

Abstract
Lung injury from pulmonary contusion is a common traumatic injury, predominantly seen after blunt chest trauma such as in vehicular accidents. The local and systemic inflammatory response to injury includes activation of innate immune receptors, elaboration of a variety inflammatory mediators, and recruitment of inflammatory cells to the injured lung. Using a mouse model of pulmonary contusion, we had previously shown that innate immune Toll like receptors 2 and 4 (TLR2 and TLR4) mediate the inflammatory response to lung injury. In this study, we used chimeric mice generated by adoptive bone marrow (BM) transfer between TLR2−/− or TLR4−/− and wild-type (WT) mice. We found that in the lung, both BM-derived and non-myeloid cells contribute to TLR-dependent inflammatory responses after injury in a cell type specific manner. We also show a novel TLR2 dependent injury mechanism that is associated with enhanced airway epithelial cell apoptosis and increased pulmonary FasL and Fas expression in the lungs from injured mice. Thus, in addition to cardiopulmonary physiological dysfunction, cell type specific TLR and their differential response to injury may provide novel specific targets for management of patients with pulmonary contusion.
Keywords: mouse model, toll-like receptor, neutrophil, apoptosis
INTRODUCTION
Trauma remains a leading cause of morbidity and mortality in the United States despite marked advancements in the care of injured patients. Traumatic injury results in activation of inflammation, and although essential for wound repair and healing, these mechanisms contribute to the mortality seen after injury (1,2). In its early stages, the inflammatory response to traumatic injury is similar to that seen following infection in that activation of the innate immune response has been shown to require TLR mediated signaling (3). At least 13 TLRs have been identified, recognizing conserved and essential components of bacteria (eg. bacterial lipopolysaccharide, LPS) and viruses (eg. double stranded RNA) broadly classified as “pathogen associated molecular patterns” (PAMPS) (4). All utilize the intracellular adapter proteins MyD88 or TRIF in order to induce NFκB activation and subsequent inflammatory mediator expression. The endogenous, injury associated TLR ligands have been termed “danger-associated molecular patterns” (DAMPS) or “alarmins” in that they activate innate immune mechanisms warning the host of tissue injury. Evidence supporting a role for TLR2, −3, −4, and −9 signaling after noninfectious injury has been reported by a number of groups, including our own, and a number of DAMPS have been implicated that are potentially liberated after traumatic lung injury including hyaluronan, heparin-sulfate, heat shock proteins (HSP) 60, HSP70, High Mobility Group Box-1 (HMGB-1) protein, mitochondrial DNA, and surfactant proteins A and D (5-12). In an animal model of pulmonary contusion that mimics a common traumatic lung injury in humans, we previously showed that TLR2 and TLR4 were activated and mediated an inflammatory response to injury (8,9). Both TLR2 and TLR4 are widely expressed by multiple cell types in the lung including alveolar macrophages, epithelial and endothelial cells (13). To better understand the mechanism(s) of traumatic injury, in the present report we determined cell type specific TLR responses to pulmonary contusion. This study reveals the presence of a novel TLR2 dependent, neutrophil independent mechanism of lung injury that is associated with enhanced airway epithelial cell apoptosis and Fas/FasL activation.
MATERIALS AND METHODS
Animals
Age matched (8-9 weeks) wild-type (WT), TLR2−/−, TLR4−/−, and GFP mice (C57/BL6 background) were utilized in this study. The TLR2−/− mice were provided by S. Akira (Osaka University, Osaka Japan); WT, TLR4−/−, and GFP mice were obtained from Jackson Laboratories (Bar Harbor, ME). All animals were bred and maintained under pathogen-free conditions at the animal facility at Wake Forest University Health Sciences (WFUHS). The protocol used in this study was approved by the WFUHS Animal Care and Use Committee (#A10-157).
Adoptive BM Transfer
To create BM chimeras (14), mice received a single irradiation of 900 rad and were reconstituted with 106 donor BM cells. Chimeric mice were used 8 weeks later; BM donor/host chimera genotypes used in these studies are shown in Table I. GFP mice were used as BM donors in studies to validate engraftment (n=9). Whole blood was stimulated with LPS (100ng/ml) or lipotecoic acid (LTA, 10ng/ml) to validate phenotype response in BM transfer (n=2).
TABLE I. Resultant genotype* of chimeric mice.
| DONOR | HOST | DONOR/HOST |
|---|---|---|
| WT | WT | WT/WT |
| WT | TLR2−/− | WT/TLR2- |
| WT | TLR4−/− | WT/TLR4- |
| TLR2−/− | WT | TLR2-/WT |
| TLR4−/− | WT | TLR4-/WT |
The genotype of BM donor, host recipient and resulting mouse chimeras used in this study are shown. All data is labeled with the DONOR/HOST genotype as shown.
Blunt Chest Injury Model
Injury was induced using the Cortical Contusion Impactor (CCI) as previously described in detail (9). Briefly, the right chest of an anesthetized mouse is struck with the CCI with defined force and depth of penetration; control animals received anesthetic alone. Serum, tissue, and bronchoalveolar lavage (BAL) samples were collected at various times after injury.
