

Abstract
The principal components of neuronal excitability include synaptic and intrinsic membrane parameters. While recent studies indicate that cocaine exposure can induce widespread changes in synaptic function in the neural circuits for reward, intrinsic firing properties have received much less attention. Using whole-cell recording in ex vivo brain slices from cocaine-treated mice, we studied the intrinsic firing characteristics of medium-spiny projection neurons of the nucleus accumbens—a key node in the circuit that controls reward-directed behavior. Our data demonstrate that repeated in vivo cocaine (5 × 15 mg/kg, i.p., once daily, 5 d) induces opposite changes in neurons of the two main subdivisions of the accumbens, the shell and the core. While shell neurons exhibit an initial depression in firing capacity (1–3 d abstinence) that persists for at least 2 weeks, core neurons exhibit increased firing capacity during early abstinence (1–3 d) that declines to basal levels within 2 weeks. Shared adaptations between addictive drugs may mediate core processes of addiction. We find that amphetamine exposure (5 × 5 mg/kg, i.p., once daily, 5 d) that induced a similar degree of locomotor sensitization as cocaine also induced an indistinguishable pattern of NAc intrinsic plasticity. Finally, we provided evidence that opposite regulation of A-type potassium current is an important factor in this bidirectional intrinsic plasticity for both cocaine and amphetamine. We propose that a persistent disparity in core/shell excitability might be an important mediator of the changes in reward circuit activity that drive drug-seeking behavior in animal models of addiction.
Introduction
Medium-spiny projection neurons (MSNs), the principal cells of the nucleus accumbens (NAc), adapt to repeated cocaine exposure through widespread and persistent changes in parameters that determine a neuron's excitability. For example, glutamate and dopamine signaling change and dendritic arbors gain new branches and spines (Robinson and Kolb, 2004; Kauer and Malenka, 2007). However, knowledge is lacking about the ability of cocaine and other drugs to produce long-lasting changes in a fundamental determinant of neuronal excitability: intrinsic membrane properties. Plasticity in intrinsic excitability is recognized as an important means through which experience modifies neural circuits (Alkon, 1984; Zhang and Linden, 2003), and is thought to play an important role in models of multiple CNS disorders (Bonci and Carlezon, 2005; Krishnan et al., 2007; Beck and Yaari, 2008; Santini et al., 2008).
Repeated in vivo cocaine alters several sodium, calcium, and potassium conductances in NAc MSNs during the first few days of abstinence, each of which is consistent with a decrease in depolarization-induced firing (Zhang et al., 1998, 2002; Hu et al., 2004; Dong et al., 2006)—an effect observed during days to weeks of cocaine abstinence (Dong et al., 2006; Kourrich et al., 2008; Ishikawa et al., 2009). Decreasing NAc MSN excitability by viral enhancement of potassium currents produces a hypersensitivity to cocaine's psychomotor effects—a characteristic cocaine-induced adaptation in behavior (Dong et al., 2006). On the other hand, increased cocaine psychomotor sensitization and conditioned-place preference appears to be associated with increased excitability in NAc MSNs in another genetic manipulation study (Benavides et al., 2007). These intriguing, apparently dichotomous, results suggest a need for further investigation of how intrinsic excitability of accumbens neurons is modified by in vivo cocaine experience itself. First, do neurons in the two major subdivisions of the NAc, core and shell, respond similarly to repeated cocaine? Previous studies did not explicitly compare firing adaptations between core and shell. Although these two types of MSNs share many electrophysiological and morphological properties (Meredith et al., 1992; O'Donnell and Grace, 1993), they have different afferent inputs and efferent projections and are thought to serve different functions in shaping reward-related behavior (Zahm, 2000; Ito et al., 2004). Second, one approach to establishing cellular substrates for addiction-related behavior is to identify shared adaptations between different drugs that produce a common behavioral output. Thus, we tested whether cocaine and amphetamine produce similar effects on NAc intrinsic excitability. Finally, we investigated whether mechanisms for intrinsic plasticity induced by these drugs might also be shared.
To address these issues, we used whole-cell recording from NAc MSNs in an ex vivo slice preparation from mice treated with repeated cocaine or amphetamine. To exclusively assess intrinsic membrane properties, we blocked synaptic transmission and measured the firing produced by depolarizing current injection in MSNs from NAc shell and core at early (1–3 d) and late (10–14 d) time points following cessation of drug treatment.
Materials and Methods
Drug treatment regimen.
Male C57BL/6J mice (4–6 weeks of age) were habituated to the animal colony for 1 week before testing. On each of the five consecutive testing days (between 11:00 A.M. and 2:00 P.M.), mice were transferred from the animal colony to a testing room and placed individually into activity boxes (clear rectangular tubs, 8.5 × 17.5 × 9 inches). They were habituated for 20 min before receiving an intraperitoneal injection of cocaine (COC; 15 mg/kg, n = 10), amphetamine (AMPH; 5 mg/kg, n = 10), or saline (SAL; 0.9% NaCl, n = 13) in a volume of 5 ml/kg. Immediately after each injection, horizontal locomotor activity was monitored for 40 min via a digital video-based tracking system (ANY-maze, Stoelting).
Electrophysiology.
Sagittal slices of the NAc shell and core (240 μm) were prepared as described previously (Thomas et al., 2001) from psychostimulant- and saline-treated mice (7–10 weeks of age). Shell recordings were made from medial shell slices that did not contain any dorsal striatal tissue (∼0.44–0.52 mm lateral), while core recordings were made from slices between ∼0.72 and 0.96 mm (see Fig. 1B for additional details) (Paxinos and Franklin, 2001). Slices recovered in a holding chamber for at least 1 h before use. During recording they were superfused with ACSF (22–23°C) saturated with 95% O2/5% CO2 and containing (in mm) 119 NaCl, 2.5 KCl, 1.0 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26.2 NaHCO3, and 11 glucose. Picrotoxin (100 μm) was added to block GABAA receptor-mediated IPSCs. Either a combination of CNQX (10 μm) and d-AP5 (50 μm) or kynurenic acid (2 mm) was used to block glutamatergic transmission during recording. Cells were visualized using infrared-differential interference contrast optics. Medium spiny neurons were identified by morphology and the presence of a hyperpolarized resting membrane potential (−75 to −85 mV). In a small number of cases (15 of 477 cells), these identification characteristics yielded cells that showed the clear electrophysiological signature of fast-spiking interneurons (no inward rectification, short duration spikes and a fast, irregular firing pattern (Belleau and Warren, 2000)). These cells were excluded from further investigation in this study.
Figure 1.
