

Background: Dysregulation of endoplasmic reticulum homeostasis elicits various stress responses.
Results: Endoplasmic reticulum stress activates lipolytic cascade in rat adipocytes.
Conclusion: The lipolysis response to endoplasmic reticulum stress is mediated via cAMP/PKA and ERK1/2 signaling.
Significance: Increased lipolysis promotes fatty acid efflux from adipocytes to other tissues and thus may contribute to lipotoxicity and insulin resistance in obesity and diabetes.
Keywords: Adipocyte, Adipose Triglyceride Lipase, Endoplasmic Reticulum Stress, Insulin Resistance, Lipid Droplets, Lipolysis, Free Fatty Acids, Lipase, Perilipin
Abstract
In obesity and diabetes, adipocytes show significant endoplasmic reticulum (ER) stress, which triggers a series of responses. This study aimed to investigate the lipolysis response to ER stress in rat adipocytes. Thapsigargin, tunicamycin, and brefeldin A, which induce ER stress through different pathways, efficiently activated a time-dependent lipolytic reaction. The lipolytic effect of ER stress occurred with elevated cAMP production and protein kinase A (PKA) activity. Inhibition of PKA reduced PKA phosphosubstrates and attenuated the lipolysis. Although both ERK1/2 and JNK are activated during ER stress, lipolysis is partially suppressed by inhibiting ERK1/2 but not JNK and p38 MAPK and PKC. Thus, ER stress induces lipolysis by activating cAMP/PKA and ERK1/2. In the downstream lipolytic cascade, phosphorylation of lipid droplet-associated protein perilipin was significantly promoted during ER stress but attenuated on PKA inhibition. Furthermore, ER stress stimuli did not alter the levels of hormone-sensitive lipase and adipose triglyceride lipase but caused Ser-563 and Ser-660 phosphorylation of hormone-sensitive lipase and moderately elevated its translocation from the cytosol to lipid droplets. Accompanying these changes, total activity of cellular lipases was promoted to confer the lipolysis. These findings suggest a novel pathway of the lipolysis response to ER stress in adipocytes. This lipolytic activation may be an adaptive response that regulates energy homeostasis but with sustained ER stress challenge could contribute to lipotoxicity, dyslipidemia, and insulin resistance because of persistently accelerated free fatty acid efflux from adipocytes to the bloodstream and other tissues.
Introduction
In mammals, fatty acids stored as triacylglycerols in adipose cells constitute the primary energy reserves. Triacylglycerol hydrolysis in adipocytes, termed lipolysis, produces glycerol and free fatty acids (FFAs).2 Because of the paucity of glycerol kinase in normal adipocytes (1), glycerol is rarely re-utilized for triglyceride resynthesis; rather, it is released in the plasma along with FFAs to supply energy to various tissues. Adipose lipolysis is an important process that controls circulating FFA concentrations (2) and governs energy homeostasis through the Randle glucose-fatty acid cycle. Dysregulation of the lipolysis pathway may result in elevated levels of circulating FFAs, which is the major basis for the development of insulin resistance in obesity and diabetes mellitus (3, 4).
Lipolysis is stimulated by various hormones and effectors. Catecholamines are major hormones that stimulate acute lipolysis. Thyronines, glucocorticoids (5), TNF-α (6–8), and lipopolysaccharides (9) induce a chronic lipolytic reaction. The lipolysis is closely associated with the production of cAMP and activation of cAMP-dependent protein kinase A (PKA) (2, 10). cAMP/PKA along with extracellular signal-regulated kinase-1/2 (ERK1/2) (6, 11) are the major early lipolytic signals. PKC may modulate lipolysis through a different pathway (12). In the downstream lipolytic cascade, the phosphorylation of both lipid droplet-associated protein perilipin A (13–17) and hormone-sensitive lipase (HSL) and the subsequent translocation of HSL from the cytosol to the lipid droplet surface are critical to confer full activation of HSL during PKA-stimulated lipolysis (14, 18, 19). Adipose triglyceride lipase (ATGL) is another important enzyme (20) that predominantly modulates basal lipolysis (21, 22) and may be indirectly activated during PKA-regulated lipolysis.