BAL
After sacrifice, the trachea was cannulated and lavage performed. BAL cell counts were performed after trypan blue exclusion and cytospins were prepared to obtain differential cell counts as previously described (9). For protein analysis, an initial 0.5ml of BAL was collected and an aliquot of the supernatant reserved for ELISA.
FACS analysis
GFP expression on F4/80 positive cells was assessed by FACS as previously described (15). Briefly, BAL was collected and F/80 staining was done according to manufacturer’s instructions (eBioscience, Inc., San Diego, CA). GFP expression on F4/80 positive cells was analyzed by flow cytometry using a BD FACSCalibur™ system (BD Biosciences) and BD CellQuest™ Pro software (BD Biosciences). Histograms were generated using WinMDI 2.9 software (J Trotter, TSRI).
Cytokine and Chemokine expression
Serum levels of IL-6 and CXCL1 were measured using commercially available ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Samples were assayed in duplicate.
Arterial Blood Gas
Arterial blood gas (ABG) was determined as previously described (16). Arterial blood was collected and used to determine the partial pressure of arterial oxygen (PaO2). PaO2 was measured in each sample using a Stat Profile pHOx Blood gas Analyzer (Nova Biomedical, Waltham, MA) according to the manufacturer’s instructions. Data are expressed as PaO2/FiO2 as an index of lung dysfunction.
Immunohistochemistry
Lung tissue samples were fixed in 10% formalin, embedded in paraffin and sectioned as previously described (16). DNA fragmentation in apoptotic cells was detected using the DeadEnd™ Colorimetric dUTP nick end labeling (TUNEL) system (Promego, Madison WI) following manufacturer’s instructions. Fas in lung tissue sections was visualized using Fas antibody, X-20 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), an avidin/biotin blocking kit, SP-2001, the VectaStain ABC Kit, PK-4001 and peroxidase substrate DAB, SK-4001 according to manufacturer’s instructions (Vector Laboratories, Inc., Burlingame, CA). Counterstaining was performed using Mayers Hematoxylin.
Immunoblot
Protein levels of FAS in lung tissue samples were visualized using anti-FAS antibody (Santa Cruz Biotechnology, CA) and enhanced chemiluminescence (ECL, Pierce) according to the manufacturer’s instructions. FAS protein was quantitated on immunoblots using ImageJ (NIH, http://imagej.nih.gov/ij).
Statistical analysis
All data are analyzed using GraphPad Prism (v4.03, San Diego, CA) and expressed as the mean ± SD of independent observations as indicated in the Figure Legends. Student t-test or one-way analysis of variance (1way ANOVA) with Bonferroni multiple comparison post test was used to compare injury groups as appropriate. p-values <0.05 were considered to be significant.
RESULTS
Validation of adoptive transfer in mouse chimeras
To increase our understanding of the mechanisms of traumatic lung injury, we created TLR deficient chimeras to determine cell type specific responses of lung non-myeloid and BM-derived TLR2 and TLR4. To assess BM cell engraftment in the lungs after adoptive transfer, we used GFP mice as BM donors into WT hosts and found expression of GFP in more than 90% of BAL alveolar macrophages (Fig. 1A, GFP/WT). To test the phenotypic response of BM transfer, we stimulated whole blood with TLR ligands LTA (TLR2) or LPS (TLR4) to test for loss of response with TLR deficient BM donors in TLR positive hosts. As shown in Figure 1, whole blood leukocytes had a response to LTA (Fig. 1B) and LPS (Fig. 1C) stimulation that was phenotypically identical to BM donors’ genotype. These data show that the adoptive transfer resulted in BM engraftment and localization of BM derived cells in the lung, and acquisition of phenotypic response of BM derived cells in whole blood according to BM donor genotype. The genotypes of mouse chimeras generated for this study are summarized in Table 1. Chimeric WT hosts with WT BM did not show any significant difference from intact WT mice (data not shown) in all injury responses assessed.
Figure 1. Validation of adoptive transfer in BM chimeras.