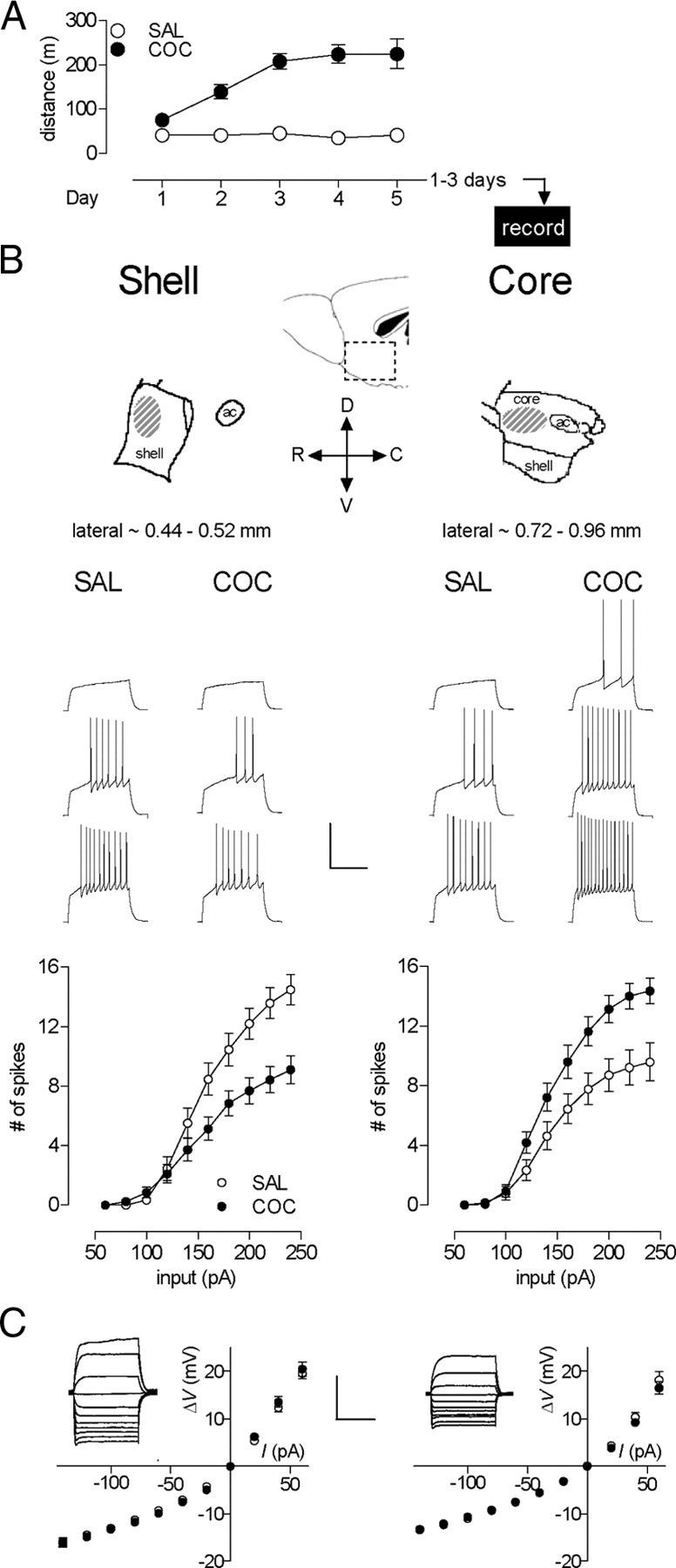
Repeated in vivo cocaine induced opposite firing adaptations in accumbens shell versus core MSNs during early abstinence. A, Cocaine-induced sensitization of locomotor activity. Data indicate distance traveled during 15 min following intraperitoneal injection of either saline (n = 13) or 15 mg/kg cocaine (n = 10). The x-axis also represents the experimental timeline. B, Top, Diagram of the sagittal view of the NAc shell and core showing the recording sites with the dorsoventral and rostrocaudal directions (adapted from Paxinos and Franklin, 2001). Middle, Sample traces from shell (left, at 100, 140, and 180 pA; SAL, n = 10, 4 mice; COC, n = 31, 6 mice) and core (right, at 120, 160, and 200 pA; SAL, n = 16, 5 mice; COC, n = 14, 6 mice) for SAL- and COC-treated animals. Calibration: 500 ms, 50 mV. Bottom, Mean spike number decreased in shell (left) and increased in core (right) in cocaine- compared with saline-treated animals. C, No cocaine-induced differences in steady-state voltage responses to a series of current pulses in either shell (left) or core (right). Insets, Sample traces (stimulation = −140 pA to 60 pA) from neurons in SAL groups. Calibration: 500 ms, 20 mV. Error bars represent SEM. In some cases, data symbols obscure small error bars.
To quantify firing properties, whole-cell current-clamp recordings were performed with electrodes (3–5 MΩ) containing 120 K-gluconate, 20 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 Na2ATP, and 0.3 Tris–GTP. Data were filtered at 5 kHz, digitized at 10 kHz, and collected and analyzed using custom software (Igor Pro; Wavemetrics). Membrane potentials were held at approximately −80 mV. Series resistances ranged from 10 to 18 MΩ and input resistances (Ri) were monitored on-line with a 40 pA, 150 ms current injection given before every 800 ms current injection stimulus. Only cells with a stable Ri (Δ < 10%) for the duration of the recording were kept for analysis. To measure “steady-state” voltage responses for each subthreshold pulse in a series of current injections, the voltage values were taken 780 ms following the onset of each current injection. Spike measurements for a given cell are the mean values measured from 1 to 3 cycles of current steps (800 ms duration at 0.1 Hz, −160 to +260 pA range with a 20 pA step increment). To quantify firing patterns, we measured the first action potential latency, the train duration, and the mean interspike interval at three representative current injection values (160, 200, and 240 pA), except as noted in the text.
Statistics.
Data acquisition and analysis were performed blind to experimental condition. On >90% of recording days, data were collected from both saline and cocaine groups. Results are presented as mean ± SEM with n representing the number of cells in each group. Statistical significance was assessed using either two-tailed Student's t tests or two-way repeated-measures ANOVAs. In Figure 2, two-way ANOVAs were used for comparison between saline- and psychostimulant-treated mice.
Figure 2.
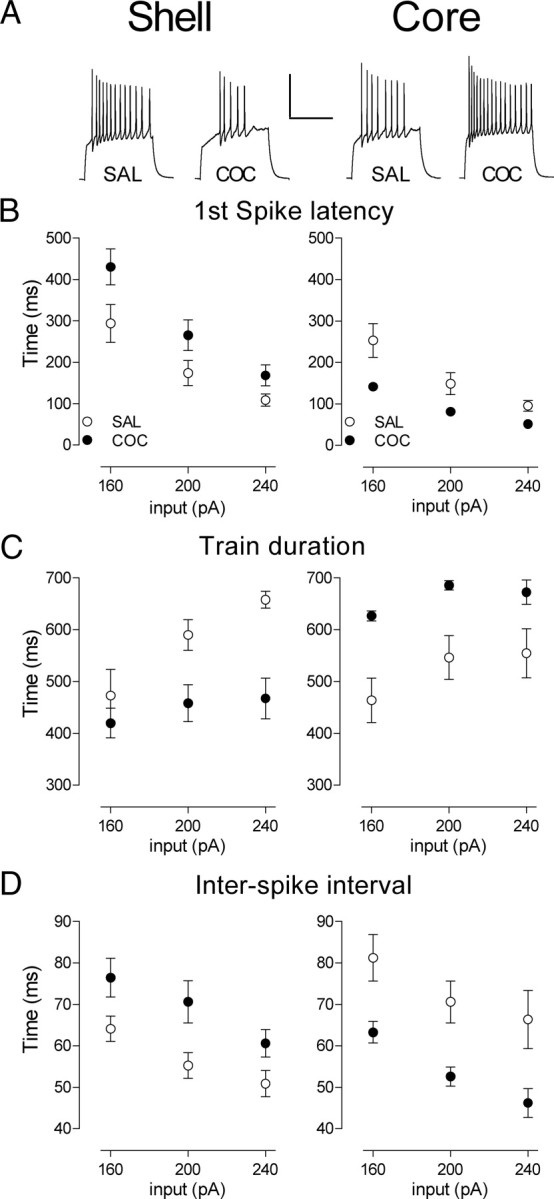
Repeated in vivo cocaine induces opposite firing pattern adaptations in NAc shell versus core MSNs. A, Sample traces obtained in shell (left) and core (right) at 240 pA for SAL- and COC-treated groups. B, Latency to the first action potential in shell (left) and core at three representative levels of depolarizing current injection (160, 200, and 240 pA). C, Train duration in shell (left) and core (right). D, Interspike interval in shell (left) and core (right). Error bars represent SEM.
