

Abstract
The c-Jun NH2-terminal protein kinase (JNK) plays important roles in a broad range of physiological processes. JNK is controlled by two upstream regulators, mitogen-activated protein kinase kinase (MKK) 4 and MKK7, which are activated by various MAPKKKs. Studies employing knockout mice have demonstrated that the JNK signaling pathway is involved in diverse phenomena in the brain, regulating brain development and maintenance as well as animal metabolism and behavior. Furthermore, examination of single or combined knockout mice of Jnk1, Jnk2, and Jnk3 has revealed both functional differences and redundancy among JNK1, JNK2, and JNK3. Phenotypic differences between knockouts of MKK4 and MKK7 have also been observed, suggesting that the JNK signaling pathway in the brain has a complex nature and is intricately regulated. This paper summarizes the functional properties of the major JNK signaling components in the developing and adult brain.
1. Introduction
The c-Jun NH2-terminal protein kinases (JNKs, also called stress-activated protein kinases (SAPKs)) are members of the evolutionarily conserved mitogen-activated protein kinase (MAPK) family [1, 2]. The JNK subfamily consists of three related genes: Jnk1, Jnk2, and Jnk3. In mammals, the JNK1 and JNK2 proteins are ubiquitously expressed, whereas JNK3 is found almost exclusively in the brain and testis. JNKs are activated by many types of external stress, including heat shock, UV irradiation, and inflammatory cytokines. JNKs phosphorylate numerous important substrates, including the transcription factors AP-1 and c-Jun, various apoptotic proteins, and microtubule-associated proteins (MAPs) [3–7]. Through phosphorylation of these substrates, JNKs regulate gene expression governing stress responses as well as the normal physiological processes of cell proliferation, apoptosis, differentiation, and cell migration.
Activation of JNK is catalyzed by two kinases, mitogen-activated protein kinase kinase (MKK) 4 and MKK7 [8–10]. Although MKK4 and MKK7 are both dual-specificity Thr and Tyr kinases, previous studies of JNK activation have shown that MKK4 preferentially phosphorylates the Tyr residue of the TPY motif in the activation loop of JNKs, whereas MKK7 preferentially phosphorylates the Thr residue [11, 12]. The activation of MKK4 and MKK7 is mediated by various MAPKKKs, including mixed lineage protein kinases (MLKs), apoptosis signal-regulating kinases (ASKs) and dual leucine zipper kinase (DLK) [13]. In addition to regulation by these upstream kinases, the JNK signaling pathway is modulated by various scaffold proteins, including JNK-interacting protein (JIP) 1, JIP2, and JIP3 (also known as JNK/SAPK associated protein-1 (JSAP1)) [14–17]. These scaffold proteins assemble multienzyme complexes that involve a specific triad of a MAPKKK, a MAPKK, and a MAPK and provide an insulated physical conduit for signal transduction. The resulting linkage of these kinases forms a functional signaling module that performs transduction duties for a particular purpose.
In most mammalian cell types, JNK activation is tightly controlled and employed at moderate levels in specific circumstances. In contrast, JNK signaling in the brain is highly and constitutively activated. Studies of gene knockout (KO) mice lacking JNKs or their upstream kinases have shown that this sustained activation is necessary to fulfil the diverse and essential roles that JNK signaling plays in the brain (Figure 1). This paper summarizes the phenotypes of these mutant animals and discusses what they reveal about the regulation and various functions of JNK and MKK signaling in the brain.
Figure 1.
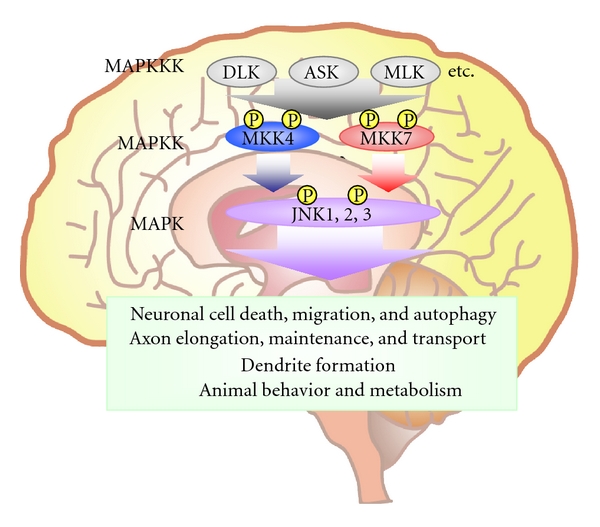
The JNK signaling pathway in the mammalian brain. A MAPKKK, such as DLK, ASK, or MLK, phosphorylates (P) and activates a MAPKK such as MKK4 or MKK7. An activated MAPKK in turn phosphorylates and activates a MAPK such as JNK1, JNK2, or JNK3. Activated JNKs then regulate various phenomena in the brain.
2. Brain Phenotypes of JNK Knockout Mice
2.1. Jnk1/2 DKO Mice: JNK1/2 Induces Programmed Cell Death during Early Brain Development
Knockout mice lacking one, two, or all JNK isoforms have been generated over the past decade, and their diverse phenotypes have been reported (Table 1). Importantly, embryonic lethality is not caused by the single KO of any Jnk gene. Two strains of double KO mice, Jnk1−/−Jnk3−/−(Jnk1/3 DKO) and Jnk2−/−Jnk3−/−(Jnk2/3 DKO), are viable, but Jnk1 −/− Jnk2−/− (Jnk1/2 DKO) animals exhibit early embryonic lethality.
