

Summary
BACE1 (β-secretase) plays a central role in the β-amyloidogenesis of Alzheimer’s disease (AD). The ubiquitin–proteasome system, a major intracellular protein quality control system, has been implicated recently in BACE1 metabolism. We report that the SCFFbx2-E3 ligase is involved in the binding and ubiquitination of BACE1 via its Trp 280 residue of F-box-associated domain. Physiologically, we found that Fbx2 was expressed in various intracellular organelles in brain neurons and that BACE1 is colocalized with Fbx2 and the amyloid precursor protein (APP), mainly at the early endosome and endoplasmic reticulum. The former are believed to be the major intracellular compartments where the APP is cleaved by BACE1 and β-amyloid is produced. Importantly, we found that overexpression of Fbx2 in the primary cortical and hippocampal neurons derived from Tg2576 transgenic mice significantly promoted BACE1 degradation and reduced β-amyloid production. In the search for specific endogenous modulators of Fbx2 expression, we found that PPARγ coactivator-1α (PGC-1α) was capable of promoting the degradation of BACE1 through a mechanism involving Fbx2 gene expression. Interestingly, we found that the expression of both Fbx2 and PGC-1α was significantly decreased in the brains of aging Tg2576 mice. Our in vivo studies using a mouse model of AD revealed that exogenous adenoviral Fbx2 expression in the brain significantly decreased BACE1 protein levels and activity, coincidentally reducing β-amyloid levels and rescuing synaptic deficits. Our study is the first to suggest that promoting Fbx2 in the brain may represent a novel strategy for the treatment of AD.
Keywords: Alzheimer’s disease, BACE1, Fbx2, synaptic function
Introduction
The major cause of Familial Alzheimer’s disease (FAD) neuropathology is associated with the increased processing of the amyloid precursor protein (APP) caused by autosomal dominant mutations (APP, presenilin 1 and 2) along the β-and ξ-secretase pathways (Selkoe, 2001), resulting in increased Aβ production (Nishitomi et al., 2006). The mechanism associated with Aβ generation in sporadic AD (SAD), which constitutes approximately 95% of all AD cases, is currently unknown (Corder et al., 1993; Hardy & Selkoe, 2002). Protein levels of the β-site APP-cleaving enzyme 1 (BACE1), a rate-limiting enzyme that cleaves APP at the N-terminus of the Aβ domain (Vassar et al., 1999; Yan et al., 1999), have been found to be significantly increased in the brains of SAD cases (Fukumoto et al., 2004; Yang et al., 2003). Experimental evidence suggests that BACE1 deficiency in the brain significantly attenuates AD-type neuropathology, indicating that BACE1 inhibition could be an ideal target for AD treatment (O’Connor et al., 2008; Ohno et al., 2004; Singer et al., 2005).
The ubiquitin–proteasome system (UPS) is a major protein quality control system for intracellular protein degradation (Ciechanover & Brundin, 2003; Oddo, 2008) in which E3 ligases play an essential role in substrate recognition, ubiquitination, and degradation (Hershko et al., 2000). Recent evidence supports the hypothesis that the UPS is involved in BACE1 degradation, coincidentally with the attenuation of Aβ accumulation in the brain (Qing et al., 2004). However, the molecular mechanisms involved in this process are currently unknown. A newly identified, neuron-specific F-box protein, Fbx2, which binds the Skp1 domain of the Skp1-Cullin-1-Fbox-forming SCFFbx2 E3-ligase complex, has been found to specifically recognize N-glycoproteins by recognizing N-glycosylation motifs through its F-box-associated (FBA) domain (Mizushima et al., 2004; Nelson et al., 2007; Staub & Rotin, 2006; Yoshida et al., 2002). SCF E3 ligase complexes are well known to contribute to the rapid elimination of neuron-specific membrane proteins by promoting substrate ubiquitination (Cardozo & Pagano, 2004; Jin et al., 2003). The great similarity of BACE1 N-glycosylation motifs with other discovered substrates suggests that BACE1 may be a potential target of the Fbx2-E3 ligase.
The transcriptional coactivator PGC-1α, a key regulator of energy metabolism, exerts potent effects on nuclear gene transcription (Puigserver & Spiegelman, 2003). PGC-1α expression decreases significantly in aging and AD brains as part of the progression of AD dementia, and it is involved in the amyloidogenic processing of APP (Qin et al., 2009; Wu et al., 2006). Recently, researchers have reported that PGC-1α is involved in regulating the expression of certain E3 ligase genes, such as atrogin-1 E3 ligase and FOXO3A, in cardiac and skeletal muscles (Hanai et al., 2007; Olson et al., 2008; Williamson et al., 2009). Manipulation of atrogin-1 expression through PGC-1α affects muscle dystrophy. Thus, it would be useful to know whether the effects of PGC-1α on AD-type amyloidosis are linked with the regulation of the Fbx2-E3-ligase-mediated BACE1 degradation.
Based on this consideration, in this study, we tested the hypothesis that the Fbx2-E3 ligase would modulate BACE1 ubiquitination and degradation in the brain, which may beneficially influence AD-type amyloid neuropathology and synaptic dysfunction in the Tg2576 mouse, a well-characterized AD mouse model that demonstrates impairments in synaptic function and amyloid neuropathology comparable to those of human patients (Chapman et al., 1999; Lesne et al., 2006). We also explored the relationship between the expression of both Fbx2-E3 ligase and PGC-1α and BACE1 degradation in the brains of Tg2576 mice and in human AD cases. Finally, using a stereotaxic injection technique, we delivered adenoviral vectors carrying Fbx2 complementary DNA (cDNA) into the brains of Tg2576 mice and observed the role of Fbx2 on BACE1 degradation and synaptic function in vivo. Our study is the first to demonstrate that the modulation of BACE1 degradation through the UPS could be a potential target for AD therapies.
Results
Fbx2 bound with BACE1 via its FBA domain
The major function of Fbx2 is to bind with neuronal membrane N-glycoproteins for ubiquitination. We tested whether Fbx2 interacts with BACE1 in mammalian cells by generating a stable human embryonic kidney (HEK293) cell line overexpressing Myc-tagged-BACE1 and by cloning FLAG-tagged full-length Fbx2 from a mouse brain cDNA library. Using this cell line, we found that FLAG-tagged Fbx2 physically bound to Myc-tagged BACE1, as assessed by co-immunoprecipitation with an anti-FLAG antibody followed by Western blot detection with an anti-Myc antibody (Fig. 1A, lanes 2, 3). We detected no noticeable binding of Fbx2 to other membrane proteins, such as APP (data not shown).
Fig. 1.

Interaction of Fbx2 with BACE1. (A) HEK293 cells stably expressing Myc-BACE1 were cotransfected with FLAG-Fbx2 plasmids. After immunoprecipitation of Fbx2–FLAG from whole-cell extracts, bound BACE1 was analyzed using an antibody against Myc. Tissue lysates from mouse (B) and human (C) brains were subjected to coimmunoprecipitation with anti-Fbx2 antibody and detected with anti-BACE1 antibody. In both tissue co-immunoprecipitations, anti-mouse or anti-rabbit IgG antibody were used as negative controls. (D) Depiction of Fbx2 and mutants. (E, F) Fbx2 binds to BACE1 through FBA (ΔC) domain. Two hundred and ninety-three cells were transiently transfected with FLAG-tagged Fbx2 variants. Cell lysates were immunoprecipitated with goat anti-FLAG antibody and the bound BACE1 was analyzed using an antibody against Myc. (G) FBA domain point mutations, Trp280, Tyr279, and K281 were transfected into 293 cells. Immunoprecipitation shows that only W280 is required for the binding. (H-T) Colocalization of Fbx2 and BACE1. 293 stable cells co-expressing Fbx2 and its variants were immunostained with anti-Myc and –FLAG antibodies and examined via confocal microscopy. White arrows indicate colocalization of Fbx2 and BACE1 (panels J and T). FBA domain deletion (panel K-M) and the W280A mutation were not colocalized with BACE1 (panels N-P) Scale bar: 10 μm. FBA, F-box-associated.