Results
Cocaine treatment induces opposite firing adaptations in accumbens core versus shell MSNs
The cocaine treatment (five once-daily injections of 15 mg/kg, i.p.) was identical to that used in our previous studies examining synaptic parameters (Kourrich et al., 2007). This regimen induced robust psychomotor sensitization (Fig. 1A) (two-way ANOVA, treatment, F(1,21) = 206.6, p < 0.0001; interaction treatment × day, F(4,84) = 11.98, p < 0.0001). To examine early effects of cocaine exposure on intrinsic excitability of NAc MSNs, we prepared acute NAc brain slices for whole-cell recording from cocaine- and saline-treated mice 1–3 d after the last injection (Fig. 1A). One straightforward means of assessing intrinsic excitability is to measure the pattern of spikes elicited by each pulse in a series of depolarizing current injections. By measuring neuronal output, this approach reveals the overall functional effect of any significant changes in passive and active membrane properties. First, we measured the number of depolarization-evoked spikes in whole-cell recordings from MSNs in the NAc shell. Notably, there were no significant changes in basic membrane properties such as the input resistance, resting membrane potential, single action potential characteristics (Table 1) and the current/voltage relationship (Fig. 1C). However, the total number of spikes elicited (sum of spikes at all current injection values) in cocaine-treated animals was reduced by 35% compared with saline-treated animals (Fig. 1B) (two-way ANOVA, treatment, F(1,39) = 5.07, p = 0.030; treatment × input interaction, F(9,351) = 7.41, p < 0.0001).
Table 1.
Repeated in vivo cocaine altered membrane properties of NAc shell and core neurons
| Early abstinence (1–3 d) |
Late abstinence (10–14 d) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Shell |
Core |
Shell |
Core |
|||||
| SAL | COC | SAL | COC | SAL | COC | SAL | COC | |
| Passive membrane properties | ||||||||
| RMP (mV) | −80.5 ± 0.4 | −80.7 ± 0.5 | −78.6 ± 0.50 | −79.0 ± 0.5 | −78.8 ± 0.5 | −78.5 ± 0.4 | −79.4 ± 0.8 | −78.3 ± 0.8 |
| Ri (MΩ) | 180.2 ± 10.1 | 174.1 ± 8.5 | 141.6 ± 6.8 | 135.6 ± 8.8 | 174.8 ± 8.4 | 188.0 ± 7.1 | 151.9 ± 11.6 | 165.4 ± 8.6 |
| Active membrane properties | ||||||||
| Rheobase (current togenerate a spike, pA) | 124 ± 5.8 | 139.4 ± 7.6 | 137.5 ± 10.0 | 116.2 ± 4.4 | 130.0 ± 9.7 | 135.5 ± 7.8 | 132.7 ± 11.8 | 134 ± 8.8 |
| Spike threshold (mV) | −35.3 ± 0.8 | −33.7 ± 0.8 | −33.2 ± 0.5 | −35.2 ± 0.6* | −35.2 ± 0.8 | −32.6 ± 0.6** | −34.9 ± 0.7 | −34.4 ± 0.9 |
| Spike amplitude (mV) | 69.4 ± 2.2 | 69.7 ± 1.5 | 77.1 ± 1.0 | 83.8 ± 1.8** | 76.8 ± 1.6 | 72.0 ± 1.4* | 78.3 ± 1.6 | 79.6 ± 2.2 |
| Spike duration (ms) | 2.60 ± 0.06 | 2.69 ± 0.06 | 2.57 ± 0.06 | 2.45 ± 0.04 | 2.39 ± 0.03 | 2.57 ± 0.04*** | 2.56 ± 0.06 | 2.62 ± 0.06 |
| AHP amplitude (mV) | 15.2 ± 0.6 | 14.2 ± 0.4 | 15.3 ± 0.4 | 15.6 ± 0.4 | 15.0 ± 0.2 | 15.8 ± 0.4 | 16.7 ± 0.6 | 15.2 ± 0.4 |
| Spike rise (10%–90%, ms) | 0.76 ± 0.04 | 0.82 ± 0.04 | 0.72 ± 0.04 | 0.60 ± 0.02* | 0.62 ± 0.03 | 0.74 ± 0.03*** | 0.69 ± 0.04 | 0.69 ± 0.04 |
Values represent the mean± SEM (*p < 0.05; **p < 0.01; ***p < 0.001; with two-tailed Student's t test used to compare with their respective saline control group). The RMP and the spike threshold were not adjusted for the liquid junction potential (∼8 mV). Spike characteristics were obtained from the first spike evoked by the minimal depolarizing current pulse in every NAc neuron recorded; the spike threshold was obtained by using the maximum of the third differential during the rising phase. The other characteristics were measured as follows: spike amplitude, difference between spike threshold and peak; spike duration, measured at 20 pA above threshold; AHP amplitude, difference between spike threshold and maximum hyperpolarization after the spike; spike rise, time difference between 10 and 90% of the spike peak.
Unexpectedly, and in sharp contrast to the shell, the firing capacity in core MSNs 1–3 d following repeated cocaine treatment was increased by 33% (Fig. 1B) (two-way ANOVA, treatment, F(1,28) = 8.34, p = 0.0074; interaction treatment × input, F(9,252) = 5.26, p < 0.0001). This firing increase occurs in the absence of changes in the input resistance (Table 1) and the current/voltage relationship (Fig. 1C), but in the presence of changes in several single-spike characteristics such as threshold, amplitude, and rise time in the direction of increased excitability (Table 1). We also noticed in drug-naive mice that core MSNs were significantly less excitable than shell MSNs. One likely contributor to this difference is the lower input resistances (Ri) in core compared with shell MSNs (Table 1; core 141.6 ± 6.8 MΩ, shell 180.2 ± 10.1 MΩ; t(24) = 3.153, p = 0.0043). This difference in Ri has been described previously (Pennartz et al., 1992) and may be due, in part, to the increased surface area of core MSNs (up to 50% greater than shell) (Meredith et al., 1992).
Repeated drug exposure is likely to be necessary to induce intrinsic plasticity in either shell or core MSNs, as neither a single injection of cocaine (15 mg/kg) nor saline induced any change in firing properties compared with a naive group (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) [two-way ANOVA between naive, SAL, and COC in shell, treatment, F(2,430) = 0.015, p = 0.99; interaction treatment × input, F(20,430) = 0.16, p = 1.0; in core, treatment, F(2,280) = 0.143, p = 0.867; interaction treatment × input, F(20,280) = 1.0, p = 1.0].
To quantify the observed differences in spiking patterns, we analyzed fundamental characteristics of spike trains elicited at three representative current injection values (Fig. 2). The decrease in the shell MSN firing is characterized by an increase in first-spike latency (Fig. 2B) (two-way ANOVA, treatment, F(1,117) = 6.214, p = 0.014), a decreased spike train duration (Fig. 2C) (two-way ANOVA, treatment, F(1,104) = 11.79, p = 0.0009), and an increase in the interspike interval (Fig. 2D) (two-way ANOVA, treatment, F(1,102) = 8.173, p = 0.005). In contrast, in core MSNs, we observed the opposite adaptations: a decrease in the first spike latency (Fig. 2B) (two-way ANOVA, treatment, F(1,75) = 16.41, p = 0.0001), an increased spike train duration (Fig. 2C) (two-way ANOVA, treatment, F(1,75) = 21.78, p < 0.0001), and a decrease in the interspike interval (Fig. 2D) (two-way ANOVA, treatment, F(1,75) = 20.04, p < 0.0001). One caveat to this analysis pertains to the absolute magnitude of the train duration. The duration of our current injection step (800 ms) puts a theoretical limit on this parameter. Of course, significant increases in the first spike latency could result in apparent decreases in the spike train duration between groups. This is not the case here. In both the core and shell, the groups with the shorter trains (cocaine in shell, saline in core) exhibit an abrupt truncation of firing, on average, before the end of the current pulse whereas the groups with longer trains do not (time between last spike and end of current pulse (240 pA)—shell: SAL, 34.1 ± 3.6 ms, COC, 179.6 ± 39.4 ms, p = 0.04; core: SAL, 221.7 ± 51.8, COC, 63.2 ± 18.6, p = 0.02). Thus, if anything, the absolute magnitude of the train duration effect presented here may be an underestimate of the true effect. In total, these results demonstrate that repeated cocaine exposure induces opposite adaptations in shell versus core MSNs not only in firing capacity, but also in firing pattern.