Table 1.
Phenotypes of JNK knockout mice.
| Mouse model | Phenotypes | References |
|---|---|---|
| Jnk1−/− | Accelerated radial migration Shorter dendrites with increased branching Degeneration of anterior commissure Enhanced glucocorticoid- or insulin-induced food intake |
[20–22, 31] |
|
| ||
| Jnk2−/− | Resistance to MPTP-induced neuronal cell death | [24] |
|
| ||
| Jnk3−/− | Resistance to MPTP-induced neuronal cell death Resistance to kainic acid-induced neuronal cell death Resistance to ischemia-induced neuronal apoptosis Resistance to 6-hydrozydopamine-induced neuronal apoptosis |
[24–27, 29] |
|
| ||
| Jnk1−/− Jnk2−/− | Embryonic lethal at E11.5 Defective neural tube closure Dysregulated apoptosis in brain (increased in forebrain, reduced in hindbrain) |
[18, 19] |
|
| ||
| Jnk2−/− Jnk3−/− | Resistance to MPTP-induced neuronal cell death | [24] |
|
| ||
|
Jnk1flox/flox
Nestin-Cre |
Animal protected from diet-induced glucose intolerance and insulin resistance Reduced serum IGF-1 and GH Increased serum thyroid hormones |
[30] |
|
| ||
|
Jnk1flox/floxJnk2−/−
Jnk3−/− Nestin-Cre |
Early embryonic lethal | [53] |
|
| ||
|
Jnk1flox/floxJnk2−/−
Jnk3−/− Pcp2-Cre |
Loss of dendritic arborization Axon hypertrophy Increased autophagic vacuoles |
[53] |
During early brain development in mice, the neural folds emerge at embryonic day (E) 7.5 in the cephalic region, close over at E8.5–E9 (neural tube closure), and form the neural tube. The Jnk1/2 DKO mouse is lethal at E11.5 due to dysregulation of apoptosis in the neural tube [18, 19]. Specifically, there is a marked reduction in cell death in the lateral edges of the hindbrain prior to neural tube closure. In contrast, increased apoptosis and caspase activation are found in the mutant forebrain, leading to precocious degeneration. Interestingly, about 25% of Jnk1−/−Jnk2+/− fetuses display excencephaly that is likely the result of failed neural tube closure, whereas Jnk1+/−Jnk2−/− mice are normal [19]. These results suggest that JNK1 and JNK2 are redundant regulators of programmed cell death during early brain development and that JNK1 is dominant in this particular function of the JNK signaling pathway.
2.2. Jnk1−/− Mice: JNK1 Is Required for Neuronal Migration, Dendrite Formation, and Axon Maintenance during Later Brain Development
After neural tube formation at E8.5–E9, three of its vesicles give rise to various specialized regions of the brain. Neuronal progenitors proliferate and generate immature neurons. These cells subsequently migrate from the proliferative zones to their final positions, where they extend neurites. For example, in the developing cortex, neural progenitors proliferate from E11 to E17 then generate immature neurons that migrate and form the cortical layers, the cortical plate, subplate, intermediate zone, and ventricular zone. The extension and maturation of neurites by these cells then continues during the remaining embryonic and postnatal stages. Jnk1−/− embryos display thicker cortical plates and thinner ventricular zones than wild-type animals [20]. Jnk1−/− embryos also show abnormally accelerated migration of cortical neurons (radial migration), a process whose precise regulation is required for the correct formation of cortical layers. These findings indicate that JNK1 regulates the rate of neuronal migration during cortical development.
Other studies have shown that JNK1 also regulates neurite formation and maintenance. Neurons extend two types of neurites: dendrites and axons. Compared to wild-type mice, dendrites in the cortex and cerebellum of Jnk1−/− mice are shorter and have more processes [21], indicating that JNK1 plays an important role in defining dendritic architecture during brain development. In contrast, axon formation is not affected by JNK1 disruption. Axon tracts such as the corpus callosum and anterior commissure appear normal in Jnk1−/− mice until postnatal day (P) 6. From P6-P12, however, the anterior commissure degenerates, demonstrating that JNK1 is required for axon maintenance [22].
One of the molecular mechanisms mediating JNK pathway functions during brain development and maintenance is the phosphorylation of MAPs such as MAP1B, MAP2, and superior cervical ganglion 10 (SCG10) [20–22]. MAPs bind to microtubules (MTs) and modulate their stability and structure. Importantly, the phosphorylation of MAPs by one of several protein kinases (including JNK) regulates the binding of these enzymes to MTs and thus the regulation of MT modification. In Jnk1−/− brain, the phosphorylation of MAP1B, MAP2, and SCG10 is reduced. Moreover, the introduction of a mutated SCG10 protein that mimics the JNK1-phosphorylated form restores normal neuronal migration in the brains of Jnk1−/− embryos [20]. These results demonstrate that, in addition to regulating radial migration, dendrite formation, and axon maintenance in the developing brain, JNK1 controls MT structure through MAP phosphorylation.