Next, we determined the physiologic relevance of this finding by assessing the endogenous binding of Fbx2 to BACE1 in mouse and human brains. Membrane fractions from brain tissue lysates were co-immunoprecipitated with a monoclonal antibody raised against mouse or human Fbx2 and probed by anti-BACE1 antibody. We found that Fbx2 and BACE1 were physically associated, as assessed by Western blot detection of Fbx2 and BACE1 from immunoprecipitated complexes in either mouse or human brains (Fig. 1B,C).
The Fbx2 protein contains three major function domains: the FBA domain, which directly binds with N-linked high-mannose oligosaccharides; the N-terminal Pro-Glu-Ser-Thr (PEST) domain, which interacts with the C-terminus of Hsc-70-interacting domain (CHIP) (Dickey et al., 2006); and the F-box domain, which binds with the SCF ubiquitin ligase (Mizushima et al., 2004; Yoshida et al., 2002; Fig. 1D). To map the binding domains of Fbx2 with BACE1, FLAG-tagged Fbx2 and variants – including the deletions of FLAG-Fbx2-ΔFBA (ΔC), FLAG-Fbx2-ΔPEST (ΔP), FLAG-Fbx2-ΔFbox (ΔF) and FLAG-Fbx2-ΔPEST/Fbox (ΔP/ΔF) domains – were transiently transfected into HEK293 cells constitutively expressing Myc-tagged BACE1. We found that only the FBA domain is required for the interaction of Fbx2 with BACE1 (Fig. 1E,F).
Recently, Fbx2 X-ray crystallography and NMR spectroscopy had suggested that the hydrophobic pocket at the edge of the ‘β-sandwich’ is essential for binding Fbx2 with chitobiose moieties of N-glycans (Mizushima et al., 2004). To verify the specific binding site of FBA for BACE1 in vitro, we transfected FLAG-tagged Fbx2 variants with point mutations localized within the hydrophobic pocket at residues Y279A, W280A, and K281A of the FBA domain into HEK293 cells constitutively expressing Myc-tagged BACE1. We found that mutations at positions Lys281 and Tyr279 of the FBA domain did not interfere with BACE1 binding, whereas the mutation at position Trp280 selectively prevented BACE1 binding (Fig. 1G), suggesting that the tryptophan residue in the hydrophobic pocket of the FBA domain is crucial for FBA/BACE1 binding. Based on this evidence, we then performed double immunofluorescence staining and confocal microscopy analysis to visualize the subcellular colocalization of Fbx2 and BACE1, which confirmed that the FBA domain and its W280 residue, but not the K281 residue, are required for this colocalization (Fig. 1P,T).
Fbx2 was colocated with BACE1 and APP in neurons
Based on this finding of Fbx2’s specific binding to BACE1, we continued to survey the subcellular distribution of the Fbx2/BACE1 binding complex in mouse brains using sucrose-gradient ultracentrifugation separation assays from mouse brain extracts. We found that, similarly to APP, Fbx2 immunoreactive material was broadly distributed in various intracellular organelles, including early (EEA1 marker) and late (adaptin marker) endosome, lysosome (cathepsin marker). BACE1 overlaps with Fbx2, as well as APP, mainly at early endosomes (Fig. 2A, lanes 7, 8, 9); only trace amount of BACE1 could be identified in late endosomes and lysosomes (Fig. 2A, lanes 1, 2, and lanes 3, 4, respectively). A significant signal from synaptophysin (a specific neuronal marker) showed in this probe, indicating that this preparation was neuron enriched. We further probed the expression of Fbx2 in brain slices with avidin–biotin–peroxidase complex (ABC) staining (Fig. 2B). The colocalization of Fbx2 with BACE1 in vivo (Fig. 2C–E) was confirmed by fluorescence immunostaining in neurons, based on size and distributions with neuron-specific immunoreactivity to synaptophysin on semi-adjacent slices. These results demonstrated that Fbx2 physiologically binds and colocalizes with BACE1 in the same intracellular compartment.
Fig. 2.

Subcellular localizations of the Fbx2 and BACE1 complex in brain neurons. (A) A stepwise sucrose gradient ultracentrifugation for the subcellular fraction of brain tissue. Nine hundred and fifty microliter fractions were collected and analyzed by Western blot. Fbx2 and APP are broadly expressed in lysosomes (cathepsin D) and early and late endosomes (EEA1 and adaptin) and are colocalized with BACE1, mainly in the early endosome (EEA1, lanes 7, 8, 9) and much less in the late endosome (lanes 3, 4) and lysosome (lane 1, 2). (B) Sections of brain slices immunostained by the ABC procedure with Fbx2 antibody. (C–E) Immunofluorescence staining of BACE1 and Fbx2, showing the colocalization of BACE1 and Fbx2 in brain slices (white arrow). (F) In vitro ubiquitination of BACE1 by the SCFFbx2 E3-ligase system. Fbx2 enhances WT BACE1 ubiquitination (left pane) but not mutant BACE1 ubiquitination (right panel). (G) N-glycosylation of BACE1 is required for the Fbx2-mediated binding. Cell lysate from cells treated (+) with PNGase F at 37 °C for 1 h or from controls (−) were immunoprecipitated with anti-FLAG antibody and probed with anti-Myc antibody. Only glycosylated BACE1 can be immunoprecipitated by Fbx2. ABC, avidin–biotin–peroxidase complex; APP, amyloid precursor protein. WT, wild-type.
Fbx2-facilitated BACE1 ubiquitination and proteasomal degradation
To study the physiologic relevance of the binding of Fbx2 and BACE1, we employed an in vitro ubiquitination assay system (Yoshida et al., 2002) that mimics in vivo ubiquitination conditions containing the Fbx2-E3 ligase complex. We found that Fbx2 causally increases ubiquitinated BACE1 levels (Fig. 2F, left panel), indicating that Fbx2 promotes activated ubiquitin attached to lysine residue of BACE1. The Fbx2-mediated ubiquitination of BACE1 was rather specific, as mutations of seven of its lysine residues (replaced by alanine; Costantini et al., 2007) significantly attenuated its ubiquitination (Fig. 2F, right panel). Because the proposed function of the Fbx2 complex is specifically to recognize certain N-glycans, we then tested whether the N-glycosylation of BACE1 was required for Fbx2 binding. We found that treating lysate with PNGase-F, an amidase that cleaves between the innermost GlcNAc and asparagine residues of high mannose from N-linked glycoproteins (Yoshida et al., 2002), significantly abolished the binding of Fbx2 to BACE1 (Fig. 2G).