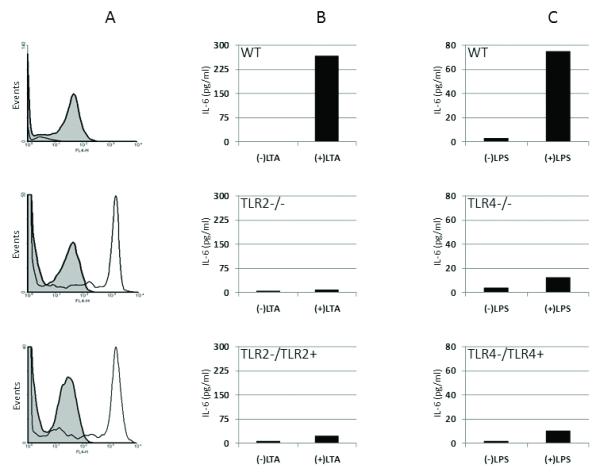
BM chimeras were generated as described in the methods. A. BAL from WT (top), GFP (middle) and a WT mouse chimera with GFP BM (GFP/WT) (bottom) was analyzed by FACS as described in Methods. F4/80+ cells (shaded histogram) in the BAL were analyzed for GFP expression (open histogram). GFP expression on F4/80+ cells present in GFP or GFP/WT mouse BAL was not observed in WT BAL. B. Whole blood from WT (top), TLR2 deficient (TLR2−/−) (middle) and a TLR2 positive mouse chimera with TLR2 deficient BM (TLR2-/TLR2+) (bottom) was stimulated ex vivo for 6H to test for blood leukocyte response to purified TLR2 ligand, LTA. Increased IL-6 levels in blood leukocytes in WT is absent in TLR2−/− or TLR2 deficient BM chimera. C. Whole blood from WT (top), TLR4 deficient (TLR4−/−) (middle) and a TLR4 positive mouse chimera with TLR4 deficient BM (TLR4-/TLR4+) (bottom) was stimulated ex vivo for 6H to test for blood leukocyte response to purified TLR4 ligand, LPS. Increased IL-6 levels in blood leukocytes in WT is absent in TLR4−/− or TLR4 deficient BM chimera. Data shown validates adoptive transfer (n=9) and BM donor phenotype (n=2) each experimental group.
TLR2 and TLR4 dependent injury response: Neutrophil recruitment in traumatic lung injury
Recruitment of activated neutrophils (PMN) to the injured lung is a hallmark of injury and inflammation and closely predicts posttraumatic lung dysfunction. As shown in Figure 2, we used mouse BM chimeras to identify cell type specific contribution to PMN recruitment to the injured lung after traumatic injury. We found that non-myeloid cell and BM derived cell TLR2 and TLR4 dependent activities potentially coordinate to elicit PMN migration to the injured lung. Reconstitution of WT neutrophilia (8,9) (data not shown) was observed in WT/WT chimeras after injury (Fig. 2, WT/WT). We found that absence of TLR2 on either non-myeloid (Fig. 2, WT/TLR2-) or BM cells (Fig. 2, TLR2-/WT) will decrease PMN recruitment independent of TLR4. In contrast, TLR4 expression by pulmonary non-myeloid cells in the presence of TLR2 is required for PMN entry into the alveolar space (Fig. 2, compare WT/TLR4- and TLR4-/WT). These findings suggest that cell type specific TLR response has distinct and potential complementary functions in PMN transmigration and activation.
Figure 2. TLR2 and TLR4 dependent injury response: Neutrophil recruitment in traumatic lung injury.
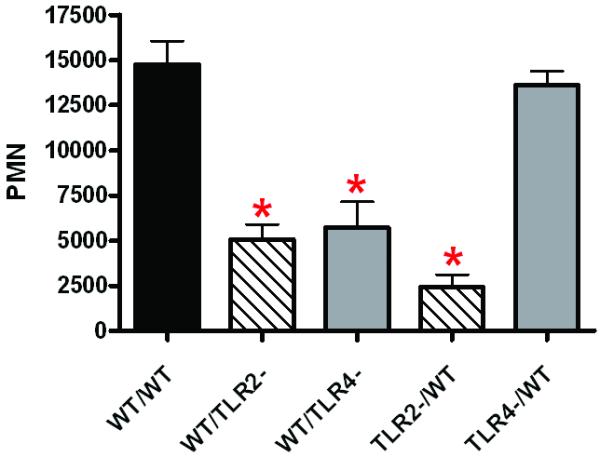
Mouse BM chimeras were generated as described in the methods and genotypes created as shown in Table I. BAL was collected at 24H after injury and PMN counted as described in the methods. Total PMN in the BAL of the experimental groups (n=5 mice in each experimental group) are shown. Significant differences (*p<0.05) from WT/WT are indicated.
TLR2 and TLR4 dependent injury response: Inflammatory mediators in traumatic lung injury
After traumatic injury in humans, serum levels of CXC chemokines and IL-6 have been associated with injury severity and increased mortality. Using our mouse model, we previously found increased levels of CXCL1 and IL-6 after pulmonary contusion similar to human pulmonary contusion (8,9). In this study and as shown in Figure 3, we used chimeric mice to determine the cell type specific contribution to elaboration of inflammatory mediators after lung injury. We found that animals deficient in either non-myeloid cell TLR2 (Fig 3A and B, WT/TLR2-) or TLR4 (Fig 3A and B, WT/TLR4-) expression showed a modest, reproducible decrease in serum levels of CXCL1 (Fig 3A) and IL-6 (Fig 3B). Reconstitution of WT cytokine and chemokine expression (8,9) (data not shown) was observed in WT/WT chimeras after injury (Fig. 3A and B, WT/WT). These data suggest that non-myeloid cell TLR2 and TLR4 participate and potentially play redundant or cooperative roles in activation of inflammatory mediators after injury. In contrast, we found significantly decreased CXCL1 and IL-6 serum levels in injured animals with TLR2 deficient BM cells (Fig 3A and B, TLR2-/WT); injured animals with TLR4 deficient BM cells showed significant decreases in only serum IL-6 expression (Fig 3B, TLR4-/WT). Taken together, the results suggest that TLR2 on BM derived cells is necessary and sufficient for CXCL1 expression and PMN recruitment to the injured lung.