The endoplasmic reticulum (ER) is an organelle that functions to synthesize, fold, and transport proteins. Dysregulation of ER homeostasis leads to accumulation of misfolded proteins in the ER lumen (23). Under stress conditions, cells activate the unfolded protein response, which includes transcriptional induction of ER chaperones and translational attenuation, thus evoking a series of ER stress responses (23, 24). Recent studies reveal that ER stress occurs in pancreatic β-cells (25–27), hepatocytes (28), and adipocytes (28–30) in obesity and diabetes and is one of the mechanisms involved in triggering insulin resistance. The ER is also the site of triglyceride synthesis and nascent lipid droplet formation, although the process is far from clarified. One prevalent model proposes that the triglyceride droplet originates between the two leaflets of the ER membrane and then buds or patches to the cytoplasm, with a phospholipid monolayer along with resident proteins of the ER on the droplet surface (31). An alternative model assumes that the triglyceride droplet forms alongside the ER membrane but with the ER facilitating biosynthesis and holding the droplet like an egg (32). Regardless of the model, morphological evidence suggests that ER bilayers wrap closely around the phospholipid monolayer surface of lipid droplets (31–34). Thus, given the ER origin of lipid droplets, the close association of the two organelles, and the occurrence of lipolysis at the lipid droplet surface, we were interested in investigating whether ER stress involves modulation of the lipolysis of triglyceride droplets in adipocytes.
This study aimed to investigate the lipolysis in rat adipocytes stimulated by ER stress inducers. We revealed that the lipolysis action of ER stress was mediated by cAMP/PKA and ERK1/2 signaling and was accompanied by phosphorylation of perilipin and HSL and activation of cellular lipases. Our findings suggest a novel pathway of lipolysis response to ER stress that accelerates FFA efflux from adipose cells to the bloodstream and other tissues and thereby may contribute to lipotoxicity, dyslipidemia, and insulin resistance in obesity and diabetes.
EXPERIMENTAL PROCEDURES
Materials
Tunicamycin and H89 were from Alexis Biochemicals (San Diego), and thapsigargin, PD98059, U0126, and phenol red-free DMEM were from Sigma. Polyclonal antibody against rat perilipin was a gift from C. Londos (National Institutes of Health, Bethesda). Antibodies recognizing HSL, ATGL, protein kinase-like eIF2α kinase (PERK), eIF2α, and their phosphorylated species were from Cell Signaling Technology (Boston, MA). Defatted bovine serum albumin, modified Lowry protein assay kit, and enhanced chemiluminescence (ECL) detection reagent were from Applygen Technologies.
Culture of Primary and Differentiating Rat Adipocytes
Primary adipocytes were isolated from epididymal fat pads of normal male Sprague-Dawley rats (160–180 g) according to our laboratory method (9, 15, 35). The minced fat pads were digested in Krebs-Ringer solution containing 0.75 mg/ml type I collagenase, 200 nm adenosine, 25 mm Hepes, pH 7.4, and 1% FFA-free BSA. After digestion, primary adipocytes floating in the tube were collected, washed, and packed by centrifugation at 200 × g for 3 min for determining the packed cell volume of adipocytes (15). Adipocytes were preincubated in phenol red-free and serum-free DMEM in an atmosphere of 5% CO2 at 37 °C for 1 h before treatments.
After the isolation of primary adipocytes, rat preadipocytes residing in the digestion mixture were collected by centrifugation at 800 × g for 10 min and then plated and differentiated into adipocytes for 3 days in serum-free DMEM/F-12 (1:1) supplemented with 5 μg/ml insulin, 33 μm biotin, and 200 pm triiodothyronine, as we described previously (15, 35). At day 4, the differentiating adipocytes were transferred to phenol red-free and serum-free DMEM and incubated for 24 h before experiments.