To further determine whether Fbx2 facilitates BACE1 degradation via proteasomal degradation, full-length and Fbx2 variants were cotransfected into HEK293 cells stably expressing BACE1. We found that BACE1 protein levels were significantly reduced in cells overexpressing full-length Fbx2, but not mutant Fbx2 (Fig. 3A). The application of lactacystin, a specific proteasome inhibitor, abolished Fbx2 effects, suggesting that BACE1 degradation occurs via UPS. In parallel studies, we explored BACE1 functional activity by assessing fluorescence resonance energy transfer (FRET) using an 18 amino-acid peptide containing the Swedish mutant APP as a substrate (Vassar et al., 1999). We found that the cleavage of the synthetic APP peptide was reduced by Fbx2, as expected (Fig. 3B).
Fig. 3.

Fbx2 promotes BACE1 protein degradation. (A) HEK293 cells stably expressing human BACE1 protein were cotransfected with Fbx2 and treated with lactacystin. The levels of BACE1 protein were probed with anti-BACE1 antibody. Equal sample loading was normalized with antibody to β-tubulin (right panel). Protein amounts were quantified by Image J and represented in the graph. Data are means ± SEM (error bar) of the results from three independent experiments. One-way anova reveals the differences between each condition with control group; *P < 0.05. (B) BACE1 enzyme activity from the same cell lysate was measured using a BACE1-FRET kit (EMD biology). Data are means ± SEM, one-way anova,*P < 0.05 n = 5. (C) Hippocampal neurons derived from Tg2576 embryos infected with adenoviral GFP or retroviral Fbx2 shRNA. BACE1 protein levels were increased by silencing endogenous Fbx2. Data represent means ± SEM; n = 5 per culture; *P < 0.05 relative to adeno-GFP-infected cell cultures. (D) BACE1 stability was measured by cycloheximide (CHX) chase followed by Western blot. After 48 h of transfection, cells were treated with CHX (50 μm) and chased at the indicated time points. (E) The quantified relative levels of BACE1 against levels of β-tubulin and plotted as a percentage of the level of BACE1 at chase time t = 0. Two-way anova reveals the differences between Fbx2 and control group; *P < 0.05 compared with control group. (F) Stable HEK293 cells were transiently transfected with human APPswe and Fbx2, and treated with lactacystin. The β-secretase-cleaved APP C-terminal fragments CTF99 were analyzed by Western blot using anti-APP C-terminal antibody (APP 8344). Fbx2 reduces C99 cleavage (lanes 3, 4, 5). APP, amyloid precursor protein; FRET, fluorescence resonance energy transfer.
To investigate the effects of Fbx2 on BACE1 degradation, we constructed retroviral vectors encoding Fbx2 short hairpin RNAs (shRNAs) that target mouse Fbx2 (Figure S1a) and studied the effects of silencing endogenous Fbx2 on BACE1 degradation in primary neuron cultures. We found that while the endogenous Fbx2 was efficiently silenced by retroviral Fbx2 shRNA, BACE1 levels were stabilized, confirming that Fbx2 is essential for the UPS-mediated BACE1 degradation (Fig. 3C). To further study the time course of the effects of Fbx2 on BACE1 degradation, we performed BACE1 cycloheximide chase assays using HEK293 cells constitutively expressing Myc-BACE1 and coexpressing wild-type (WT) or Fbx2 mutants. Similarly, we found that Fbx2 significantly increased BACE1 degradation over time (Fig. 3D; two-way anova analysis for the control and Fbx2 overexpression groups, P < 0.05), and the degradation of BACE1 was abolished by lactacystin and the Fbx2-FBA mutant (Fig. 3D,E).
Next, we further tested the role of Fbx2 in AD-type β-amyloidosis. We generated an adenoviral vector expressing Fbx2 cDNA (Figure S1b) and delivered it into hippocampal neuron cultures derived from Tg2576 embryos. We found that Fbx2 significantly attenuated the BACE1-mediated β-cleavage of APP, as assessed by generation of the C-terminal fragment C99 (Fig. 3F).
PGC-1α-promoted Fbx2 gene expression and BACE1 ubiquitination
We previously reported that PGC-1α gene expression is reduced both in patients with AD and in an AD mouse model (Qin et al., 2009) and is involved in amyloidogenic processing of APP by repressing BACE1 gene promoter activity through the activation of PPARγ (Sastre et al., 2006). Recently, it has been reported that the SCFCdc4-E3 ligase is involved in PGC-1α-regulated gene expression and that PGC-1α is an important regulator of certain E3 ligase genes in cardiac and skeletal muscle (Olson et al., 2008). Based on this consideration, we tested whether PGC-1α could influence Fbx2-E3 ligase expression and, further, whether it could influence BACE1 degradation in neurons.
We found that overexpression of PGC-1α in primary cultured neurons derived from embryonic Tg2576 mouse brains significantly increased both Fbx2 protein levels (Fig. 4A) and gene expression (Fig. 4B), as analyzed by Western blot and qRT-PCR, respectively. Conversely, silencing retroviral shRNA PGC-1α attenuated Fbx2 protein levels and gene expression (Fig. 4A,B).
Fig. 4.

PGC-1α-mediated BACE1 degradation through Fbx2. Hippocampal neurons derived from Tg2576 embryos were infected with adenoviral GFP, adenoviral PGC-1α, PGC-1α shRNA or scrambled shRNA, respectively. (A) Forty-eight hours after infection, Fbx2 protein levels were probed using anti-Fbx2 antibody. PGC-1α markedly increases Fbx2 protein expression (lane 2), while adenoviral-PGC-1α shRNA reduces the Fbx2 levels (lane 3) (N = 5, means ± SEM, *P < 0.05. (B) Total RNA was extracted from transfected cells. Levels of Fbx2 mRNA were quantified by qRT-PCR (n = 5 independent studies; *P < 0.05). (C) PGC-1α promotes BACE1 ubiquitination. HEK293 cells stably expressing Myc-BACE1 were infected with adenoviral-PGC-1α or adenoviral-GFP constructs and treated with 5 μm lactacystin. BACE1 was immunoprecipitated with an antibody against Myc, The ubiquitinated BACE1 was probed with anti-ubiquitin antibody. The levels of ubiquitinated BACE1 levels in the square area were quantified and represented in the graph (right panel). Data are means ± SEM of the results from two independent experiments; *P < 0.05 compared with adenoviral-GFP/lactacystin-treated cells (control). (D) Silencing endogenous PGC-1α blocks BACE1 degradation. Neurons were infected with adenoviral GFP, adenoviral PGC-1α, and adenoviral PGC-1α shRNA; (E) lactacystin blocked the effects of PGC-1α on BACE1 degradation. (F) Silencing Fbx2 diminishes the effects of PGC-1α on BACE1 degradation. (n = 5, data are means ± SEM; n = 5 per culture; *P < 0.05 relative to adenoviral-GFP-infected cell cultures).
As Fbx2 facilitates BACE1 ubiquitination, we then explored whether PGC-1α also enhances BACE1 ubiquitination. Using an HEK293 cell line stably expressing Myc-BACE1 and expressing exogenous viral PGC-1α, we found that the ubiquitinated BACE1 protein levels markedly increased in PGC-1α overexpressing HEK293 cells relative to control cells (Fig. 4C) in the presence of lactacystin, suggesting a mechanism through which PGC-1α-enhanced Fbx2 expression could facilitate BACE1 ubiquitination in the UPS.