Cocaine-induced decreases in shell MSN firing are long lasting
One hallmark of drug addiction is its persistence despite long periods of drug abstinence. To assess the persistence of excitability changes, we measured MSN firing properties following a prolonged drug-free period (10–14 d after the last injection) (Fig. 3A). As above, we measured the number of spikes elicited by pulses of depolarizing current. While firing in core neurons returned to control levels (Fig. 3B) (two-way ANOVA, treatment, F(1,21) = 0.00003, p = 0.9957; interaction treatment × input F(9,189) = 0.315, p = 0.969), the decreased firing capacity in shell MSNs was maintained (Fig. 3B) (44% decrease; two-way ANOVA, treatment, F(1,39) = 9.662, p = 0.0035; interaction treatment × input F(9,351) = 8.24, p < 0.0001). This decrease is accompanied by several changes in single-spike parameters, including threshold, amplitude, duration, and rise time—each of which is altered in manner consistent with decreased excitability (Table 1). In addition, shell spike train parameters are altered identically to those during early withdrawal (Fig. 3C) (first spike latency: t(33) = 2.40, p = 0.022; train duration: t(37) = 3.44, p = 0.0014; interspike interval: t(37) = 2.38, p = 0.023). Thus, core and shell neurons show opposite regulation not only in firing capacity and pattern, but also in the persistence of these adaptations.
Figure 3.
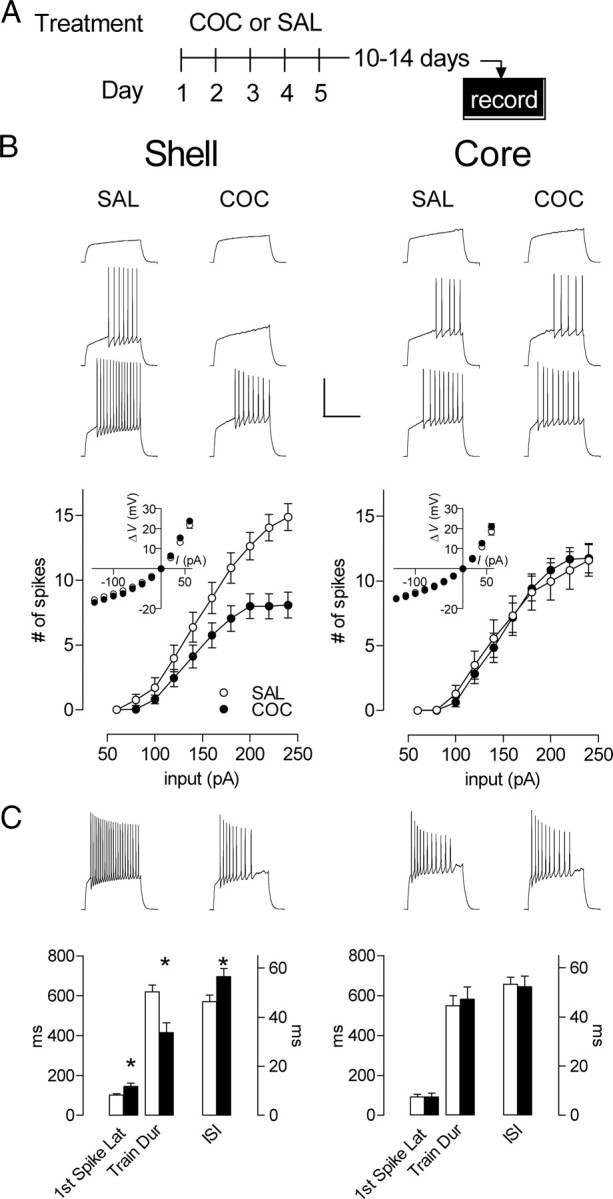
During prolonged abstinence, the shell firing depression is maintained while the core firing potentiation is lost. A, Experimental timeline. B, Top, Sample of traces obtained in shell (left, at 80, 120, and 160 pA; SAL, n = 20, 7 mice; COC, n = 22, 4 mice) and core (right, at 140, 180, and 220 pA; SAL, n = 11, 4 mice; COC, n = 12, 4 mice). Responses from neurons in saline-treated and injection-naive mice were similar. Thus, SAL group is composed of an equal number of neurons (10/10 for shell, 6/5 for core) from saline-pretreated and injection-naive mice. Bottom, The mean spike number for a given magnitude of current injection was still decreased in shell (left) and returned to basal levels in core (right) in cocaine- compared with saline-treated animals. Inset, no cocaine-induced differences in steady-state voltage responses to a series of current pulses in either shell (left) or core (right). C, Top, Samples of traces from shell (left) and core (right) at 240 pA. Bottom, Latency to the first action potential, train duration and interspike interval at 240 pA for the shell (left) and core (right). In shell, while train duration is shorter, latency for the first spike and the interspike interval are longer in the cocaine group. In contrast, cocaine induced no significant changes the core. *p < 0.05. Error bars represent SEM.
Amphetamine exposure induces similar intrinsic plasticity as cocaine
One approach to establishing the cellular substrates for addiction-related behavior is to look for shared adaptations between different drug treatments that produce a common behavioral output. For example, although cocaine and amphetamine exposure both can induce psychomotor sensitization, the neuroadaptations induced by these closely related psychostimulants are only partially overlapping (Vanderschuren and Kalivas, 2000). Here, we also evaluated the effect of amphetamine exposure on intrinsic plasticity in NAc shell and core using a treatment (five once-daily injections of 5 mg/kg, i.p.) (Fig. 4A, left) that triggers an indistinguishable degree of behavioral sensitization (Fig. 4A, right) (two-way ANOVA, treatment, F(2,30) = 136.9, p < 0.0001; interaction treatment × day, F(8,120) = 8.43, p < 0.0001). No interaction was found between COC and AMPH groups (F(4,72) = 0.564, p = 0.689).
Figure 4.