Figure 3. TLR2 and TLR4 dependent injury response: Inflammatory mediators in traumatic lung injury.

Serum was collected at 3H after injury and assayed for CXCL1 and IL-6 by ELISA as described in the methods. CXCL1 (A) and IL-6 (B) levels in the serum of the experimental groups (n=6 for each experiment group) are shown. Significant differences (*p<0.05) from WT/WT are indicated.
TLR2 and TLR4 dependent injury response: Lung dysfunction in traumatic lung injury
Clinically significant lung dysfunction can be quantitated by the PaO2/FiO2 (P/F) ratio with reduced ratios being indicative of lung injury. We had previously found significantly improved lung function in TLR4 and TLR2 deficient injured mice with values that were near uninjured PaO2/FiO2 ratios in TLR2 and MyD88 deficient injured mice (8,9). These results supported the concept of TLR4 and TLR2 mediated lung dysfunction after chest injury and suggest a greater role for TLR2 in the pathogenesis of lung injury after contusion. To confirm and extend these studies, we now show that WT/WT and chimeras with either TLR4 deficient BM or pulmonary non-myeloid cells (Fig 4,) showed significant lung dysfunction after injury (Fig. 4, WT/WT, TLR4-/WT, WT/TLR4-). We further found that PaO2/FiO2 values in injured WT/TLR2-mice were indistinguishable from uninjured mice (8,16) (PaO2/FiO2=433; data not shown) suggesting that post-injury lung dysfunction is largely dependent on pulmonary non-myeloid TLR2 expression (Fig. 4, WT/TLR2-).
Figure 4. TLR2 and TLR4 dependent injury response: Lung dysfunction in traumatic lung injury.
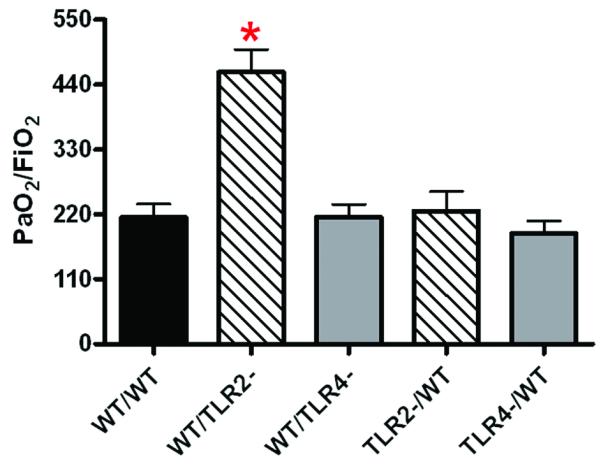
Lung dysfunction at 24H after injury was assessed by arterial blood gas oxygenation as described in the methods and reported as PaO2/FiO2 for the experimental groups (n=7 for each experiment group) as shown. Significant differences (*p<0.05) from WT/WT are indicated.
TLR2 specific injury response: Non-myeloid cell apoptosis and Fas/FasL activation
As we previously reported that post-injury lung dysfunction is dependent on pulmonary PMN recruitment (16), we sought an explanation for the observation that non-myeloid cell TLR2 expression plays a dominant role in lung dysfunction in absence of significant pulmonary neutrophilia. This result suggested the presence of a second mechanism of lung injury that is TLR2 mediated and PMN independent. In adult respiratory distress syndrome (ARDS) and in other models of blunt chest trauma (17,18), enhanced alveolar epithelial cell apoptosis via activation of the Fas/FasL system has been implicated as a mechanism of lung injury. To test for non-myeloid cell apoptosis in our model of pulmonary contusion, we used TUNEL staining to detect apoptotic cells in injured lungs after contusion. As shown in Figure 5, pulmonary contusion induced apoptosis of lung non-myeloid cells in WT injured mice. To establish a specific role for TLR2, we assessed non-myeloid cell apoptosis in TLR2 deficient mice and found that apoptosis was markedly reduced in injured TLR2−/− mice. To identify apoptotic pathways activated after pulmonary contusion, we determined FasL levels in the BAL after injury and found a significant increase at 24H after injury in WT mice (Figure 6A) that was significantly reduced in TLR2 deficient mice (Figure 6B). Using immunohistochemistry, we observed reduced Fas expression in non-myeloid cells of TLR2 deficient mice after injury, when compared to the lung of injured WT mice (Figure 7A). This observation was confirmed and quantitated by immunoblot analysis that demonstrated a significant increase in Fas levels in lung tissue lysates at 24 hours after injury (Figure 7B). Injury induced Fas was significantly decreased in TLR2 deficient mice. These data suggest that pulmonary cell apoptosis contributes to postinjury lung dysfunction, and can act independent of PMN mediated lung injury. We further conclude traumatic lung injury activates innate immune responses (like TLR mediated inflammation) as well as apoptosis pathways (FasL:Fas). We suggest that TLR2 activation may have a unique role in coordinating the injury response to pulmonary contusion.