PGC-1α-promoted BACE1 degradation via Fbx2-regulated degradation
In light of the evidence that PGC-1α increases BACE1 ubiquitination, we assessed whether PGC-1α is capable of promoting BACE1 degradation in primary neuron cultures. We found that exogenous overexpression of PGC-1α in primary neurons significantly decreased BACE1 levels (Fig. 4D) compared to overexpression of adenoviral Green Fluorescent Protein (GFP) (control). Conversely, PGC-1α silencing by adenoviral PGC-1α shRNA neuronal infection resulted in significantly increased BACE1 levels. Treatments with lactacystin (5 μm, 4 h) blocked BACE1 degradation (Fig. 4E), thereby strongly supporting the hypothesis that PGC-1α is involved in BACE1 degradation via the UPS. To further confirm that PGC-1α-mediated degradation of BACE1 requires Fbx2, we tested whether Fbx2 silencing using an shRNA technique would attenuate PGC-1α-mediated responses. We found that silencing Fbx2 shRNA significantly attenuated PGC-1α-mediated BACE1 degradation, again implying that PGC-1α-mediated BACE1 degradation occurs through Fbx2-mediated UPS ubiquitination (Fig. 4F).
Fbx2 acted downstream of PGC-1α in the regulation of BACE1 degradation
To further explore the correlation between Fbx2 and PGC-1α in the degradation of BACE1, we tested whether the silencing of PGC-1α could affect Fbx2-regulated BACE1 degradation. Interestingly, we found that while Fbx2 reduced BACE1 levels in primary cortical cultures obtained from Tg2576 embryos, conversely, silencing PGC-1α using shRNA technique had no effect on BACE1 degradation in neurons overexpressing Fbx2 (Fig. 5A). We also found that silencing PGC-1α did not change the effects of overexpressing Fbx2 on BACE1 activity (Fig. 5B), assessed by probing full-length APP and APP β-site fragment (C99) (Fig. 5C). This evidence strongly suggests that Fbx2 acts downstream of PGC-1α in the UPS-BACE1 degradation pathway and that the manipulation of Fbx2 expression may overcome the deleterious amyloidosis process caused by the downregulation of PGC-1α.
Fig. 5.

Fbx2 acts on the downstream of PGC-1α. (A) Silencing endogenous PGC-1α did not affect the role of Fbx2 in the BACE1 degradation in primary cultured neurons from E16 Tg2576 embryos. BACE1 protein was probed by Western blot with anti-BACE1 antibody and normalized by β-tubulin (left panel). The right panel shows quantification of BACE1 protein levels. Data are means ± SEM; n = 5, *P < 0.05 compared with control. (B) BACE1 activity was measured in (A) and quantified as a percent of control; *P < 0.05. (C) Full APP and BACE1 cleavage product APP-C terminal fragment C99 were analyzed by immunoblots with 6E10 antibody. APP, amyloid precursor protein.
Fbx2 and PGC-1α expressions were altered in AD brains
Based on the aforesaid studies showing that PGC-1α promoted BACE1 degradation and induction of the Fbx2-E3 protein, we explored the association of Fbx2 and PGC-1α protein expression with BACE1 in the brains of Tg2576 mice and postmortem human AD subjects. We found that significantly reduced levels of PGC-1α (Fig. 6A) coincided with significantly reduced levels of Fbx2-E3 ligase (Fig. 6B) and significantly increased levels of BACE1 (Fig. 6C) in the brains of 12- to 14-month-old Tg2576 mice relative to age- and gender-matched control WT littermates. Consistent with the suggested potentially central role of Fbx2 in the development of AD amyloid pathology, we found that AD brains (BA36) were characterized by significantly reduced levels of Fbx2 protein (Fig. 6D) compared to age-matched neurologic control cases, suggesting an inverse correlation between the expression of Fbx2 and PGC-1α proteins with abnormal post-translational modification of BACE1 in AD brains. We found no significant changes in Fbx2 mRNA expression (Figure S2a) and BACE1 (Figure S2b) as analyzed by qRT–PCR in postmortem human AD brains, implying that, in addition to PGC-1α, other factors may be involved in Fbx2 gene expression (Kato et al., 2005).
Fig. 6.

Fbx2 and PGC-1α expression are compromised in Tg2576 mice and human AD brains. (A) PGC-1α protein levels are reduced in the hippocampal formation of 12- to 14-month-old Tg2576 mice compared to age- and gender-matched WT littermates. (B and D) Fbx2 protein levels are decreased in 12-month-old Tg2576 mice and in the postmortem brains of patients with AD (parahippocampal gyrus; BA 36), respectively. (C) BACE1 protein levels are elevated in the hippocampal formation of 12- to 14-month-old Tg2576 mice compared to age- and gender-matched WT littermates. The lower panels in D, E, F, and G showed the quantification of immunoreactivity. Student’s t-test was used for statistical analysis. *P < 0.05, n = 5 per group. AD, Alzheimer’s disease; WT, wild-type.
Exogenous Fbx2 reduced BACE1 and improved synaptic function in vivo
To further investigate whether exogenous expression of Fbx2 can reduce BACE1 protein levels and eventually AD-type amyloidogenesis in vivo, we performed stereotaxic bilateral injections of control adenoviral-GFP or adenoviral-GFP-Fbx2 virus (2 μL) into the hippocampal formations of 12- to 14-month-old Tg2576 mice and age-, gender-, and strain-matched WT littermates. We first confirmed expression of the GFP-Fbx2 construct in the hippocampus surrounding the injection site 4 weeks after injection, as visualized by GFP-fluorescent microscope (Figure S3a–c). Excitingly, we found that exogenous adenoviral GFP-Fbx2 expression in the hippocampal formation of Tg2576 mice resulted in a significant attenuation of BACE1 immuno-reactivity in the CA1 pyramidal layer of the hippocampal formation (Fig. 7B,D) relative to the BACE1 immunoreactive signal in the hippocampal formation of control Tg2576 mice injected with adenoviral-GFP (Fig. 7A,C; analyzed by Student’s t-test, n = 5 per group, P < 0.05, Fig. 7E). Analysis of parallel tissue sections from the same Tg2576 mice revealed that exogenous adenoviral Fbx2 expression led to an approximate 30% reduction in Aβ1-42 peptide levels relative to the control adenoviral-GFP-treated group, as assessed by ELISA analysis (Fig. 7F).
Fig. 7.

Stereotaxic injection of Fbx2 reduces BACE1 levels and improves synaptic function. Mouse brain specimens were analyzed 4 weeks after injection of either adenoviral GFP-Fbx2 or control adenoviral-GFP constructs. (A, B) Representative micrographs of the BACE1 immunoreactive signal in the CA1 pyramidal layer of the hippocampal formation of Tg2576 mice 4 weeks after injection. Boxed areas of the CA1 region shown in (A) and (B) are enlarged in (C) and (D), respectively. Arrows show BACE1 immunostaining signal. (E) Quantitative assessment of the BACE1 immunoreactive signal in (C) and (D) (n = 15 slices/group, three consecutive sections/mouse, five mice in each group; *P < 0.05). (F) ELISA determination of Aβ1-42 levels in the hippocampal formation of Tg2576 mice 4 weeks after injection. Values are expressed as means ± SEM, n = 5 per group; *P < 0.05, Student t-test relative to control adenoviral GFP injection. (G) Analysis of long-term potentiation (LTP) in the hippocampal Schaffer collateral/CA1 region of Tg2576 mice shows that Fbx2 significantly increased LTP compared with adenoviral-GFP-treated Tg mice (these experiments were interleaved with those of adenoviral-GFP-treated mice). Arrows indicate time and pattern of the tetani. All data shown are means ± SEM; n = 10 slices from five mice in each group; two-way anova following post hoc analysis reveals P < 0.01. (H) Basal synaptic transmission at the connection of hippocampal slices from 12- to 14-month-old Tg2576 mice was not affected by Fbx2 4 weeks after injection (n = 10 slices from five mice in each group, two-way anova; P > 0.05).