2.3. Jnk2−/− and Jnk3−/− Mice: JNK2 and/or JNK3 Are Required for Neuronal Cell Death Induced by Neuronal Stresses in Adult Brain
The redundancy of many JNK functions is highlighted by the fact that Jnk2−/− and Jnk3−/− single KO mice show none of the defects observed in Jnk1−/− mice. However, Jnk2−/− and Jnk3−/− mice do show alterations in neuronal stress-induced neuronal cell death that are rarely observed in Jnk1−/− mice, implying that each JNK enzyme has a unique function. For example, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a neurotoxin whose administration induces dopaminergic cell demise and so results in symptoms that replicate most of the neuropathological hallmarks of Parkinson's disease [23]. Jnk2−/− or Jnk3−/− mice (but not Jnk1−/− mice) display resistance to MPTP-induced neuronal cell death in vivo [24]. Moreover, this resistance to MPTP is enhanced by double mutation of Jnk2 and Jnk3, indicating that JNK2 and JNK3 play partially overlapping roles in this process. Jnk3−/− mice also display resistance to kainic acid-induced cell death (excitotoxicity-induced apoptosis) and ischemia-induced cell death in vivo [25–27], as well as reduced susceptibility to apoptosis induced by the Alzheimer's disease-related protein beta-amyloid in vitro [28]. Finally, Jnk3−/− mice intrastriatally injected with 6-hydroxydopamine, which provokes the death of dopaminergic neurons, show a transient prolongation of dopaminergic neuron survival in the substantia nigra compacta compared to injected control mice [29]. These results suggest that the physiological function of both JNK2 and JNK3 in the brain is to induce apoptosis in response to neuronal stress.
2.4. Conditional Jnk1−/− Mice: JNK1 Activity in Adult Brain Regulates Animal Metabolism
Intact JNK signaling in the adult brain is required for normal animal metabolism. A high-fat diet induces JNK activation in the hypothalamus and pituitary, which regulate body weight control, glucose homeostasis, and secretion of hormones. Conditional KO (cKO) mice have been generated in which Jnk1 expression is controlled by Nestin-Cre, which induces Cre recombinase expression in neural stem cells [30]. Upon high-fat feeding, Jnk1flox/flox Nestin-Cre mice exhibit increased insulin sensitivity (compared to wild-type controls) both in the CNS and in peripheral tissues, improved glucose metabolism and protection from hepatic steatosis and adipose tissue dysfunction. Jnk1flox/flox Nestin-Cre mice also display reduced somatic growth and altered secretion of growth hormone (GH), insulin-like growth factor (IGF), and thyroid hormones. JNK activity in the brain is thus required for normal metabolism.
JNK signaling in the brain is also involved in the control of feeding. The hypothalamus governs food intake in response to nutrient status, cytokines, and hormones. Jnk1−/− mice exhibit enhanced food intake and weight gain upon hypothalamic administration of glucocorticoid [31]. Moreover, JNK1 disruption increases mouse sensitivity to the anorexigenic effects of hypothalamic insulin administration. These data show that JNK signaling in the brain regulates not only hormone secretion and peripheral metabolism but also feeding and thus contributes to the maintenance of energy homeostasis. It remains unclear whether JNK2 and/or JNK3 are also involved in these functions, but improved insulin resistance has been reported for both Jnk1+/−Jnk2−/− mice and Jnk1−/− mice fed a high-fat diet [32, 33]. These results suggest that JNK2 may be involved in the central and/or peripheral regulation of glucose homeostasis. Analyses of cKO mice lacking Jnk1 and/or Jnk2 will resolve this issue.
3. Brain Phenotypes of MKK Knockout Mice
Because JNK isoforms are required for brain development, and MKK4 and MKK7 activate all JNK isoforms, it is not surprising that disruption of Mkk4 and/or Mkk7 results in severe defects in mouse development. Mkk4−/−Mkk7−/− (Mkk4/7 DKO) mice die at E8.5 before neural tube formation, and both Mkk4−/− and Mkk7−/− single KO mice are embryonic lethal at around E11.5 due to impaired liver formation [9, 34–36]. To circumvent this limitation, the functions of MKK4 and MKK7 during brain development have been investigated using Nestin-Cre cKO mice lacking MKK4 and/or MKK7. Analyses of these mutants have proven very helpful in overcoming the conundrum posed by the redundancy of JNK isoforms.
3.1. Mkk4flox/flox Nestin-Cre Mice: MKK4 Is Required for Neuronal Migration and Axon Maintenance in the Developing Brain
In Mkk4flox/flox Nestin-Cre mice, total JNK activation in the brain is reduced to 20% of normal but these animals are not embryonic lethal and dysregulated apoptosis is not observed [37]. At birth, Mkk4flox/flox Nestin-Cre mice are indistinguishable from their control littermates, but the mutants stop growing a few days later and die at around 3 weeks of age. The brains of Mkk4flox/flox Nestin-Cre mice display misaligned Purkinje cells in the cerebellum and delayed radial migration in the cerebral cortex. In their commissural axon tracts, axonal degeneration is observed not only in the anterior commissure, the site of a similar defect in Jnk1−/− brain [22], but also in the corpus callosum. At the molecular level, hypophosphorylation of MAP1B is observed in Mkk4flox/flox Nestin-Cre mice, suggesting that dysregulation of MT dynamics is involved in these phenotypes. The altered neuronal cell migration and axon maintenance observed in Mkk4flox/flox Nestin-Cre mice are also seen in Jnk1−/− mice, but these phenotypes are less severe in the latter. Thus, MKK4 is a regulator of radial migration and axon maintenance in the brain, and MKK4's effects are mediated not only by JNK1 but also by JNK2 and/or JNK3.