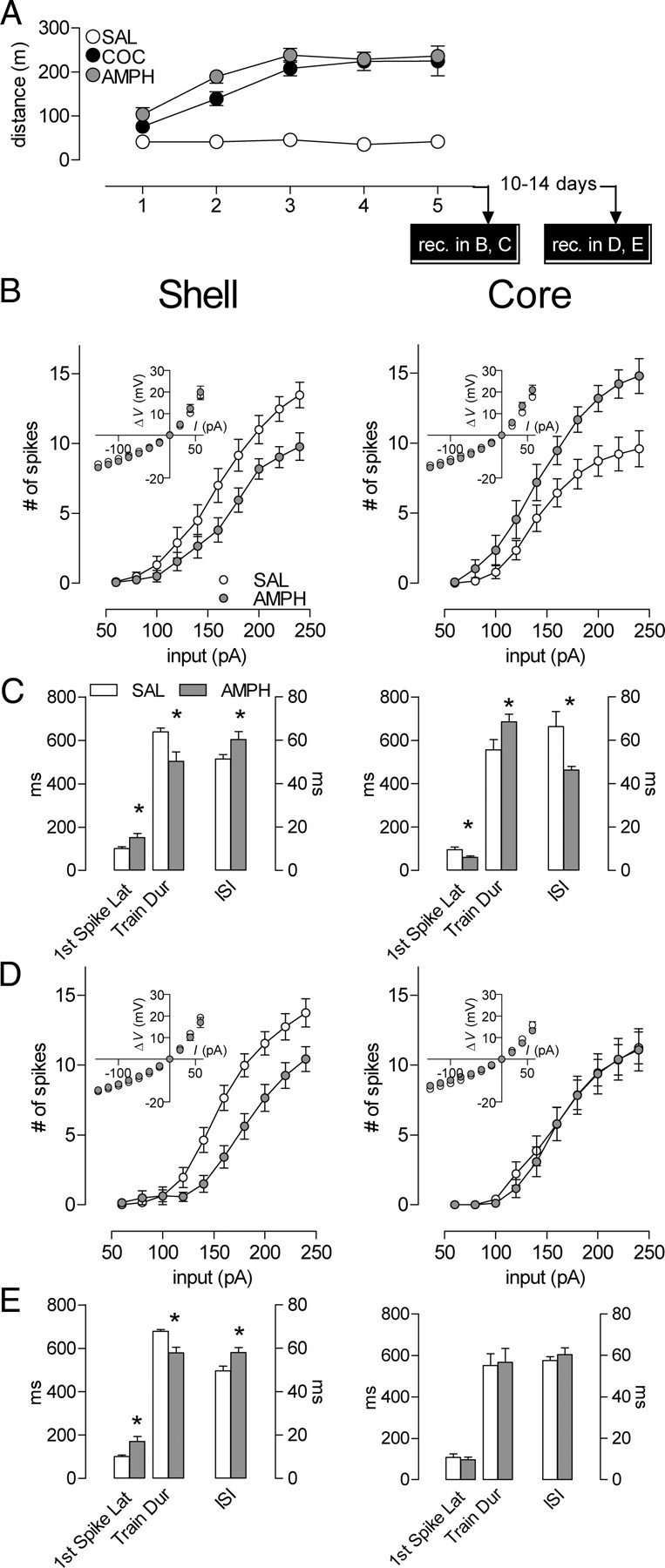
Amphetamine exposure produces similar intrinsic plasticity to that of cocaine. A, Cocaine- and amphetamine-induced behavioral sensitization (SAL, n = 13 mice; COC, n = 10; AMPH, n = 10). The first 15 min of postinjection locomotor activity from day 1 to day 5 are represented here. No interaction between COC and AMPH groups. The cocaine- and saline-treated groups are the same as the ones used in Figure 1. The x-axis also represents the experimental timeline. B, Early abstinence (1–3 d), mean spike number decreased in shell (left) (SAL, n = 19, 9 mice; AMPH, n = 20, 7 mice) and increased in core (right) (SAL, n = 16, 9 mice; AMPH, n = 14, 6 mice) in amphetamine- compared with saline-treated animals. Insets, No amphetamine-induced differences in steady-state voltage responses to a series of current pulses in either shell (left) or core (right). C, Latency to the first spike, train duration, and interspike interval at 240 pA for the shell (left) and core (right). In shell, while train duration is shorter, latency for the first spike and the interspike interval are longer in the amphetamine group. In contrast, amphetamine induces opposite changes in the core. D, The mean spike number for a given magnitude of current injection was still decreased in shell (left, SAL, n = 21, 9 mice; AMPH, n = 14, 4 mice) and returned to basal levels in core (right, SAL, n = 11, 9 mice; AMPH, n = 12, 4 mice) in amphetamine- compared with saline-treated animals. Inset, no cocaine-induced differences in steady-state voltage responses to a series of current pulses in either shell (left) or core (right). E, Latency to the first spike, train duration, and interspike interval at 240 pA for the shell (left) and core (right). In shell, while train duration is shorter, latency for the first spike and the interspike interval are longer in the amphetamine group. In contrast, amphetamine induced no significant changes in the core. *p < 0.05. Error bars represent SEM.
As with cocaine exposure, in early (1–3 d) abstinence, we observed an amphetamine-induced decrease in shell and an increase in the core firing capacity (Fig. 4B) (shell: two-way ANOVA, treatment, F(1,37) = 5.58, p = 0.024; interaction treatment × input, F(9,333) = 3.14, p = 0.0012; core: two-way ANOVA, treatment, F(1,28) = 9.73, p = 0.0042; interaction treatment × input, F(9,252) = 2.88, p = 0.0029). The characteristics of MSN firing patterns are also altered in an similar way as with cocaine exposure (Fig. 4C) (shell: first spike latency: t(35) = 2.46, p = 0.019; train duration: t(37) = 2.82, p = 0.0077; interspike interval: t(35) = 2.16, p = 0.0375; core: first spike latency: t(27) = 2.24, p = 0.034; train duration: t(27) = 2.13, p = 0.042; interspike interval: t(27) = 2.523, p = 0.0178). The data during late abstinence (10–14 d) for amphetamine also duplicate those seen with cocaine (Fig. 4D) (shell: two-way ANOVA, treatment, F(1,33) = 9.06, p = 0.0050; interaction treatment × input, F(9,297) = 5.63, p < 0.0001; core: two-way ANOVA, treatment, F(1,21) = 0.0492, p = 0.827; interaction treatment × input F(9,189) = 0.149, p = 0.998) (Fig. 4E) (shell: first spike latency: t(29) = 3.19, p = 0.003; train duration: t(29) = 4.17, p = 0.0003; interspike interval: t(29) = 2.66, p = 0.0125). We also observed the spike train truncation effect, as mentioned above for cocaine (Time between last spike and end of current pulse (240 pA)—shell: SAL, 48.4 ± 8.8 ms, AMPH, 168.6 ± 52.9 ms, p = 0.04; core: SAL, 162.7 ± 51.4, AMPH, 24.6 ± 4.2, p = 0.02). Thus, although cocaine and amphetamine have been reported to show many differences in biochemical and physiological adaptations, here we demonstrate that their effect on intrinsic plasticity, like their ability to induce psychomotor sensitization, is shared.
Differential sensitivity to A-type potassium current (IA) blockade
To begin to investigate the mechanisms for the opposite changes in intrinsic excitability in shell and core MSNs, we used a pharmacological strategy. Previous studies identified several cocaine-modulated sodium, calcium, and potassium currents in a mixture of shell and core NAc MSNs (Zhang et al., 1998, 2002; Hu et al., 2004). These adaptations are potential “building blocks” for changes in firing properties. However, extrapolation from changes in individual (or even several) currents to changes in firing pattern is problematic (Marder and Prinz, 2002; Marder and Goaillard, 2006), and thus, direct measurement of firing properties is necessary to determine experience-dependent changes in neuronal output. Previous studies suggest that the fast activating transient potassium current, IA, might be enhanced by repeated cocaine (Hu et al., 2004) and is acutely modulated by dopamine receptor activation (Hopf et al., 2003; Perez et al., 2006)—an indirect effect of psychostimulant exposure. We also observed two changes in intrinsic excitability that suggest a potential contribution of IA to the opposite firing changes we observed in NAc shell versus core: (1) a first spike latency shift (Figs. 2B, 4C) and (2) alterations in repetitive firing (Figs. 2C, 4C). IA is an important contributor to these properties of cellular excitability in MSNs (Bargas et al., 1989; Nisenbaum et al., 1994). We investigated a potential role for IA in psychostimulant-induced adaptations by using a dose of 4-aminopyridine (4-AP; 100 μm) that selectively blocks this current (e.g., Bargas et al., 1989; Nisenbaum et al., 1994).
The relatively long spike latency in MSNs is controlled by a competition between depolarizing ionic currents (Na+, Ca2+) and a hyperpolarizing current, IA, that is activated at subthreshold voltages (Kita et al., 1985; Calabresi et al., 1987; Bargas et al., 1989; Galarraga et al., 1989; Nisenbaum et al., 1994). In principal, then, a change in any one (or a combination) of these conductances could produce a change in the spike latency. To test whether IA is a contributor to the differential latency effects we observed, we applied 4-AP and found that, as expected, it produced a modest reduction in first spike latency in the shell saline group in early abstinence (Fig. 5B, left). In psychostimulant groups, however, the magnitude of the reduction was significantly larger (Fig. 5B, left) (two-way ANOVA, interaction treatment × 4-AP, F(2,25) = 4.01, p = 0.031). Interestingly, in 4-AP, the difference in latency between saline and psychostimulant groups was abolished (Fig. 5B, left) (treatment effect p > 0.05). In the core, during early abstinence, we observed the opposite pattern of 4-AP modulation of first spike latency. Here, it was the saline group that showed a larger latency reduction in 4-AP than the psychostimulant groups (Fig. 5C, left) (two-way ANOVA, interaction treatment × 4-AP, F(2,29) = 3.81, p = 0.034). Again, 4-AP abolished any latency difference between the groups (Fig. 5C, left) (treatment effect p > 0.05).
Figure 5.