Because reducing BACE1 levels in the brain is expected to attenuate the development of amyloid neuropathology (Nishitomi et al., 2006), we hypothesized that Fbx2-mediated promotion of BACE1 degradation by the UPS may attenuate the deficits of synaptic function in AD. We thus assessed long-term potentiation (LTP) in the hippocampal formations of 12- to 14-month-old Tg2576 mice, an age when LTP is severely impaired in both the CA1 and dentate gyrus regions of the hippocampus (Chapman et al., 1999; Hsiao et al., 1996). We found that compared to control Tg2576 mice, which were injected with the adenoviral GFP construct, exogenous adenoviral GFP-Fbx2 injection significantly improved the impaired LTP in the CA1 region of the hippocampus of Tg2576 mice, recorded 4 weeks after adenoviral injection (Fig. 7G, P < 0.01). The adenoviral Fbx2 did not affect the basal synaptic transmission (Fig. 7H). The delivery of adenoviral Fbx2 did not affect the LTP in WT littermates, and the LTP deficits in Tg2576 mice treated with adenoviral Fbx2 were nearly completely eliminated compared with those in WT control animals (data not shown). No impaired behavior was observed. These results indicated that exogenous Fbx2 expression may reduce AD-type amyloidosis through regulation of BACE1 degradation, coinciding with the rescue of AD-type synaptic dysfunction.
Discussion
We found that Fbx2-E3 ligase is crucial for UPS-mediated BACE1 degradation. In particular, we found that Fbx2 specifically targets BACE1 through its F-box domain at the early endosome, promoting BACE1 ubiquitination and degradation. Exogenous application of Fbx2 using an adenoviral gene delivery system attenuated AD-type β-amyloidogenesis and improves synaptic function in an AD mouse model. In addition, we found that PGC-1α was capable of promoting BACE1 degradation through the regulation of Fbx2 gene expression. This article largely extends our current knowledge about the molecular mechanisms underlying the regulation of BACE1 degradation by the UPS (Fig. 8).
Fig. 8.
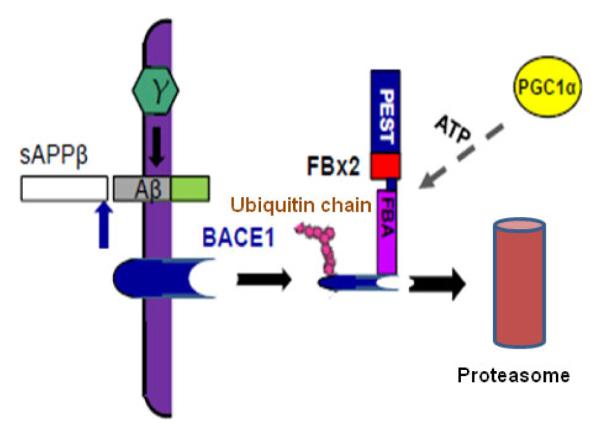
Working hypothesis of Fbx2 facilitating BACE1 degradation. In the AD brain, BACE1 is essential to cleave APP to generate Aβ. Fbx2 binds BACE1 through the F-box domain and promotes BACE1 ubiquitination and proteasomal degradation. PGC-1α mediated BACE1 metabolism through the enhancement of Fbx2 gene/protein expression and further reduced Aβ production. AD, Alzheimer’s disease; APP, amyloid precursor protein.
UPS-regulated protein quality control and BACE1 degradation
The important correlation between increases in BACE1 protein levels and increased amyloidogenesis in SAD patients, along with the failure to find efficient BACE1 inhibitors, highlights the importance of understanding the regulatory mechanisms underlying BACE1 degradation. Research has identified several proteins that interact with BACE1 and are linked with BACE1 degradation, including GGA3 (He et al., 2005; Tesco et al., 2007), reticulon (He et al., 2004), and sorting protein-related receptor/lipoprotein receptor L (Dodson et al., 2008). It has been suggested that the GGA3-targeted protein is ubiquitination-dependent (Puertollano & Bonifacino, 2004; Tesco et al., 2007), and more direct later evidence demonstrated that frame mutant ubiquitin (UBB+1) greatly slows BACE1 degradation (Zhang et al., 2010). However, the particular E3 ligases responsible for recognizing and targeting BACE1 and for initiating BACE1 ubiquitination have not yet been identified. The evidence reported here indicates that N-glycosylated BACE1 interacts with the Trp280 residue at the hydrophobic pocket of the F-box domain in Fbx2 and that the lysine residues in BACE1 are required for its ubiquitination.
Proper BACE1 N-glycosylation, processed in the Golgi and trans-Golgi network, plays a key role in the formation of the functional BACE1 enzyme. Fbx2 recognizes the innermost chitobiose of neuron-specific N-glycan proteins through the FBA domain. We found that similar to other Fbx2 substrates, BACE1 N-glycosylation is required for Fbx2-regulated binding and ubiquitination and that the Try280 residue in the hydrophobic pocket of the FBA domain is essential for interaction with the high-mannose oligosaccharides of BACE1, suggesting that BACE1 glycosylation is not only linked with its maturation in the endoplasmic reticulum, but also is involved in BACE1 ubiquitination in the endosome. The fact that mutations in Lys281 and Tyr279 residues have no effect on in this binding, suggesting that N-glycosylation sites among these substrates are important for the substrate reorganization. In addition, Fbx2 only accelerates WT BACE1 ubiquitination but not BACE1 with lysine mutations, suggesting that the lysine residues are not only important for acetylation modification (Costantini et al., 2007) but are also crucial for ubiquitination binding (Gong et al., 2006; Wilkinson et al., 1989).
PGC-1α-mediated signaling pathways in amyloid pathogenesis
PPARγ plays an important role in repressing BACE1 gene expression by directly binding with the BACE1 promoter (Sastre et al., 2006). An inhibition of PPARγ caused by inflammation factors, such as interleukins and interferons, increases BACE1 gene expression, providing evidence that links BACE1 increases with inflammatory reactions in AD brains (Sastre et al., 2006). PGC-1α, the transcriptional coactivator of PPARy involved in cellular energy metabolism and linked with numerous neurodegenerative diseases, has also been reported to play a role in amyloidogenic processing in the brain, though with unclear mechanisms (Qin et al., 2009; Wu et al., 2006). Here, we found that overexpression of PGC-1α is capable of promoting BACE1 degradation through the enhancement of Fbx2 gene/protein expression and of further reducing Aβ production. The gene silencing of PGC-1α by shRNA did not prevent Fbx2-regulated BACE1 degradation, suggesting that Fbx2 acts downstream of PGC-1α in the ubiquitin-BACE1 degradation pathway. Our finding that the decreased Fbx2 and increased BACE1 levels in the brains of old Tg2576 mice (12–14 months of age) and in postmortem AD brains of aging humans coincide with decreased PGC-1α expression (Qin et al., 2009) further supports this hypothesis. Precisely how PGC-1α regulates Fbx2 and causes further BACE1 degradation, and which nuclear factor is involved in its expression, are not yet understood. However, the enhanced turnover of PGC-1α proteins under oxidative stress conditions largely depends on the UPS (Anderson et al., 2008), suggesting an intimate link between the PGC-1α and E3 ligases in the UPS.