3.2. Mkk7flox/flox Nestin-Cre Mice: MKK7 Is Required for Neuronal Migration and Axon Elongation in Developing Brain
Our group has generated Mkk7flox/flox Nestin-Cre mice [38]. Unlike Mkk4flox/flox Nestin-Cre mice, which survive until age 3 weeks, Mkk7flox/flox Nestin-Cre mice die at birth without breathing. Like Mkk4flox/flox Nestin-Cre mice, JNK activation is reduced to 20% of normal in the developing brain of Mkk7flox/flox Nestin-Cre mutants, and a delay in neuronal migration in the cerebrum is observed. However, other phenotypes do not overlap between Mkk7flox/flox Nestin-Cre and Mkk4flox/flox Nestin-Cre mice (Table 2). At E18.5, Mkk7flox/flox Nestin-Cre mice display enlarged brain ventricles, diminished striatum, decreased forebrain axon tracts, and reduced corticofugal axons; none of these defects has been found in Mkk4flox/flox Nestin-Cre mice. In addition, ultrastructural alterations such as abnormal accumulations of filamentous structures and autophagic vacuoles are observed in Mkk7flox/flox Nestin-Cre brain but not in Mkk4flox/flox Nestin-Cre brain. Thus, Mkk7 has unique functions in the developing brain that differ from those of MKK4.
Table 2.
Comparison of phenotypes of Mkk4 flox/flox Nestin-Cre and Mkk7 flox/flox Nestin-Cre mice.
| Phenotype | Mkk4flox/flox Nestin-Cre [37] | Mkk7flox/flox Nestin-Cre [38] |
|---|---|---|
| Neuronal JNK activity | Suppressed | Suppressed |
| Age of lethality | Around 3 weeks old | At birth |
| Brain ventricle size at E18.5 | Not reported | Enlarged |
| Striatum | Not reported | Reduced |
| Axon tracts (E18.5) | Unaffected | Greatly reduced |
| Axon tracts (postnatal) | Less fasciculated | Not determined |
| Intermediate filaments | Not reported | Accumulate in axons |
| Autophagic vacuoles | Not reported | Accumulate |
| JNK phosphorylation in axons | Not reported | Suppressed |
| L1-positive axons (E18.5) | Unaffected | Reduced |
| TAG-1-positive axons (E18.5) | Not reported | Reduced |
| Apoptosis | Unaffected | Unaffected |
| Cortical layer markers | Not reported | Expressed |
| Radial migration | Delayed | Delayed |
| Phosphorylation of c-Jun | Suppressed | Suppressed |
| Phosphorylation of NF-H | Suppressed | Suppressed |
| Phosphorylation of MAP1B | Suppressed | Suppressed |
| Phosphorylation of DCX | Unaffected | Suppressed |
Differences between MKK7 and MKK4 functions also appear at the molecular level. In Mkk4flox/flox Nestin-Cre brain, phosphorylation levels of MAP1B are reduced but DCX phosphorylation is not altered. In contrast, phosphorylation levels of both MAP1B and DCX are decreased in Mkk7flox/flox Nestin-Cre brain, suggesting that the MKK7-JNK and MKK4-JNK signaling modules in this organ are not identical. In line with this hypothesis, the scaffold protein JIP1 binds to JNK, MKK7, and DCX but not to MKK4. We therefore propose that differences in scaffold proteins and/or substrates involved in the MKK7-JNK versus MKK4-JNK pathways could cause the phenotypic divergence observed between Mkk7flox/flox Nestin-Cre and MKK4flox/flox Nestin-Cre mice (Figure 2).
Figure 2.