IA blockade with 4-AP (100 μm) differentially affects psychostimulant-induced firing adaptations in shell versus core. A, Sample of traces obtained in cocaine-treated groups at 240 pA in shell (left) and core (right) before and during 4-AP application. Calibration: 200 ms, 50 mV. B, Latency to the first spike during early (left) and prolonged (right) abstinence before and during 4-AP application at 240 pA in shell (early abstinence: SAL, n = 9, 5 mice; COC, n = 11, 6 mice; AMPH, n = 8, 4 mice; prolonged abstinence: SAL, n = 10, 8 mice; COC, n = 10, 6 mice; AMPH, n = 11, 4 mice). C, Latency to the first spike during early (left) and prolonged (right) abstinence before and during 4-AP application at 240 pA in core (early abstinence: SAL, n = 8, 5 mice; COC, n = 14, 6 mice; AMPH, n = 10, 5 mice; prolonged abstinence: SAL, n = 10, 6 mice; COC, n = 15, 6 mice; AMPH, n = 10, 4 mice). D, The mean spike number during early (left) and prolonged (right) abstinence before and during 4-AP application at 240 pA in shell (early abstinence: SAL, n = 31, 11 mice; COC, n = 31, 6 mice; AMPH, n = 8, 4 mice; prolonged abstinence: SAL, n = 13, 9 mice; COC, n = 13, 7 mice; AMPH, n = 12, 4 mice). E, The mean spike number during early (left) and prolonged (right) abstinence before and during 4-AP application at 240 pA in core (early abstinence: SAL, n = 16, 5 mice; COC, n = 14, 6 mice; AMPH, n = 14, 6 mice; prolonged abstinence: SAL, n = 10, 6 mice; COC, n = 15, 6 mice; AMPH, n = 9, 4 mice). Error bars represent SEM.
We considered the possibility that 4-AP may simply increase the overall excitability and readjust the spike latency to a minimal value even if IA is not a key contributor to effects observed here. However, 4-AP did not increase the input resistance (supplemental table, available at www.jneurosci.org as supplemental material) as an agent that simply increases overall excitability would be expected to do, and the latency is not at a minimal value in 4-AP, as even strong depolarizing current pulses that elicit near-maximal spiking (240 pA) show a remaining latency of 35–60 ms (Fig. 5B,C). Moreover, studies of the mechanism for delayed spiking in MSNs indicate that the broad-spectrum potassium channel blocker, TEA, does not affect the first spike latency (Bargas et al., 1989). This demonstrates that simple increases in overall excitability do not necessarily reduce the latency, while IA blockade does. Thus, the normalization of spike latency differences by 4-AP provides evidence that the differential engagement of IA in psychostimulant versus control cells plays a key role in spike latency changes.
Given that IA blockade abolished the drug-induced difference in first spike latency, we also tested for a differential effect of 4-AP between psychostimulant and saline groups on firing capacity—another factor that is modulated by IA. Consistent with our latency observations, IA blockade in psychostimulant groups enhanced the number of spikes per input in shell MSNs to the level seen in the saline group (Fig. 5D, left) (two-way ANOVA, interaction treatment × 4-AP, F(2,24) = 5.386, p = 0.012; treatment effect during 4-AP, p > 0.05). Also consistent with the latency effects, in core MSNs, IA blockade in the saline group enhanced firing capacity while in the psychostimulant groups it did not (Fig. 5E, left) (two-way ANOVA, interaction treatment × 4-AP, F(2,29) = 3.969, p = 0.030; treatment effect during 4-AP, p > 0.05). In each case in shell and core, the abolishment of firing level differences was present across the range of depolarizing current values tested (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Again, these effects occur in the absence of any 4-AP-induced changes in input resistance in any group (see supplemental table, available at www.jneurosci.org as supplemental material).
In each case where there is a greater reduction in spike latency with IA blockade, we also see a larger increase in the number of spikes per current value. What is the relationship between these effects? One trivial possibility is that the reduced latency provides more time for spiking during the current pulse. While, of course, it does provide more time for spiking, this does not fully explain the readjustment in spike number. First, the changes in spike number are too great to be explained by the latency shift. Second, on average, in cases where fewer spikes are generated (i.e., core saline and shell COC/AMPH), the train is truncated before the end of the current step (see Figs. 1–3, 5 for examples). We find that 4-AP reduces or completely eliminates this truncation, and thus permits the neuron to fire to the end of the current step. In summary, 4-AP does not simply decrease the latency for the first spike, but also eliminates the cocaine-induced truncation in shell, enabling more spikes to occur. Thus, our data support the idea that IA is a key contributor to the opposite changes in psychostimulant-induced firing properties observed in shell versus core MSNs.
To test whether the differential sensitivity to IA blockade between shell and core in psychostimulant groups persists, we tested the modulation of firing properties with 4-AP during late abstinence (10–14 d). As in early abstinence, in shell MSNs, 4-AP induces significantly greater modulation of latency and number of spikes in psychostimulant groups compared with saline (Fig. 5B, right, two-way ANOVA, interaction treatment × 4-AP, F(2,28) = 3.52, p = 0.043; Fig. 5D, right, two-way ANOVA, interaction treatment × 4-AP, F(2,35) = 10.63, p = 0.0002). In core MSNs, however, the differential effect of IA blockade between psychostimulant groups and saline on latency and number of spikes is absent (Fig. 5C, right; two-way ANOVA, interaction treatment × 4-AP, F(2,32) = 0.164, p = 0.849; Fig. 5E, right; two-way ANOVA, interaction treatment × 4-AP, F(2,31) = 0.111, p = 0.895). Thus, in each case where psychostimulants induced a change in MSN firing properties, we also observed a concurrent change in the ability of IA blockade to modulate firing. When IA modulation returned to normal (i.e., late abstinence core), the firing properties were also readjusted to drug-naive levels.
Discussion
Repeated in vivo psychostimulants induce opposite changes in intrinsic excitability in two closely related populations of neurons—the principal neurons of the accumbens core and shell. In shell, we observed an immediate and persistent decrease in intrinsic excitability manifested by a reduction in depolarization-induced firing. In contrast, the same psychostimulant regimen produced an opposite effect in MSNs of the core. During early abstinence (1–3 d), core neurons exhibited an increase in firing properties. Unlike the shell, core adaptations were transient. By 10–14 d following the last psychostimulant injection, firing in core MSNs had returned to control levels whereas decreases in shell firing were maintained. These changes are opposite not only in terms of firing capacity, but also in firing patterns. Finally, our data suggest that opposite regulation of IA is an important factor in the bidirectional intrinsic plasticity induced by psychostimulants in shell versus core.
Parallel regulation of NAc intrinsic plasticity by cocaine and amphetamine
Although cocaine and amphetamine have similar pharmacological and behavioral effects and produce many neuroadaptations in common, other effects are distinct (Vanderschuren and Kalivas, 2000). For example, repeated cocaine increases the overall surface levels of AMPA-type glutamate receptors (AMPARs) in NAc, whereas amphetamine does not (Boudreau and Wolf, 2005; Nelson et al., 2009). Here, in the first report of amphetamine's effects on NAc intrinsic excitability, we show that these effects appear not distinct, but similar to those of cocaine. The cocaine and amphetamine regimens we used also produced an indistinguishable degree of behavioral sensitization. Thus, it is possible that these shared excitability changes play a functional role in this model of addiction-related change in behavior.
A-type potassium current (IA) contributes to NAc intrinsic plasticity
We observed specific psychostimulant-induced changes in shell and core firing that suggested IA as a potential mediator. IA in MSNs is a known regulator of both the first spike latency and repetitive firing (Bargas et al., 1989; Nisenbaum et al., 1994), two parameters that are clearly modified by repeated psychostimulants. The effects of IA blockade in shell and core were opposite. In the shell, IA blockade produced robust decreases in first spike latency and increases in repetitive spiking in psychostimulant groups with only modest effects in controls. In contrast, in the core, IA blockade produced robust effects in the control group. Psychostimulant groups in the core were resistant to IA blockade during early abstinence—the time point when intrinsic excitability was enhanced. By late abstinence in the core, both the diminished effects of IA blockade and the increase in excitability were gone. In total, our data indicating that the effects of IA blockade correlate with changes in intrinsic excitability in both presence and direction supports the idea that IA is a key contributor to the bidirectional psychostimulant-induced intrinsic plasticity in NAc MSNs.