Figure 5. TLR2 specific injury response: Alveolar cell apoptosis.

Immunohistochemistry and TUNEL staining was used to detect apoptotic cells in uninjured or injured lungs at 3H or 24H after contusion as described in the methods. WT and TLR2−/− sections are shown at 63X and 100X and are representative of at least 3 independent observations. For comparison, TUNEL staining in DNAse treated samples from WT uninjured mice are shown as a positive control.
Figure 6. TLR2 specific injury response: FasL expression.
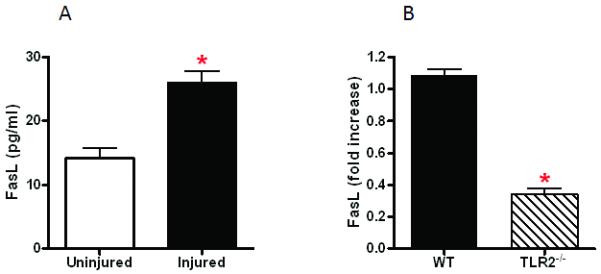
FasL protein in the BAL was measured as described in the methods. FasL levels in uninjured and injured (n=3 in each experimental group) mice at 24H after pulmonary contusion are shown (A). The relative level of FasL (expressed as fold increase over uninjured) in WT and TLR2 deficient mice (n=10 in each experimental group) at 24H after pulmonary contusion are shown (B). Significant differences (*p<0.05) are indicated.
Figure 7. TLR2 specific injury response: Fas expression.
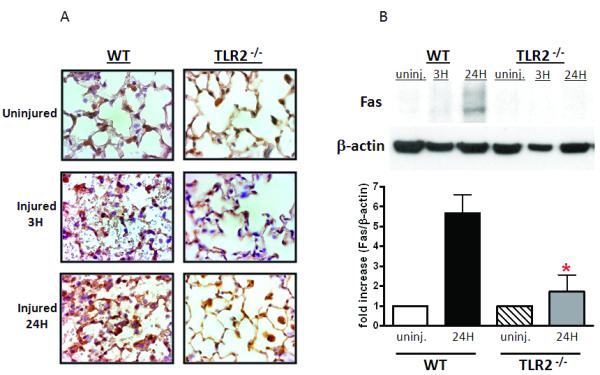
Immunohistochemistry (A) and immunoblot analysis (B) was used to visualize Fas expression in uninjured or injured lungs at 3H or 24H after contusion as described in the methods. WT and TLR2−/− sections are shown at 100X and are representative of at least 3 independent observations. Immunoblot of WT and TLR2−/−lung lysates are shown (B, top) and Fas protein levels quantitated and normalized to β-actin (B, bottom, n=3). Significant differences (*p<0.05) are indicated.
DISCUSSION
Pulmonary contusion is a potentially lethal traumatic lung injury predominantly seen after blunt mechanisms of injury such as car accidents. In addition to its known affects on cardiopulmonary physiology, innate immune mechanisms are activated with elaboration of various inflammatory mediators (IL-1b, IL-6, IL-8, prostanoids) and recruitment of activated neutrophils (PMN) to the injured lung (19). In order to better understand the inflammatory response to lung contusion, we use a murine model to study the activation of innate immunity after injury. We (8,9,20) and others (21,22) have found that cytokines and CXC chemokines are expressed, endothelial expression of adhesion receptors (ICAM-1) is increased, and systemic PMN are activated and recruited, in a manner similar to what is seen after human injury. We also found that the innate response is initiated by TLR2 and TLR4 activation and is MyD88 dependent (8). Further studies (16) identified a primary role for PMN recruitment after pulmonary contusion where we showed decreased injury with inhibition of CXC chemokines and found that neutrophil NADPH oxidase contributes to post injury lung dysfunction. In the studies presented here, we determine cell type specific TLR responses to pulmonary contusion.
TLR2 and TLR4 dependent injury response: PMN recruitment in traumatic lung injury
We used our mouse BM chimeras to identify cell type specific contribution to PMN recruitment to the injured lung after traumatic injury. Our findings suggest that non-myeloid cell and BM derived cell TLR2 and TLR4 dependent activities coordinate to elicit PMN migration to the injured lung. PMN recruitment is dependent on the presence of TLR2 on either non-myeloid or BM derived cells while TLR4 expression by pulmonary non-myeloid cells in the presence of TLR2 is required for PMN entry into the alveolar space. These findings indicate that the spatial and cell type specific TLR response has distinct and complementary function in PMN transmigration and activation. This concept is supported by other studies: Sabroe et al (23) used highly purified PMN and TLR2 or TLR4 specific ligands to identify a cell type specific role for TLR4 in regulating PMN lifespan. In this study, PMN co-culture with purified monocytes revealed that TLR4 activation of monocytes, but not TLR2, was associated with the release of neutrophil survival factors; TLR2 activation in monocyte/neutrophil co-cultures did not prevent neutrophil apoptosis. The authors conclude that TLR mediated responses need to be understood both at the level of highly purified and mixed cell populations. Our studies begin to extend these experimental models to examine TLR biology in the animal in response to traumatic injury.