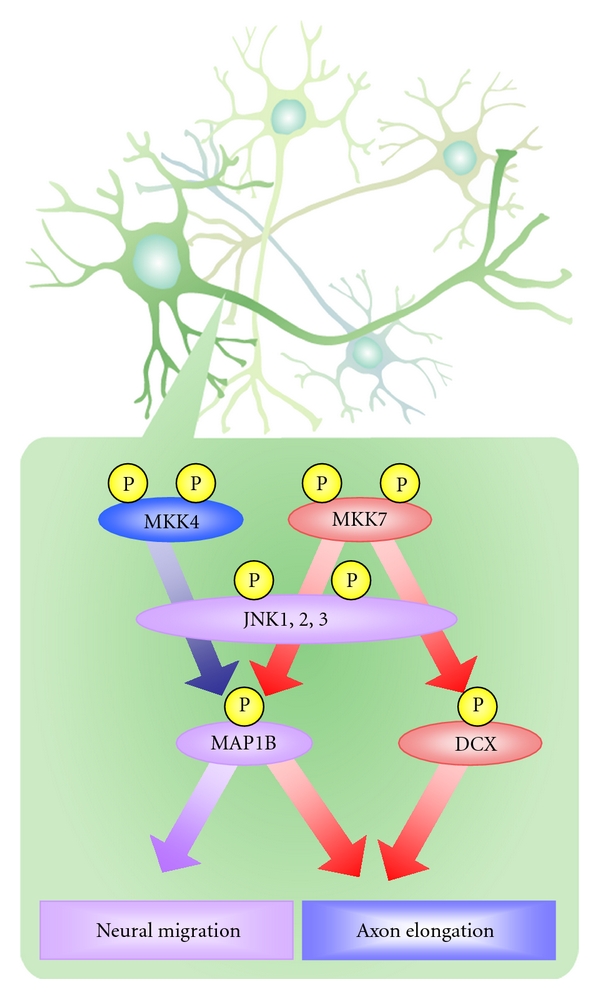
MKK4 and MKK7 have different functions in the developing brain. MKK4 and MKK7 both activate JNKs, which phosphorylate MT-associated proteins such as MAP1B and DCX. Activated MAP1B and DCX regulate neuronal migration and axon elongation in the developing brain. However, JNK activated by MKK4 regulates radial migration but not axon elongation, whereas JNK activated by MKK7 controls both radial migration and axon elongation. These differences between MKK7 and MKK4 functions also appear at the molecular level. The phosphorylation of MAP1B requires that JNK be activated by both MKK4 and MKK7. However, the phosphorylation of DCX requires JNK activation only by MKK7 and not by MKK4.
4. Brain Phenotypes of MAPKKK Knockout Mice
Compared with MAPKKs, MAPKKKs comprise a much larger family of related proteins. Indeed, at least 12 MAPKKKs have been identified as regulating various steps of the JNK signaling pathway [13]. This multiplicity of related functions suggests that each MAPKKK has a specific spatiotemporal role in controlling JNK signaling. However, the precise mechanisms by which most MAPKKKs regulate JNKs and their biological roles in the brain have yet to be fully elucidated. We summarize below evidence supporting the importance of two MAPKKKs, DLK, and ASK1, in the JNK signaling pathway in the brain.
4.1. Dlk−/− Mice: DLK Is Required for Axon Elongation and Neuronal Migration in Developing Brain
DLK is known to be critical in the developing mammalian brain. Dlk−/− mice die perinatally, with no homozygous mutant surviving until weaning. Dlk−/− mice display retarded radial migration and impaired fiber tract development by neocortical pyramidal neurons in the cerebrum [40]. Direct and quantitative analysis of pyramidal neuron radial migration using slice culture and a time-lapse imaging system has revealed that Dlk disruption affects acceleration around the cortical subplate. Furthermore, in vitro culture of Dlk−/− neurons has shown that DLK is involved in the establishment of neuronal polarity and regulates the MT dynamics driving the transition of stage 1 (nonpolar) neurons to stage 2 (multipolar) neurons and then the transition of stage 2 to stage 3 (axon-forming) neurons [41]. These results demonstrate that DLK regulates radial migration and axon formation during brain development. However, a reduction of only 30% in total phosphorylated JNK is observed in Dlk−/− brain, indicating that other MAPKKKs must be involved in JNK activation in this organ. The identities of these enzymes are under active investigation.
4.2. Ask1−/− Mice: ASK1 Drives Neuronal Cell Death in Adult Brain Following Ischemia or Neurodegeneration and Regulates Animal Behavior
ASK1 is well known as a proapoptotic MAPKKK that is involved in responses to diverse stresses and activates both the JNK and p38 signaling pathways [42]. Ask1−/− mice are born at the expected Mendelian frequency and show no developmental abnormalities as determined by histological analysis [43]. Retinal ganglion cells of Ask1−/− mice do not readily undergo ischemia-induced apoptosis in vivo [44], and Ask1−/− primary neurons display resistance to neuronal cell death triggered by polyglutamine in vitro [45]. In mice transgenic for a mutation of Cu/Zn-superoxide dismutase, which serve as a model of human amyotrophic lateral sclerosis, deletion of ASK1 mitigates motor neuron loss and extends mouse lifespan [46]. These data indicate that ASK1's proapoptotic functions extend to neurons in vivo. ASK1 also has nonapoptotic functions in the brain, since Ask1−/− mice exhibit temporary hyperactivity in an open-field test [47]. Interestingly, this hyperactivity is specific to the novel environment, with Ask1−/− mice displaying normal activities in the familiar field. Ask1−/− mice also show impaired novelty preference at 24 hours after training but superior performance on the rotarod test. These results demonstrate that ASK1 is involved in locomotor activity, novelty preference, and motor coordination requiring dopaminergic transmission.
5. Other Evidence Supporting Roles for JNK Signaling in the Nervous System
5.1. Regulation of Axonal Transport
In Drosophila, an absence of the function of either Bsk (Drosophila JNK) or Hep (Drosophila MKK7) causes a failure of lateral epithelial cells to stretch such that the embryo develops a hole in the dorsal cuticle [48–50]. These data indicate that the MKK7-JNK signaling pathway mediates cell migration in Drosophila and regulates dorsal closure during early morphogenesis.