Reports have identified the most potent activator of PGC-1α as the transducer of regulated CREB (cAMP response element-binding protein) (TORC) (Kelly & Scarpulla, 2004; Wu et al., 2006). Previously, our group reported that the cAMP-PKA-pCREB signaling pathway is downregulated in the AD brain and that augmenting this pathway by increasing PKA activity improves the deficits of synaptic function in an AD mouse model (Gong et al., 2004, 2006; Smith et al., 2009). Again, our data here have consistently shown that delivering exogenous Fbx2 into the hippocampi of Tg2576 mice lessened amyloidosis and is capable of eliminating LTP deficits, supporting the hypothesis that Aβ has direct toxic effects on synaptic plasticity. As CREB phosphorylation is required for the expression of certain E3 ligase genes, we speculate that some of the nuclear actions of the cAMP/PKA/CREB pathway may be involved in PGC-1α-mediated Fbx2 expression.
It has been reported that the expression of the major components in UPS and PGC-1α is downregulated in aging brains in both animal models and human subjects and that this contributes to the formation of intracellular inclusion, to deficits of neuronal energy metabolism, and to neurodegenerative diseases (Grillari et al. 2006; Dröge & Schipper, 2007). This work has provided insights into the mechanisms of BACE1 ubiquitination and degradation by the UPS. Thus far, the search for efficient BACE1 inhibitors to treat AD is far from successful. On the other hand, completely blocking the activity of BACE1 would bring deleterious neurologic dysfunctions (Cole & Vassar, 2007; Hemming et al., 2009), because BACE1 also plays important physiologic roles in maintaining normal neuronal function. Our findings that upregulating Fbx2 in the AD brain can biologically reduce BACE1 levels/activity and Aβ production and improve synaptic function provides a novel molecular target for the future intervention of AD, as well as other age-related dementias.
Experimental procedures
Constructs, transfection, BACE1 HEK293 cell line, immunoprecipitation, and immunoblotting
Plasmids (pcDNA3) encoding WT and domain deletions of FLAG-tagged Fbx2-full, and ΔF, ΔP, ΔC and ΔP, as well as the Fbx2 point mutations W280A, K281A, and Y279, have been described (Mizushima et al., 2004; Yoshida et al., 2002). HEK293 cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum [Gibco (Invitrogen), Carlsbad, CA, USA] and penicillin/streptomycin (Invitrogen). The cell line (HEK293) stably expressing BACE1 was generated by expressing Myc-BACE1 plasmid (kindly provided by Dr Puiligri) into HEK293 cells and selected with G418 at 400 μg mL−1. Transfections were performed with Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. Forty-eight hours after transfection, cells were lysed using lysis buffer [20 mm Tris-HCl (pH 7.4), 1 m NaCl, 1 mm dithiothreitol, 1.0% Triton X-100], immunoprecipitated using the FLAG Immunoprecipitation kit (Sigma, St. Louis, MO, USA) or the c-Myc Tag IP kit (Pierce, Waltham, MA, USA) according to the manufacturers’ manuals. Proteins were probed using standard Western blotting procedures. The following antibodies were used: FLAG polyclonal (Sigma), mouse anti-BACE1 (Sigma), rabbit anti-BACE1 (Rockland Immunochemicals, Gilbert, PA, USA), anti-ubiquitin [Chemicon (Millipore), Billerica, MA, USA], mouse anti-Myc [Upstate Biotech (Millipore), Billerica, MA, USA], mouse anti-Fbx2 (Millipore) and monoclonal anti-Uch-L1 (Novocastra, Newcastle Upon Tyne, UK). In quantification experiments, cell extracts were usually normalized on the basis of total protein concentrations, and an equal amount of individual protein was loaded and finally normalized with β-tubulin or β–actin.
Sucrose density gradient fraction
Fresh mouse brain tissue was homogenized with PBS and centrifuged at 1700 g for 5 min, and the pellet was resuspended in lysis buffer [50 mm TrisHCl (pH 7.6), 1 mm EDTA, 10 μL of protease inhibitor cocktail (Sigma)] on ice and centrifuged at 1500 g for 5 min. The supernatant was centrifuged at 21 000 g for 30 min at 4 °C. The membrane pellet was resuspended in 0.2 mL of solubilization buffer [50 mm TrisHCl (pH 7.6), 1 mm EDTA, 150 mm NaCl, 10 mm iodoacetamide, 5 μL protease inhibitor cocktail, 10 mm 3-[(3-Cholamidopropyl)dimethylammonio]-1 propanesulfonate], and a 1 mL sample was placed on the top of a centrifuge tube prepared with solutions with successively decreasing sucrose densities (15–50%) layered upon one another and centrifuged in a swinging bucket SW 50.1 type rotor (Beckman Coulter, Brea, CA, USA) at RCFav 132 000 g for 16 h and 30 min at 4 °C. Twelve 1-mL fractions were collected from the top of the gradient. Equal volumes of solution from each fraction were separated on an SDS gel and subjected to Western blot analysis.
Production of adenoviral vectors
The adeno-GFP-PGC-1α and PGC-1α shRNA were kind gifts from Dr Puigserver. For the adenoviral-GFP-Fbx2, rat Fbx2 cDNA was subcloned into a pShuttle-IRES-hrGFP-2 vector [Stratagene (Agilent Technologies), Santa Clara, CA, USA], following an experimental protocol described previously (Puigserver & Spiegelman, 2003). After it was linearized with Pme I, it was cotransfected into BJ5183 cells with pAdEasy-1 DNA plasmid. A transformation colony was selected for kanamycin resistance. Purified plasmid was transfected to AD-293 cells to produce adenoviral Fbx2 using the calcium phosphate method. Infectious adenovirus containing GFP-Fbx2 cDNA was harvested at 48 and 72 h post-transfection and concentrated by ultracentrifugation (2 h at 50 000 g) and subsequently purified on a sucrose 20% gradient (2 h at 46 000 g). Vector concentrations were analyzed by using an immunocapture p24-gag ELISA (Alliance, DuPont/NEN) following the manufacturer’s instructions. The pShuttle-IRES-hrGFP-2 blank vector, used as the control, was linearized and cotransfected into BJ5183 cells with pAdEasy-1 DNA plasmid to produce the adenoviral GFP control vector. The transfection efficacy and protein expression of adeno-Fbx2 in HEK293 cells and primary neurons were characterized by fluorescence microscope and blotting using anti-Fbx2 antibody.
Retroviral Fbx2 shRNA and validation
Four shRNA sequences and a control were inserted into the pEGFP-V-RS shRNA Vector U6-shRNA expression cassette (OriGene, Rockville, MD, USA). The pGFP-V-RS Fbx2 shRNA constructs targeting different Fbx2 regions were transfected into packaging cells (Phoenix cell) at 70–80% confluence. On day two post-transfection, the media was collected and centrifuged at 2000 g for 5 min to remove cell debris. The supernatant was used directly for viral titering through infection of HEK293 cells. High-titer retroviral shRNA was used to infect HEK293 cells and primary cultured neurons. Forty-eight hours after infection, cell lysate was collected; Fbx2 levels were tested by Western blot analysis with anti-Fbx2 antibody.