Other studies have suggested distinct TLR functions in the setting of a uniform stimulus. In models of pulmonary infection (24) and ischemia/reperfusion (25), TLR2 was shown to be critical for PMN activation and transmigration into the alveolar space. Whereas in mice lacking TLR4 exclusively on endothelial cells, TLR4 expression was found to be required for pulmonary PMN sequestration in response to intratracheal LPS (26). Moreover, it has been shown that TLR4 dependent NADPH oxidase activation will upregulate TLR2 expression after ischemia/reperfusion, suggesting that is possible that TLR4 may, in part, augment TLR2 dependent responses (27). These feedback mechanisms may play an important role in the “second hit” phenomenon due to innate immune priming that is often observed after severe injury and studies to understand the cell type specificity of this response can be explored in our chimeric mouse models.
The differences in TLR functional discrimination may further be explained by ligand availability at site(s) of injury and receptor affinity. Within the lung, after noninfectious injury, a number of endogenous TLR ligands are liberated including low molecular weight hyaluronan, HMGB-1, surfactant-A and -D, heat shock proteins, biglycan, and CpG DNA (3,28). Many of these DAMPS have been demonstrated to activate both TLR2 and TLR4. As such, TLR ligands may be localized to areas of injury and may have varying affinity for TLR2 or TLR4 expressed by effector cells. However, we have consistently observed distinct dependence on TLR2, although activation of TLR4 is also a major contributor to the injury and inflammatory response after contusion (8,9). TLR 2 and 4 are expressed on myeloid cells, alveolar epithelial and pulmonary endothelial cells; however, surface availability/distribution of TLRs may vary. For example, TLR2 has been demonstrated to localize to the apical surface of airway epithelial cells while TLR4 is present in a more basolateral distribution suggesting a selective participation of TLRs in the airway inflammatory response (29).
TLR2 and TLR4 dependent injury response: Inflammatory mediators in traumatic lung injury
In human studies, elevated levels of IL-6 and CXC chemokines after traumatic injury have been associated with increased mortality and the onset of acute respiratory distress syndrome (ARDS) and multiple organ dysfunction (MODS) (30,31). In this study, our data suggest that non-myeloid TLR2 and TLR4 participate and play potentially redundant or cooperative roles in activation of inflammatory mediators after injury. However, we found significantly decreased CXCL1 and IL-6 serum levels in injured animals with TLR2 deficient BM cells. In contrast, only serum IL-6 expression was significantly decreased in injured animals with TLR4 deficient BM cells. These results suggest a hierarchical function for TLRs in BM derived cell response to pulmonary contusion where unlike TLR4, TLR2 on BM derived cells appears to be necessary and sufficient for PMN recruitment to the injured lung mediated at least in part, by CXCL1. Our study supports the inflammatory character (in addition to physical damage) of acute trauma and highlights the role of BM derived cells in the circulation and tissue in the response to lung injury.
TLR2 and TLR4 dependent injury response: Lung dysfunction in traumatic lung injury
While consistent with previous studies, our present study reveals a second, complementary, TLR2 dependent injury mechanism that is independent of pulmonary PMN recruitment and associated with enhanced airway epithelial cell apoptosis. Furthermore, an association between airway epithelial cell apotosis and elevated BAL Fas and caspase-8 is demonstrated after injury. This response is absent in TLR2 deficient mice, suggesting the presence crosstalk between TLR2 and the Fas/FasL system. The alveolar epithelial cell is increasingly being recognized as an active participant in the lung’s response to injury and infection. Specifically, apoptosis of distal airway epithelial cells has been proposed as an important mechanism of traumatic lung injury (32). Recently, elevated levels of FasL in the BAL after blunt chest injury have been described, implicating the Fas-FasL in the pathogenesis of lung injury following pulmonary contusion (18,33). These studies demonstrated an association between Fas-FasL system activation and postinjury inflammation and alveolar epithelial cell apoptosis. Furthermore, studies by Matute-Bello demonstrated Fas-induced lung injury is dependent upon Fas expression by nonmyeloid cells of the lung (34). In pulmonary fibrosis models (35) and hyperoxic lung injury (36), the absence of TLR2 and TLR4 proved detrimental to survival despite reduced inflammatory mediator expression and reduced pulmonary PMN. Lung injury was associated with enhanced alveolar epithelial cell apoptosis, suggesting a link between TLR activation and epithelial cell apoptosis. These studies and our present results suggest the presence of a novel second mechanism of injury involving TLR2 dependent Fas-induced apoptosis of alveolar epithelial cells and suggest the presence of crosstalk between the FasL/Fas system and TLR2 signaling. Taken together, our previous and current studies reveal PMN dependent and independent injuries after pulmonary contusion. The modest improvement in lung function we observed in TLR4 deficient animals (8) may be explained by the reduction PMN recruitment to the lung. The results of our current study support a pulmonary non-myeloid cell specific, TLR2 dependent mechanism that appears to play a principal role in traumatic lung injury. We suggest that a second injury mechanism, dependent on non-myeloid cell TLR2, may activate FAS mediated apoptosis and initiate lung dysfunction and respiratory distress. Future studies will address the role evaluating epithelial cell apoptosis in our model of lung injury.