Axonal transport in Drosophila is driven along MT by the kinesin motor system, and APLIP (Drosophila JIP1) is known as a “cargo linker” that joins kinesin-1 to various vesicle proteins such as the Drosophila equivalent of the Alzheimer's APP protein [51]. Drosophila axonal transport is believed to be regulated by the JNK signaling pathway because mutation of Wnd (Drosophila DLK), Hep, or Bsk results in the abnormal accumulation of synaptic vesicles in the nerves of third instar larvae [39]. Mutation of Wnd or Hep also disrupts the binding of kinesin-1 to APLIP1. Thus, the JNK signaling pathway regulates the attachment of APLIP1 to kinesin-1 in Drosophila, ensuring that the dissociation of kinesin-1 from its vesicle protein cargo occurs at the appropriate position (Figure 3(a)).
Figure 3.
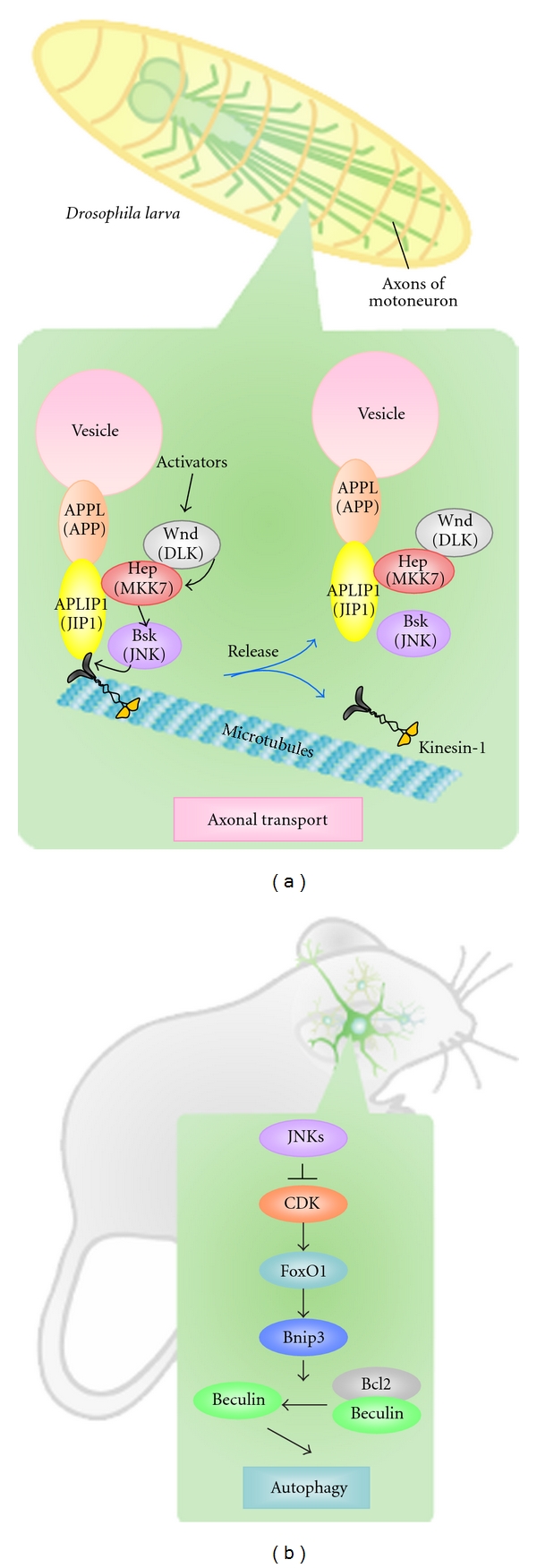
The JNK signaling pathway regulates axonal transport and autophagy. (a) Model of how the JNK signaling pathway controls axonal transport in Drosophila. Axonal transport is driven along MT by kinesin-1, which binds to the vesicles that make up its cargo through APLIP1 (Drosophila JIP1) and APPL (APP). This process is driven by Wnd (DLK), which is activated by unknown upstream signals, and phosphorylates Hep (MKK7). Activated Hep then phosphorylates and activates Bsk (JNK), which then directly or indirectly modifies the linkage complex and causes APLIP1 and the cargo to dissociate from kinesin-1. (This figure is excerpted from [39] with some modifications.) (b) Model of the regulation of neuronal autophagy in mice. In normal neurons, constitutively activated JNKs suppress CDK-induced FoxO1 activation, preventing autophagy. When all three of JNK1, JNK2, and JNK3 are disrupted, CDK-mediated FoxO1 activation increases Bnip3 expression. High levels of Bnip inhibit the binding of Beclin1 to Bcl2, and this freshly released Beclin1 induces autophagy.
The involvement of the JNK signaling pathway in axonal transport has also been reported in mammals. JNK can be activated by the pathogenic polyglutamine-containing Huntingtin protein associated with human Huntington's disease. Fast axonal transport is then inhibited because JNK3 directly phosphorylates the kinesin-1 motor domain [52]. It remains to be determined how extensively this molecular mechanism is conserved between Drosophila and mammals.