Cycloheximide BACE1 degradation time course
FLAG-tagged Fbx2 or Fbx2 variants were transfected into a stable HEK293 BACE1 cell line grown in 100-mm dishes and starved overnight in serum-free DMEM medium. Forty-eight hours after transfection, the cells were treated with cyclohexomide (50 lg mL−1) at indicated time points before harvesting cells from each dish (Fig. 3D). Harvested cells were lysed, and Myc-tagged BACE1 proteins were separated on SDS-PAGE and analyzed by immunoblots.
Primary neuron culture, BACE1 activity measurements, and quantification of amyloid peptides by ELISA
In cell culture experiments, we used fetuses at embryonic day 15 from C57/B6J (control) or Tg2576 mice. We removed the uteruses and rinsed them in calcium- and magnesium-free Hanks buffered salt solution (HBSS) containing 0.27 mm pyruvate and 1× antibiotic cocktail [100 units mL−1 penicillin G, 0.25 μg mL−1 amphotericin B, and 100 units mL−1 streptomycin (Cellgro; Mediatech, Inc., Herndon, VA, USA)]. The cortex and hippocampi were dissected and then mechanically dissociated by fine-polished glass pipette after trypsin treatment, plated on six-well plates coated with poly-l-lysine and maintained in a defined serum-free medium containing minimum essential medium supplemented with glucose, albumin, pyruvic acid, antibiotics, and antimycotics. 12 day in vitro cells were used for the experiments. BACE1 activity was measured using as a substrate a secretase-specific FRET peptide conjugated with EDANS and DABCYL (EMD biology), based on the EVNLDAEF sequence of the ‘Swedish’ mutation in the APP, which is the BACE1 cleavage site. When the substrate is intact (i.e. uncleaved), the fluorescence from EDANS is effectively quenched by the DABCYL moiety. Cleavage of the peptide by β-secretase physically separates EDANS and DABCYL, allowing the emission of the fluorescence signal. The increase in fluorescence is measured at an excitation wavelength of 335–355 nm and an emission wavelength of 495–510 nm. The measurement of the Aβ levels has been described previously (Gong et al., 2004). Briefly, levels of Aβ1-40 and Aβ1-42 in primary cultured Tg2576 neurons infected with various adenoviral vectors were determined using sandwich-type ELISA (Biosource International, Camarillo, CA, USA). The background from control medium (transfected with the adeno-GFP vector) was subtracted from the sample values.
In vitro ubiquitination assay
Recombinant His-tagged rat Fbx2 complex and each SCFFbx2 (FLAG-tagged human Skp1, human Cul1-HA/His-tagged rat Fbs1 derivatives, T7-tagged human Roc1) were produced by baculovirus-infected HiFive insect cells. Each SCFFbx2 complex was obtained by simultaneously infecting four baculoviruses. These proteins were affinity-purified by a HiTrap HP Column (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Each 1 μg of WT-BACE1 or BACE1 Ala mutant was incubated in 50 μL of the reaction mixture containing ATP-regenerating system, 0.5 μg E1, 1 μg Ubc4 (E2), 2 μg SCFFbx2, 6.5 μg recombinant glutathione S-transferase-ubiquitin and NEDD8 system at 30 °C. After the reaction was terminated by adding 25 μL of 3× SDS-PAGE sample buffer, the proteins in 8 μL of the boiled supernatants were separated with 5–20% (w/v) SDS-PAGE, and ubiquitinated BACE1 [(GST-Ub)n-GTF; higher molecular weight bands] were detected by immunoblotting with anti-BACE1 antibody (Sigma).
Mouse brain surgery and stereotaxic injection of adenoviral vectors
Adenoviral Fbx2 vectors were delivered into the hippocampus, as described previously. Briefly, mice were placed on a stereotaxic apparatus under ketamine (60 mg kg−1; i.p.) and xylazine (7.5 mg kg−1; i.p.) anesthesia to minimize discomfort, pain, and injury. We implanted 24-gauge guide cannulas bilaterally into the dorsal hippocampus (2.0 mm posterior to the Bregma, ±1.5 mm lateral from the midline, 1.9 mm beneath the surface of the skull). Mice were given at least 2 weeks to recover after cannula implantation. A total of 2 μL (1 μg μL−1) of adenoviral preparation was injected at a rate of 0.5 μL min−1. To allow the diffusion of the solution into the brain tissue, the needle was left for an additional 5 min after completion of the injection. All procedures were performed in accordance with the requirements of the Animal Studies Committee at MSSM. To confirm the locations of the hippocampal injection sites, brains were fixed with 4% paraformaldehyde. Coronal sections (30 μm) were mounted on glass slides and stained with hematoxylin and eosin (HE).
Primary neuron culture, immunocytochemistry, immunohistology, and confocal analysis
HEK293 cells or primary cultured neurons were washed in PBS and fixed for 20 min in 4% paraformaldehyde and permeabilized in 0.1% Triton X-100. After being blocked in 2% BSA for at least 1 h, cells were stained with various antibodies (between 1:200 and 1:500, according to the individual antibodies) overnight in 2% BSA and 0.01% Triton X-100; cells were washed three times for 10 min each in 2% BSA and 0.01% Triton X-100; they were stained for 1 h at room temperature with goat anti– rabbit Alexa Fluor 488, goat anti–mouse Alexa Fluor 568, or goat anti–mouse IgG–TRITC (Sigma) antibody. In some samples, cells were also treated with DAPI for 5 min. For brain slices, mouse brains were infused and fixed in 4% neutral buffered formalin for 24 h; fixed brains were serially sectioned coronally at 40 μm with a vibratome and were dehydrated through a series of graded ethanol solutions to xylene. To confirm the locations of the hippocampal injection sites, the sections were mounted on glass slides and stained with HE. To verify virus infection and the BACE1 and Fbx2 probing, tissue sections were stained using the standard (ABC) method, following the company’s manual (Vector, Burlingame, CA, USA). The immunostaining densities of BACE1 or Fbx2 over pyramidal layers of the hippocampal formation were quantified from approximately six to eight frames per section encompassing the hippocampal pyramidal layers, using Bioquant computer-assisted densitometry (Biometrics, Inc., Nashville, TN, USA), as previously described (Ho et al., 2001). Data were expressed as percentages of the mean value of the control (adenoviral GFP).
Hippocampal slice preparation and electrophysiology recording
We cut 400-μm brain slices from Tg2576 mice, as well as WT littermates, and maintained them in an interface chamber at 29 °C for 90 min prior to recording, as previously reported (Gong et al., 2006). The bath solution consisted of 124.0 mm NaCl, 4.4 mm KCl, 1.0 mm Na2HPO4, 25.0 mm NaHCO3, 2.0 mm CaCl2, 2.0 mm MgSO4, and 10.0 mm glucose. The stimulating electrode, a bipolar tungsten electrode, was placed at the level of the Schaeffer collateral fibers, whereas the recording electrode, a glass electrode filled with bath solution, was placed at the level of the CA1 stratum radiatum. Basal synaptic transmission was assayed by plotting the stimulus voltages against slopes of field excitatory postsynaptic potentials (fEPSP). For the LTP experiments, a 15-min baseline was recorded every minute at an intensity that evoked a response of approximately 35% of the maximum evoked response. Long-term potentiation was induced using ø-burst stimulation (four pulses at 100 Hz, with the bursts repeated at 5 Hz and each tetanus, including three ten-burst trains, separated by 15 s). Two-way anova followed by Bonferroni’s test was used for statistical analysis. Data are means ± SEM (error bar) of the results from two independent experiments.
