS injury: lack of axonal dieback and enhanced sprouting within the inhibitory environment of the glial scar. Journal of Neuroscience. 2011;31(15):5605–5616. doi: 10.1523/JNEUROSCI.6663-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Guo G, Bhat NR. p38α MAP kinase mediates hypoxia-induced motor neuron cell death: a potential target of minocycline’s neuroprotective action. Neurochemical Research. 2007;32(12):2160–2166. doi: 10.1007/s11064-007-9408-8. [DOI] [PubMed] [Google Scholar]
- 164.Fan LW, Lin S, Pang Y, Rhodes PG, Cai Z. Minocycline attenuates hypoxia-ischemia-induced neurological dysfunction and brain injury in the juvenile rat. European Journal of Neuroscience. 2006;24(2):341–350. doi: 10.1111/j.1460-9568.2006.04918.x. [DOI] [PubMed] [Google Scholar]
- 165.Matsukawa N, Yasuhara T, Hara K, et al. Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC Neuroscience. 2009;10, article 1471:p. 126. doi: 10.1186/1471-2202-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]