5.2. Regulation of Neuronal Autophagy
Conditional Jnk1, Jnk2, Jnk3 triple knockout mice (Jnk1/2/3 cTKO mice) have been generated to eliminate the problem of functional redundancy among JNK1, 2, and 3 [53]. These animals were created by crossing Jnk1 flox mice with Jnk2−/−, Jnk3−/−, and Cre-expressing strains. Jnk1/2/3 cTKO Nestin-Cre mice are embryonic lethal at an early stage, but Jnk1/2/3 cTKO Pcp2-Cre mice, which express Cre in Purkinje cells, are viable. While numerous abnormal phenotypes have been reported for Jnk1/2/3 cTKO Pcp2-Cre mice, including axon hypertrophy, abnormal mitochondrial transport, and prolonged cellular lifespan in culture, the most striking anomaly observed has been increased neuronal autophagy [53]. Results to date indicate that, in neurons, the JNK signaling pathway suppresses autophagy, whereas, in nonneuronal cells, JNK signaling either induces autophagy or serves as an effector of autophagy-associated cell death [54, 55]. Although the JNK substrate leading to autophagy has yet to be identified, it has been shown that increased autophagy in Jnk1/2/3 cTKO neurons is mediated by FoxO1 and not by an mTORC1-dependent mechanism [53]. The combined disruption of JNK1, JNK2, and JNK3 increases Bnip3 expression through FoxO1 activation that is mediated by CDK (Figure 3(b)). Bnip3 inhibits Beclin1-Bcl2 binding and induces Beclin1 release, which in turn triggers autophagy.
6. Conclusion and Future Perspectives
Studies of KO mice lacking JNKs, MAPKKs or MAPKKKs, have revealed that the JNK signaling pathway is involved in diverse roles in the brain, including induction of neuronal cell death, radial migration, neurite formation, metabolism regulation, and behavioral control. Twelve MAPKKKs, two MAPKs, and three JNKs can be combined in triads with specific scaffold proteins to form a large array of JNK signaling modules. These various modules are believed to facilitate JNK participation in specific biological functions. Indeed, the difference in phenotypes displayed by Mkk4flox/flox Nestin-Cre and Mkk7flox/flox Nestin-Cre mice reinforces the notion that there are distinct JNK signaling modules that may be MKK4 dependent or MKK7 dependent. Thus, to truly elucidate the JNK signaling network, it will be necessary to investigate JNK signaling at every step of its hierarchy: MAPKs, MAPKKs, and MAPKKKs.
The brain contains many different cell types, including various classes of neurons, astrocytes, and oligodendrocytes. To date, cell type-specific analyses of JNK signaling have not been performed. Future reviews will no doubt focus on the use of cell type-specific cKO mice to further dissect the striking reach of JNK signaling in normal and abnormal physiology.
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Ministry of Health, Labour and Welfare of Japan, and the Japan Society for the Promotion of Science.
References
- 1.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 2.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 3.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes and Development. 1993;7(11):2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 4.Gdalyahu A, Ghosh I, Levy T, et al. DCX, a new mediator of the JNK pathway. EMBO Journal. 2004;23(4):823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawauchi T, Chihama K, Nabeshima YI, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO Journal. 2003;22(16):4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tararuk T, Östman N, Li W, et al. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. Journal of Cell Biology. 2006;173(2):265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiology and Molecular Biology Reviews. 2006;70(4):1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochimica et Biophysica Acta. 2007;1773(8):1349–1357. doi: 10.1016/j.bbamcr.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Asaoka Y, Nishina H. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. Journal of Biochemistry. 2010;148(4):393–401. doi: 10.1093/jb/mvq098. [DOI] [PubMed] [Google Scholar]
- 10.Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. European Journal of Cell Biology. 2011;90(6-7):536–544. doi: 10.1016/j.ejcb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Lawler S, Fleming Y, Goedert M, Cohen P. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Current Biology. 1998;8(25):1387–1390. doi: 10.1016/s0960-9822(98)00019-0. [DOI] [PubMed] [Google Scholar]
- 12.Kishimoto H, Nakagawa K, Watanabe T, et al. Different properties of SEK1 and MKK7 in dual phosphorylation of stress-induced activated protein kinase SAPK/JNK in embryonic stem cells. Journal of Biological Chemistry. 2003;278(19):16595–16601. doi: 10.1074/jbc.M213182200. [DOI] [PubMed] [Google Scholar]
- 13.Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26(22):3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- 14.Dickens M, Rogers JS, Cavanagh J, et al. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277(5326):693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 15.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Molecular and Cellular Biology. 1999;19(10):7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito M, Yoshioka K, Akechi M, et al. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a scaffold factor in the JNK signaling pathway. Molecular and Cellular Biology. 1999;19(11):7539–7548. doi: 10.1128/mcb.19.11.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelkar N, Gupta S, Dickens M, Davis RJ. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Molecular and Cellular Biology. 2000;20(3):1030–1043. doi: 10.1128/mcb.20.3.1030-1043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22(4):667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 19.Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mechanisms of Development. 1999;89(1-2):115–124. doi: 10.1016/s0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- 20.Westerlund N, Zdrojewska J, Padzik A, et al. Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nature Neuroscience. 2011;14(3):305–313. doi: 10.1038/nn.2755. [DOI] [PubMed] [Google Scholar]