The origin of these differential effects of IA blockade is unknown. Differences in activation or inactivation parameters such as kinetics or voltage or in the relative abundance of the channels that mediate IA are among the possible contributors. Interestingly, dopamine receptor activation transiently modulates IA in NAc slices (Hopf et al., 2003; Perez et al., 2006), although the precise origin of this modulation is not understood either. Whether multiple bouts of intense dopamine receptor activation, such as would occur with repeated psychostimulants, would produce persistent effects on IA and whether these effects might differ between shell and core are intriguing unanswered questions.
While IA appears to be important, our data do not exclude the involvement of other cocaine-associated firing modulators such as the Ca2+-activated (KCa) or inward rectifier (Kir) potassium currents (Hu et al., 2004; Ishikawa et al., 2009). Although our data do not provide strong indications of KCa or Kir involvement [see Table 1 for lack of changes in resting membrane potential (RMP), input resistance, and afterhyperpolarization (AHP) peak], an interesting recent study found an increase in an AHP component in shell MSNs following repeated cocaine (Ishikawa et al., 2009). Blocking this component with apamin partially restored firing capacity, suggesting that a small-conductance KCa channel is also involved in the cocaine-induced decrease in firing in the shell.
A “disparity” hypothesis: possible functional consequences of greater excitability in core versus shell MSNs?
Previous data have suggested that unidirectional changes in NAc neuronal excitability may drive addiction-related behavior (Kalivas and Hu, 2006; Conrad et al., 2008; Thomas et al., 2008). Combined with data from other studies, however, our excitability findings suggest a revised hypothesis. Rather than a unidirectional change in MSN activity, perhaps a psychostimulant-induced disparity between core and shell activity precipitates addiction-related behavior. Data from two recent studies—studies that had appeared to contradict one another—support this hypothesis. First, Dong et al. (2006) produced a NAc shell-specific decrease in intrinsic excitability via viral-mediated overexpression of potassium channel subunits, which resulted in amplified psychomotor responses to cocaine. Second, while investigating a forebrain-specific knock-out of the enzyme Cdk5, Benavides et al. (2007) measured enhanced intrinsic excitability in core MSNs. Surprisingly, a NAc core-specific Cdk5 knock-out—a mouse that in all likelihood has a selective increase in core excitability—exhibited a cocaine-related behavioral profile (e.g., enhanced cocaine conditioned place preference) that was consistent with the profile seen in Dong et al. (2006). Thus, opposite exogenous manipulations of excitability—a selective decrease in shell (Dong et al., 2006) and a (presumed) selective increase in core (Benavides et al., 2007)— appear to produce a common, cocaine-related behavioral phenotype. Our data demonstrate that each of these excitability effects is induced by psychostimulant experience itself. Thus, we hypothesize that the endogenous pattern of opposite core and shell intrinsic plasticity may be a mediator for psychostimulant-induced behavioral plasticity.
Integrating drug-induced intrinsic and synaptic plasticity in NAc
Two fundamental factors, intrinsic membrane and synaptic properties, determine a neuron's overall state of excitability. Although little is yet known about amphetamine-induced changes in glutamatergic transmission in NAc MSNs, recent studies have described the time course of cocaine-induced changes in this parameter (Kourrich et al., 2007). Combining our current data with these synaptic findings provides evidence for a persistent enhancement in core versus shell excitability. During early abstinence (24 h), AMPAR currents show little to no change in both core and shell MSNs (Kourrich et al., 2007; M. J. Thomas, unpublished data) and thus the state of overall excitability is determined by the intrinsic properties described here. During late abstinence (>10 d), AMPAR currents show a robust increase in both core and shell MSNs (Kourrich et al., 2007; Conrad et al., 2008; M. J. Thomas, unpublished data), while core firing has returned to a basal level, and the decrease in shell firing is maintained. This pattern also yields a shift toward a relative increase in core excitability. In summary, by combining what we know of cocaine-induced synaptic and intrinsic plasticity, the current picture is one of a persistent disparity between core and shell MSN excitability that favors the core.
Potential for a core/shell excitability disparity to drive behavior
Recent studies indicate that with repeated cocaine experience, phasic firing to a cocaine-paired cue is enhanced—an effect that is greater in core than in shell neurons (Ghitza et al., 2004; Hollander and Carelli, 2005; Ghitza et al., 2006; Hollander and Carelli, 2007). On the other hand, widespread hypoactivity develops in tonic-firing NAc neurons with repeated cocaine experience (Peoples et al., 1998, 1999; Peoples and Cavanaugh, 2003). This generalized hypoactivity is more prominent in shell than in core neurons—an effect that appears to have implications for cocaine self-administration behavior (Peoples et al., 2007). Thus, the pattern of excitability changes described here appears to map onto the pattern of drug experience-induced changes in single-unit activity in behaving animals.
How might these opposite changes combine to produce a potent behavioral effect? Activation of core MSNs appears to be important for driving motor programs needed to obtain rewards (Ghitza et al., 2004; Wood and Rebec, 2004; Ambroggi et al., 2008). It is clear that core MSNs send projections to motor-related circuits that can influence these behavioral patterns (Zahm and Brog, 1992). At the same time, a decrease in shell MSN activity has been likened to releasing a “brake” on appetitive reward-seeking behaviors (Kelley, 2004; Taha and Fields, 2006). This may be due to disinhibition of downstream reward-related structures such as the lateral hypothalamus (Harris et al., 2005). Thus, the simultaneous augmentation of core activity and diminution of shell activity that develops with repeated psychostimulant experience might be a particularly potent combination that drives abnormally strong drug-seeking behavior.
Conclusions
This study demonstrates that repeated psychostimulant experience produces opposite changes in intrinsic excitability in two related populations of reward circuit neurons—NAc MSNs in shell and core. This stresses the importance of studying shell and core neurons in separate populations. When considered with other data from the literature, our data raise the possibility that increasing core compared with shell MSN activity may be a potent driver of addiction-related behavior. These results highlight the potential importance of intrinsic membrane plasticity in long-lasting functional changes in neural circuits for reward.
Footnotes
This work was supported by funding to M.J.T. from the National Institute on Drug Abuse (R01 DA019666) and the Whitehall Foundation. We thank Patrick Rothwell and Dr. Matt Kelly in the Thomas laboratory and Drs. Paul Mermelstein and Paulo Kofuji for comments on earlier versions of this manuscript. We also thank Jason Klug, Greg Cardinal, and Kacey Rajkovich for technical assistance.