As we previously reported in TLR deficient animals, the TLR2- and TLR4-dependent injury and inflammatory response appeared straightforward (8,9); however, our results using cell type specific TLR deficient chimeric mice reveals the complexity and redundancy in the injury response. The response is likely initiated immediately at the site of injury with a number of known and unknown DAMPS that are released and react with a number of different pattern recognition receptors. The variable activation of different cells types, signaling pathways and mediators have specific, redundant, and interdependent local and systemic responses. Our reproducible injury model of pulmonary contusion is an important tool to understand the host response to trauma. Interventions that prevent or alleviate downstream adverse events, like adult respiratory distress syndrome or sepsis, are likely dependent on identifying and specifically targeting critical cellular responses. The studies presented here begin to address the challenge of identifying cell type specific responses to injury.
ACKNOWLEDGMENT
The authors would like to thank Ms. Sarah E. Jones for technical assistance.
Funding Disclosure
This work was supported by the Clowes ACS/AAST/NIGMS Mentored Clinical Scientist Development Award (JH), and NIH grants K08 GM083154 and supplement (JH), R01 AI065791 (CM), and R01 AI079144 (CM).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury multiple organ failure. Injury. 2009;40:912–918. doi: 10.1016/j.injury.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38:1336–1345. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Xiang M, Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol. Med. 2010;16:69–82. doi: 10.2119/molmed.2009.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 5.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill R, Ruan X, Menzel CL, Namkoong S, Loughran P, Hackam DJ, Billiar TR. Systemic inflammation and liver injury following hemorrhagic shock and peripheral tissue trauma involve functional TLR9 signaling on bone marrow-derived cells and parenchymal cells. Shock. 2011;35:164–170. doi: 10.1097/SHK.0b013e3181eddcab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henning LN, Azad AK, Parsa KV, Crowther JE, Tridandapani S, Schlesinger LS. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J. Immunol. 2008;180:7847–7858. doi: 10.4049/jimmunol.180.12.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoth JJ, Wells JD, Brownlee NA, Hiltbold EM, Meredith JW, McCall CE, Yoza BK. Toll-like receptor 4-dependent responses to lung injury in a murine model of pulmonary contusion. Shock. 2009;31:376–381. doi: 10.1097/SHK.0b013e3181862279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoth JJ, Hudson WP, Brownlee NA, Yoza BK, Hiltbold EM, Meredith JW, McCall CE. Toll-like receptor 2 participates in the response to lung injury in a murine model of pulmonary contusion. Shock. 2007;28:447–452. doi: 10.1097/shk.0b013e318048801a. [DOI] [PubMed] [Google Scholar]
- 10.Prince JM, Levy RM, Yang R, Mollen KP, Fink MP, Vodovotz Y, Billiar TR. Toll-like receptor-4 signaling mediates hepatic injury and systemic inflammation in hemorrhagic shock. J. Am. Coll. Surg. 2006;202:407–417. doi: 10.1016/j.jamcollsurg.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 11.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang ZL. Important aspects of Toll-like receptors, ligands and their signaling pathways. Inflamm. Res. 2010;59:791–808. doi: 10.1007/s00011-010-0208-2. [DOI] [PubMed] [Google Scholar]
- 14.Spangrude GJ. Assessment of lymphocyte development in radiation bone marrow chimeras. Curr. Protoc. Immunol. 2008 doi: 10.1002/0471142735.im0406s81. Chapter 4: Unit. [DOI] [PubMed] [Google Scholar]
- 15.Hoth JJ, Martin RS, Yoza BK, Wells JD, Meredith JW, McCall CE. Pulmonary contusion primes systemic innate immunity responses. J. Trauma. 2009;67:14–21. doi: 10.1097/TA.0b013e31819ea600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoth JJ, Wells JD, Hiltbold EM, McCall CE, Yoza BK. Mechanism of neutrophil recruitment to the lung after pulmonary contusion. Shock. 2011;35:604–609. doi: 10.1097/SHK.0b013e3182144a50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS) J. Immunol. 1999;163:2217–2225. [PubMed] [Google Scholar]
- 18.Seitz DH, Palmer A, Niesler U, Braumuller ST, Bauknecht S, Gebhard F, Knoferl MW. Altered expression of fas receptor on alveolar macrophages and inflammatory effects of soluble fas ligand following blunt chest trauma. Shock. 2011;35:610–617. doi: 10.1097/SHK.0b013e318213665d. [DOI] [PubMed] [Google Scholar]