Evaluation of Lipolysis by Glycerol Assay
Full hydrolysis of one triglyceride in adipocytes produces three fatty acids and one glycerol molecule, which are then released into the medium. Thus, we measured glycerol content in the medium as an index of lipolytic reaction (5, 9, 15). Adipocytes or minced fat tissues were incubated in serum-free and phenol red-free DMEM with or without the agents. The culture medium was collected and heated at 70 °C for 10 min to inactivate residue lipase activity. Glycerol was determined by the enzyme-coupled colorimetric assay (GPO Trinder reaction) from the absorption of 550 nm (5), with use of a colorimetric assay kit (Applygen Technologies).
cAMP Radioimmunoassay
According to our previous method (5, 8), adipocytes were lysed in 150 μl of ice-cold buffer containing 50 mm Tris-HCl, pH 7.4, and 1 mm EDTA. The lysate was centrifuged at 12,000 × g for 20 min at 4 °C. The cytosol fraction was collected from below the solidified fat cake in the tube, mixed with ⅓ volume of 40% trichloroacetic acid, and further cleaned by centrifugation. The supernatant was collected and used for cAMP assay by use of a commercial 125I radioimmunoassay kit (Isotope Laboratory of Shanghai University of Chinese Medicine).
Immunoblotting
Adipocytes were lysed in sample buffer containing 62 mm Tris-HCl, pH 6.8, 2% SDS, 0.1 mm sodium orthovanadate, and 50 mm sodium fluoride. The protein content was determined by the Lowry protein assay. Equal amounts of proteins were loaded and separated by SDS-PAGE. For most immunoblot assays, the traditional polyacrylamide gels were used. For detection of phosphorylated perilipin with the anti-perilipin antibody, we used a low-bis concentration polyacrylamide gel prepared with 10% acrylamide and 0.07% N,N′-methylenebisacrylamide (at a ratio of 142:1 versus 39:1 in the traditional gel), because this gel system provides maximal resolution of proteins in the 60–70-kDa range (9, 15, 16, 36). The proteins transferred on membranes were recognized with primary antibodies and horseradish peroxidase-conjugated secondary antibodies. The blots were developed by use of ECL reagents. If required, the antibodies bound to membranes were removed by a commercial stripping solution (Applygen Technologies), and the blots were reprobed with use of other antibodies.
Lipase Activity
Lipase activity was assayed by determining the rate of hydrolysis of emulsified triolein substrates by endogenous lipases of primary adipocytes as described previously (37) with modifications as described previously (5, 8). Adipocyte lysates were centrifuged at 12,000 × g for 15 min at 4 °C. The infranatant phase below the fat cake fraction was transferred to a new tube and further cleaned by centrifuging at 12,000 × g for 15 min at 4 °C. The supernatant was added to an emulsified triolein substrate solution (reaction A) and in parallel to a control solution without triolein substrates (reaction B). The reactions were incubated for 30 min at 37 °C, when the endogenous lipases hydrolyze trioleins to release glycerol. Reaction B contained only endogenous glycerol derived from the adipocyte extract. Next, glycerol contents of reaction A and B were determined. The calculated (A and B) value indicated a rate of glycerol release by lipase-catalyzed triolein hydrolysis and represented lipase activity.
HSL Immunostaining
The differentiated adipocytes were stimulated with thapsigargin or isoproterenol and then fixed with 4% paraformaldehyde and blocked with 5% donkey serum for 60 min. The cells were incubated with rabbit antisera against HSL at 1:400 overnight at 4 °C and then with fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG at 1:1000 for 2 h in the dark at room temperature. Immunofluorescent HSL signal was observed under a Nikon Eclipse 50i microscope.
Cell Viability Assay
MTT and lactate dehydrogenase assays involved use of commercial kits (Applygen Technologies) (8). Cell viability rates by MTT assay were calculated by optical density measured at 570 nm and presented as percentage of control values. LDH activity was measured by optical density at 440 nm. The cell viability index was presented as a percentage of LDH leakage in medium compared with total LDH activity.
Statistical Analysis
Data are expressed as mean ± S.E. Student's t test or one-way analysis of variance Turkey's test was used for statistical analysis. A p < 0.05 was considered statistically significant.
RESULTS
Induction of ER Stress in Rat Adipocytes
Thapsigargin is a commonly used agent known to rapidly elicit ER stress by inhibiting the ER Ca2+ pump (23). Exposure of differentiated rat adipocytes to 1 μm thapsigargin caused a rapid increase of glucose regulated protein 78 (GRP78) (Fig. 1), a chaperone protein located in the ER lumen whose expression is increased with ER stress (24). During ER stress, the RNA-dependent PERK is activated to phosphorylate eukaryotic initiation factor 2α (eIF2α); therefore, their phosphorylation is a key indicator of ER stress (23, 28). We examined the phosphorylation status of PERK (Thr-980) and eIF2α (Ser-51) by immunoblotting and found their phosphorylation rapidly promoted in adipocytes treated with thapsigargin, with total PERK and eIF2α expression unchanged (Fig. 1). These data confirm that ER stress was activated in adipocytes with thapsigargin stimulation.