- 21.Björkblom B, Östman N, Hongisto V, et al. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. Journal of Neuroscience. 2005;25(27):6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang L, Jones Y, Ellisman MH, Goldstein LSB, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Developmental Cell. 2003;4(4):521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- 23.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 24.Hunot S, Vila M, Teismann P, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(2):665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang DD, Kuan CY, Whitmarsh AJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389(6653):865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 26.Kuan CY, Whitmarsh AJ, Yang DD, et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(25):15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pirianov G, Brywe KG, Mallard C, et al. Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury. Journal of Cerebral Blood Flow and Metabolism. 2007;27(5):1022–1032. doi: 10.1038/sj.jcbfm.9600413. [DOI] [PubMed] [Google Scholar]
- 28.Morishima Y, Gotoh Y, Zieg J, et al. β-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of fas ligand. Journal of Neuroscience. 2001;21(19):7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brecht S, Kirchhof R, Chromik A, et al. Specific pathophysiological functions of JNK isoforms in the brain. European Journal of Neuroscience. 2005;21(2):363–377. doi: 10.1111/j.1460-9568.2005.03857.x. [DOI] [PubMed] [Google Scholar]
- 30.Belgardt BF, Mauer J, Wunderlich FT, et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(13):6028–6033. doi: 10.1073/pnas.1001796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Unger EK, Piper ML, Olofsson LE, Xu AW. Functional role of c-Jun-N-terminal kinase in feeding regulation. Endocrinology. 2010;151(2):671–682. doi: 10.1210/en.2009-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(28):10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirosumi J, Tuncman G, Chang L, et al. A central, role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 34.Nishina H, Vaz C, Billia P, et al. Defective liver formation and liver cell apoptosis in mice lacking the stress signaling kinase SEK1/MKK4. Development. 1999;126(3):505–516. doi: 10.1242/dev.126.3.505. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe T, Nakagawa K, Ohata S, et al. SEK1/MKK4-mediated SAPK/JNK signaling participates in embryonic hepatoblast proliferation via a pathway different from NF-κB-induced anti-apoptosis. Developmental Biology. 2002;250(2):332–347. [PubMed] [Google Scholar]
- 36.Wada T, Joza N, Cheng HM, et al. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nature Cell Biology. 2004;6(3):215–226. doi: 10.1038/ncb1098. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Nadarajah B, Robinson AC, et al. Targeted deletion of the mitogen-activated protein kinase kinase 4 gene in the nervous system causes severe brain developmental defects and premature death. Molecular and Cellular Biology. 2007;27(22):7935–7946. doi: 10.1128/MCB.00226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamasaki T, et al. Stress-activated protein kinase MKK7 regulates axon elongation in the developing cerebral cortex. The Journal of Neuroscience. 2011;31(46):16872–16883. doi: 10.1523/JNEUROSCI.1111-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horiuchi D, Collins CA, Bhat P, Barkus RV, DiAntonio A, Saxton WM. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Current Biology. 2007;17(15):1313–1317. doi: 10.1016/j.cub.2007.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirai SI, De FC, Miyata T, et al. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. Journal of Neuroscience. 2006;26(46):11992–12002. doi: 10.1523/JNEUROSCI.2272-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirai S-I, Banba Y, Satake T, Ohno S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-jun N-terminal kinase pathway. Journal of Neuroscience. 2011;31(17):6468–6480. doi: 10.1523/JNEUROSCI.5038-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 43.Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Reports. 2001;2(3):222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harada C, Nakamura K, Namekata K, et al. Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis in vivo. American Journal of Pathology. 2006;168(1):261–269. doi: 10.2353/ajpath.2006.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes and Development. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishitoh H, Kadowaki H, Nagai A, et al. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes and Development. 2008;22(11):1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumakura K, Nomura H, Toyoda T, et al. Hyperactivity in novel environment with increased dopamine and impaired novelty preference in apoptosis signal-regulating kinase 1 (ASK1)-deficient mice. Neuroscience Research. 2010;66(3):313–320. doi: 10.1016/j.neures.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Glise B, Bourbon H, Noselli S. hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell. 1995;83(3):451–461. doi: 10.1016/0092-8674(95)90123-x. [DOI] [PubMed] [Google Scholar]
- 49.Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E. The Drosophila jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes and Development. 1996;10(21):2759–2768. doi: 10.1101/gad.10.21.2759. [DOI] [PubMed] [Google Scholar]
- 50.Sluss HK, Han Z, Barrett T, Davis RJ, Ip YT. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes and Development. 1996;10(21):2745–2758. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- 51.Taru H, Iijima KI, Hase M, Kirino Y, Yagi Y, Suzuki T. Interaction of Alzheimer’s β-amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. Journal of Biological Chemistry. 2002;277(22):20070–20078. doi: 10.1074/jbc.M108372200. [DOI] [PubMed] [Google Scholar]
- 52.Morfini GA, You YM, Pollema SL, et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nature Neuroscience. 2009;12(7):864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes and Development. 2011;25(4):310–322. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu L, Alva A, Su H, et al. Regulation of an ATG7-beclin 1 program of autophaglic cell death by caspase-8. Science. 2004;304(5676):1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 55.Shimizu S, Konishi A, Nishida Y, et al. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29(14):2070–2082. doi: 10.1038/onc.2009.487. [DOI] [PubMed] [Google Scholar]