- 19.Raghavendran K, Notter RH, Davidson BA, Helinski JD, Kunkel SL, Knight PR. Lung contusion: inflammatory mechanisms and interaction with other injuries. Shock. 2009;32:122–130. doi: 10.1097/SHK.0b013e31819c385c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoth JJ, Stitzel JD, Gayzik FS, Brownlee NA, Miller PR, Yoza BK, McCall CE, Meredith JW, Payne RM. The pathogenesis of pulmonary contusion: an open chest model in the rat. J Trauma. 2006;61:32–44. doi: 10.1097/01.ta.0000224141.69216.aa. [DOI] [PubMed] [Google Scholar]
- 21.Knoferl MW, Liener UC, Perl M, Bruckner UB, Kinzl L, Gebhard F. Blunt chest trauma induces delayed splenic immunosuppression. Shock. 2004;22:51–56. doi: 10.1097/01.shk.0000127684.64611.5c. [DOI] [PubMed] [Google Scholar]
- 22.Raghavendran K, Davidson BA, Woytash JA, Helinski JD, Marschke CJ, Manderscheid PA, Notter RH, Knight PR. The evolution of isolated bilateral lung contusion from blunt chest trauma in rats: cellular and cytokine responses. Shock. 2005;24:132–138. doi: 10.1097/01.shk.0000169725.80068.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, Dower SK, Whyte MK. Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J. Immunol. 2003;170:5268–5275. doi: 10.4049/jimmunol.170.10.5268. [DOI] [PubMed] [Google Scholar]
- 24.Chin AC, Fournier B, Peatman EJ, Reaves TA, Lee WY, Parkos CA. CD47 and TLR-2 cross-talk regulates neutrophil transmigration. J. Immunol. 2009;183:5957–5963. doi: 10.4049/jimmunol.0900789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khandoga AG, Khandoga A, Anders HJ, Krombach F. Postischemic vascular permeability requires both TLR-2 and TLR-4, but only TLR-2 mediates the transendothelial migration of leukocytes. Shock. 2009;31:592–598. doi: 10.1097/SHK.0b013e318193c859. [DOI] [PubMed] [Google Scholar]
- 26.Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, Kubes P. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J. Clin. Invest. 2003;111:1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Neutrophil NAD(P)H oxidase is required for hemorrhagic shock-enhanced TLR2 up-regulation in alveolar macrophages in response to LPS. Shock. 2007;28:213–218. doi: 10.1097/shk.0b013e318033ec9d. [DOI] [PubMed] [Google Scholar]
- 28.Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J. Leukoc. Biol. 2010;87:989–999. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- 29.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van HA, Prince A. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004;30:777–783. doi: 10.1165/rcmb.2003-0329OC. [DOI] [PubMed] [Google Scholar]
- 30.Cuschieri J,E, Bulger V, Schaeffer S, Sakr AB, Nathens L, Hennessy J, Minei EE, Moore G, O’Keefe, Sperry J, Remick D, Tompkins R, Maier RV. Early elevation in random plasma IL-6 after severe injury is associated with development of organ failure. Shock. 2010;34:346–351. doi: 10.1097/SHK.0b013e3181d8e687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keel M, Ecknauer E, Stocker R, Ungethum U, Steckholzer U, Kenney J, Gallati H, Trentz O, Ertel W. Different pattern of local and systemic release of proinflammatory and anti-inflammatory mediators in severely injured patients with chest trauma. J. Trauma. 1996;40:907–912. doi: 10.1097/00005373-199606000-00008. [DOI] [PubMed] [Google Scholar]
- 32.Perl M, Chung CS, Lomas-Neira J, Rachel TM, Biffl WL, Cioffi WG, Ayala A. Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am. J. Pathol. 2005;167:1545–1559. doi: 10.1016/S0002-9440(10)61240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seitz DH, Perl M, Mangold S, Neddermann A, Braumuller ST, Zhou S, Bachem MG, Huber-Lang MS, Knoferl MW. Pulmonary contusion induces alveolar type 2 epithelial cell apoptosis: role of alveolar macrophages and neutrophils. Shock. 2008;30:537–544. doi: 10.1097/SHK.0b013e31816a394b. [DOI] [PubMed] [Google Scholar]
- 34.Matute-Bello G, Liles WC, Frevert CW, Dhanireddy S, Ballman K, Wong V, Green RR, Song HY, Witcher DR, Jakubowski JA, Martin TR. Blockade of the Fas/FasL system improves pneumococcal clearance from the lungs without preventing dissemination of bacteria to the spleen. J. Infect. Dis. 2005;191:596–606. doi: 10.1086/427261. [DOI] [PubMed] [Google Scholar]
- 35.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Shan P, Qureshi S, Homer R, Medzhitov R, Noble PW, Lee PJ. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J Immunol. 2005;175:4834–4838. doi: 10.4049/jimmunol.175.8.4834. [DOI] [PubMed] [Google Scholar]