Supplementary Material
Acknowledgments
We gratefully thank Dr M. Shelanski and Dr O. Arancio (Columbia University) for generously supporting the reported preliminary feasibility studies (NIH grant NS-15076 and the Columbia ADRC AG-008702); W. Zhao and S. Yemul for key contributions to the qRT–PCR experiments and for the human brain sample collection; J. Wang and L. Ho for helpful comments on this manuscript; and H. Fivecoat and I. Orozco for assistance in preparing this manuscript. The studies described here were supported in part from a grant from the Veterans Administration by the US National Institutes of Health grants and by grants from Alzheimer’s Association to (IIRG-07-59856) G.M.P and (IIRG-08-89354) to B.G.
Footnotes
Author contributions B.G and G.M.P designed and supervised all experiments and wrote the manuscript. B.G. conducted preliminary feasibility experiments and electrophysiology recordings. F.C. and Y.P. conducted the molecular biology and biochemistry experiments. I.A. conducted stereotaxic injections and immunostaining in brain slices. Y.Y. provided Fbx2 and its variants and the in vitro ubiquitination assay system. V.H. provided AD postmortem brain tissues from the brain bank at the Mount Sinai School of Medicine.
Conflict of interest The authors declare that they have no competing financial interests.
Supporting Information Additional supporting information may be found in the online version of this article:
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1αlpha localization and turnover implicates mitochondrial adaptation in longevity and the stress response. Aging Cell. 2008;7:10. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Cole S, Vassar R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007;2:22. doi: 10.1186/1750-1326-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem. J. 2007;407:383–395. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho-and caspase-3-cleaved tau species. J. Neurosci. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson SE, Andersen OM, Karmali V, Fritz JJ, Cheng D, Peng J, Levey AI, Willnow TE, Lah JJ. Loss of LR11/SORLA enhances early pathology in a mouse model of amyloidosis: evidence for a proximal role in Alzheimer’s disease. J. Neurosci. 2008;28:12877–12886. doi: 10.1523/JNEUROSCI.4582-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge W, Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007;6:361–370. doi: 10.1111/j.1474-9726.2007.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. Betasecretase activity increases with aging in human, monkey, and mouse brain. Am. J. Pathol. 2004;164:719–725. doi: 10.1016/s0002-9440(10)63159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Grillari J, Katinger H, Voglauer R. Aging and the ubiquitinome: traditional and non-traditional functions of ubiquitin in aging cells and tissues. Exp. Gerontol. 2006;41:1067–1079. doi: 10.1016/j.exger.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, Kishi S, Yamashita M, Phillips PS, Sukhatme VP, Lecker SH. The muscle-specific ubiquitin ligase atrogin1/MAFbx mediates statin-induced muscle toxicity. J. Clin. Invest. 2007;117:3940–3951. doi: 10.1172/JCI32741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat. Med. 2004;10:959–965. doi: 10.1038/nm1088. [DOI] [PubMed] [Google Scholar]
- He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE) J. Biol. Chem. 2005;280:11696–11703. doi: 10.1074/jbc.M411296200. [DOI] [PubMed] [Google Scholar]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS ONE. 2009;4:e8477. doi: 10.1371/journal.pone.0008477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A, Varshavsky A. Basic Medical Research Award. The ubiquitin system. Nat. Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- Ho L, Purohit D, Haroutunian V, Luterman JD, Willis F, Naslund J, Buxbaum JD, Mohs RC, Aisen PS, Pasinetti GM. Neuronal cyclooxygenase 2 expression in the hippocampal formation as a function of the clinical progression of Alzheimer disease. Arch. Neurol. 2001;58:487–492. doi: 10.1001/archneur.58.3.487. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–103. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. SCFβ-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Rouach N, Nicoll RA, Bredt DS. Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 2005;102:5600–5605. doi: 10.1073/pnas.0501769102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Mizushima T, Hirao T, Yoshida Y, Lee SJ, Chiba T, Iwai K, Yamaguchi Y, Kato K, Tsukihara T, Tanaka K. Structural basis of sugar-recognizing ubiquitin ligase. Nat. Struct. Mol. Biol. 2004;11:365–370. doi: 10.1038/nsmb732. [DOI] [PubMed] [Google Scholar]
- Nelson RF, Glenn KA, Zhang Y, Wen H, Knutson T, Gouvion CM, Robinson BK, Zhou Z, Yang B, Smith RJ, Paulson HL. Selective cochlear degeneration in mice lacking the F-box protein, Fbx2, a glycoprotein-specific ubiquitin ligase subunit. J. Neurosci. 2007;27:5163–5171. doi: 10.1523/JNEUROSCI.0206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, Becker AG, Hosono M, Sakaguchi I, Minami SS, et al. BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J. Neurochem. 2006;99:1555–1563. doi: 10.1111/j.1471-4159.2006.04178.x. [DOI] [PubMed] [Google Scholar]
- O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S. The ubiquitin–proteasome system in Alzheimer’s disease. J. Cell Mol. Med. 2008;12:363–373. doi: 10.1111/j.1582-4934.2008.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Olson BL, Hock MB, Ekholm-Reed S, Wohlschlegel JA, Dev KK, Kralli A, Reed SI. SCFCdc4 acts antagonistically to the PGC-1αlpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008;22:252–264. doi: 10.1101/gad.1624208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Bonifacino JS. Interactions of GGA3 with the ubiquitin sorting machinery. Nat. Cell Biol. 2004;6:244–251. doi: 10.1038/ncb1106. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 2004;18:1571–1573. doi: 10.1096/fj.04-1994fje. [DOI] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, et al. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. U.S.A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat. Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Smith DL, Pozueta J, Gong B, Arancio O, Shelanski M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. U.S.A. 2009;106:16877–16882. doi: 10.1073/pnas.0908706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub O, Rotin D. Role of ubiquitylation in cellular membrane transport. Physiol. Rev. 2006;86:669–707. doi: 10.1152/physrev.00020.2005. [DOI] [PubMed] [Google Scholar]
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science. 1989;246:670–673. doi: 10.1126/science.2530630. [DOI] [PubMed] [Google Scholar]
- Williamson DL, Butler DC, Alway SE. AMPK inhibits myoblast differentiation through a PGC-1αlpha-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 2009;297:E304–E314. doi: 10.1152/ajpendo.91007.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Huang X, Feng Y, Handschin C, Feng Y, Gullicksen PS, Bare O, Labow M, Spiegelman B, Stevenson SC. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1αlpha transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. U.S.A. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashler JR, Stratman NC, Mathews WR, Buhl AE, et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Chiba T, Tokunaga F, Kawasaki H, Iwai K, Suzuki T, Ito Y, Matsuoka K, Yoshida M, Tanaka K, Tai T. E3 ubiquitin ligase that recognizes sugar chains. Nature. 2002;418:438–442. doi: 10.1038/nature00890. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong M, Yan RQ, Sun FY. Mutant ubiquitin-mediated beta-secretase stability via activation of caspase-3 is related to beta-amyloid accumulation in ischemic striatum in rats. J. Cereb. Blood Flow Metab. 2010;30:566–575. doi: 10.1038/jcbfm.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.