References
- Alkon DL. Changes of membrane currents during learning. J Exp Biol. 1984;112:95–112. doi: 10.1242/jeb.112.1.95. [DOI] [PubMed] [Google Scholar]
- Ambroggi F, Ishikawa A, Fields HL, Nicola SM. Basolateral amygdala neurons facilitate reward-seeking behavior by exciting nucleus accumbens neurons. Neuron. 2008;59:648–661. doi: 10.1016/j.neuron.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargas J, Galarraga E, Aceves J. An early outward conductance modulates the firing latency and frequency of neostriatal neurons of the rat brain. Exp Brain Res. 1989;75:146–156. doi: 10.1007/BF00248538. [DOI] [PubMed] [Google Scholar]
- Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- Belleau ML, Warren RA. Postnatal development of electrophysiological properties of nucleus accumbens neurons. J Neurophysiol. 2000;84:2204–2216. doi: 10.1152/jn.2000.84.5.2204. [DOI] [PubMed] [Google Scholar]
- Benavides DR, Quinn JJ, Zhong P, Hawasli AH, DiLeone RJ, Kansy JW, Olausson P, Yan Z, Taylor JR, Bibb JA. Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J Neurosci. 2007;27:12967–12976. doi: 10.1523/JNEUROSCI.4061-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Carlezon WA., Jr Ion channels and intracellular signaling proteins as potential targets for novel therapeutics for addictive and depressive disorders. Pharmacol Ther. 2005;108:65–75. doi: 10.1016/j.pharmthera.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Mercuri N, Stanzione P, Stefani A, Bernardi G. Intracellular studies on the dopamine-induced firing inhibition of neostriatal neurons in vitro: evidence for D1 receptor involvement. Neuroscience. 1987;20:757–771. doi: 10.1016/0306-4522(87)90239-9. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Galarraga E, Bargas J, Sierra A, Aceves J. The role of calcium in the repetitive firing of neostriatal neurons. Exp Brain Res. 1989;75:157–168. doi: 10.1007/BF00248539. [DOI] [PubMed] [Google Scholar]
- Ghitza UE, Fabbricatore AT, Prokopenko VF, West MO. Differences between accumbens core and shell neurons exhibiting phasic firing patterns related to drug-seeking behavior during a discriminative-stimulus task. J Neurophysiol. 2004;92:1608–1614. doi: 10.1152/jn.00268.2004. [DOI] [PubMed] [Google Scholar]
- Ghitza UE, Prokopenko VF, West MO, Fabbricatore AT. Higher magnitude accumbal phasic firing changes among core neurons exhibiting tonic firing increases during cocaine self-administration. Neuroscience. 2006;137:1075–1085. doi: 10.1016/j.neuroscience.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- Hollander JA, Carelli RM. Abstinence from cocaine self-administration heightens neural encoding of goal-directed behaviors in the accumbens. Neuropsychopharmacology. 2005;30:1464–1474. doi: 10.1038/sj.npp.1300748. [DOI] [PubMed] [Google Scholar]
- Hollander JA, Carelli RM. Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J Neurosci. 2007;27:3535–3539. doi: 10.1523/JNEUROSCI.3667-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Cascini MG, Gordon AS, Diamond I, Bonci A. Cooperative activation of dopamine D1 and D2 receptors increases spike firing of nucleus accumbens neurons via G-protein βγ subunits. J Neurosci. 2003;23:5079–5087. doi: 10.1523/JNEUROSCI.23-12-05079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XT, Basu S, White FJ. Repeated cocaine administration suppresses HVA-Ca2+ potentials and enhances activity of K+ channels in rat nucleus accumbens neurons. J Neurophysiol. 2004;92:1597–1607. doi: 10.1152/jn.00217.2004. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Mu P, Moyer JT, Wolf JA, Quock RM, Davies NM, Hu XT, Schlüter OM, Dong Y. Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J Neurosci. 2009;29:5820–5831. doi: 10.1523/JNEUROSCI.5703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito R, Robbins TW, Everitt BJ. Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat Neurosci. 2004;7:389–397. doi: 10.1038/nn1217. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Hu XT. Exciting inhibition in psychostimulant addiction. Trends Neurosci. 2006;29:610–616. doi: 10.1016/j.tins.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev. 2004;27:765–776. doi: 10.1016/j.neubiorev.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Kita H, Kita T, Kitai ST. Regenerative potentials in rat neostriatal neurons in an in vitro slice preparation. Exp Brain Res. 1985;60:63–70. doi: 10.1007/BF00237019. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Klug JR, Cardinal G, Thomas MJ. Repeated in vivo cocaine induces a firing rate depression in accumbens neurons that is persistent and not affected by drug challenge. Soc Neurosci Abstr. 2008;34:661–19. [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Marder E, Prinz AA. Modeling stability in neuron and network function: the role of activity in homeostasis. Bioessays. 2002;24:1145–1154. doi: 10.1002/bies.10185. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Agolia R, Arts MP, Groenewegen HJ, Zahm DS. Morphological differences between projection neurons of the core and shell in the nucleus accumbens of the rat. Neuroscience. 1992;50:149–162. doi: 10.1016/0306-4522(92)90389-j. [DOI] [PubMed] [Google Scholar]
- Nelson CL, Milovanovic M, Wetter JB, Ford KA, Wolf ME. Behavioral sensitization to amphetamine is not accompanied by changes in glutamate receptor surface expression in the rat nucleus accumbens. J Neurochem. 2009;109:35–51. doi: 10.1111/j.1471-4159.2009.05911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum ES, Xu ZC, Wilson CJ. Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J Neurophysiol. 1994;71:1174–1189. doi: 10.1152/jn.1994.71.3.1174. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Physiological and morphological properties of accumbens core and shell neurons recorded in vitro. Synapse. 1993;13:135–160. doi: 10.1002/syn.890130206. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. Amsterdam: Elsevier Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Pennartz CM, Dolleman-Van der Weel MJ, Lopes da Silva FH. Differential membrane properties and dopamine effects in the shell and core of the rat nucleus accumbens studied in vitro. Neurosci Lett. 1992;136:109–112. doi: 10.1016/0304-3940(92)90660-y. [DOI] [PubMed] [Google Scholar]
- Peoples LL, Cavanaugh D. Differential changes in signal and background firing of accumbal neurons during cocaine self-administration. J Neurophysiol. 2003;90:993–1010. doi: 10.1152/jn.00849.2002. [DOI] [PubMed] [Google Scholar]
- Peoples LL, Uzwiak AJ, Guyette FX, West MO. Tonic inhibition of single nucleus accumbens neurons in the rat: a predominant but not exclusive firing pattern induced by cocaine self-administration sessions. Neuroscience. 1998;86:13–22. doi: 10.1016/s0306-4522(98)00116-x. [DOI] [PubMed] [Google Scholar]
- Peoples LL, Uzwiak AJ, Gee F, West MO. Tonic firing of rat nucleus accumbens neurons: changes during the first 2 weeks of daily cocaine self-administration sessions. Brain Res. 1999;822:231–236. doi: 10.1016/s0006-8993(98)01271-2. [DOI] [PubMed] [Google Scholar]
- Peoples LL, Kravitz AV, Lynch KG, Cavanaugh DJ. Accumbal neurons that are activated during cocaine self-administration are spared from inhibitory effects of repeated cocaine self-administration. Neuropsychopharmacology. 2007;32:1141–1158. doi: 10.1038/sj.npp.1301203. [DOI] [PubMed] [Google Scholar]
- Perez MF, White FJ, Hu XT. Dopamine D(2) receptor modulation of K(+) channel activity regulates excitability of nucleus accumbens neurons at different membrane potentials. J Neurophysiol. 2006;96:2217–2228. doi: 10.1152/jn.00254.2006. [DOI] [PubMed] [Google Scholar]
- Robinson T, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28:4028–4036. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha SA, Fields HL. Inhibitions of nucleus accumbens neurons encode a gating signal for reward-directed behavior. J Neurosci. 2006;26:217–222. doi: 10.1523/JNEUROSCI.3227-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology. 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Wood DA, Rebec GV. Dissociation of core and shell single-unit activity in the nucleus accumbens in free-choice novelty. Behav Brain Res. 2004;152:59–66. doi: 10.1016/j.bbr.2003.09.038. [DOI] [PubMed] [Google Scholar]
- Zahm DS. An integrative neuroanatomical perspective on some subcortical substrates of adaptive responding with emphasis on the nucleus accumbens. Neurosci Biobehav Rev. 2000;24:85–105. doi: 10.1016/s0149-7634(99)00065-2. [DOI] [PubMed] [Google Scholar]
- Zahm DS, Brog JS. On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience. 1992;50:751–767. doi: 10.1016/0306-4522(92)90202-d. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Hu XT, White FJ. Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci. 1998;18:488–498. doi: 10.1523/JNEUROSCI.18-01-00488.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Cooper DC, White FJ. Repeated cocaine treatment decreases whole-cell calcium current in rat nucleus accumbens neurons. J Pharmacol Exp Ther. 2002;301:1119–1125. doi: 10.1124/jpet.301.3.1119. [DOI] [PubMed] [Google Scholar]