FIGURE 1.
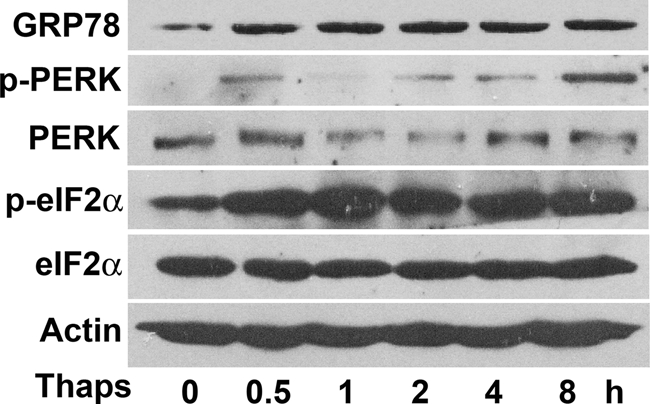
ER stress is induced by thapsigargin in rat adipocytes. Rat differentiated adipocytes were incubated for 0.5 to 8 h with or without 1 μm Thaps. Protein levels of GRP78, PERK, or its Thr-980 phosphorylation form, eIF2α, or its Ser-51 phosphorylation species, with actin as a loading control, were detected by immunoblotting analysis. The blots are representative of three separate experiments.
Lipolysis of Adipocytes Is Activated in Response to ER Stress
To investigate whether lipolysis is activated on ER stress challenge, we measured the glycerol release from differentiated or primary rat adipocytes and minced epididymal adipose tissues stimulated with ER stress inducers. Three agents, thapsigargin, tunicamycin, and brefeldin A, which elicit ER stress through different pathways (23), effectively elevated lipolytic reaction in adipocytes (Fig. 2A). The magnitude of glycerol release was lower in thapsigargin-stressed adipocytes than in adipocytes stimulated with isoproterenol, a known adrenergic lipolysis activator (Fig. 2A, inset). The lipolysis in differentiated adipocytes was time-dependently increased on stimulation with 1 μm thapsigargin (Fig. 2B). Similarly, glycerol level was increased in isolated primary adipocytes 2 h after ER stress induction (Fig. 2C). To evaluate ex vivo lipolytic action, minced rat epididymal adipose tissue was treated with 1 μm thapsigargin for 8 or 24 h. The 1-h glycerol release in the freshly changed medium significantly increased, which indicates lipolytic activation ex vivo in adipose tissues in response to ER stress (Fig. 2D).
FIGURE 2.

Lipolysis is activated in response to ER stress in rat adipose cells or tissues. Rat adipose cells or tissues were incubated in phenol red-free and serum-free DMEM. Glycerol content in the medium was assayed as an index of lipolysis. A, lipolysis responses to ER stress after rat differentiating adipocytes were incubated for 8 h with or without 1 μm Thaps, 5 μg/ml tunicamycin (Tun), or 0.5 μg/ml brefeldin A (BFA). Inset in A shows a positive stimulation of the lipolysis. After incubation with 1 μm Thaps or 100 nm Iso, an adrenergic lipolytic activator, 1-h glycerol release from the adipocytes, was assayed. B and C, time-dependent lipolysis in rat differentiated (B) and primary (C) adipocytes stimulated with 1 μm Thaps. D, ex vivo lipolysis. Minced rat epididymal adipose tissues were incubated with 1 μm Thaps for the indicated times and then 1-h glycerol release in freshly changed medium was determined. Lipolysis data are expressed as micromoles of glycerol/ml of packed cell volume (PCV) or milligram of cell proteins. Data are mean ± S.E. of at least three separate experiments performed in triplicate. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus control.
Evaluation of cell viability by LDH and MTT assay indicated no significant LDH leakage into culture medium from rat adipocytes during an 8-h incubation with thapsigargin at 0.5 and 1 μm or tunicamycin at 5 μg/ml (Table 1). Only tunicamycin at 10 μg/ml conferred cytotoxicity in adipocytes. Therefore, the agents at the tested concentrations did not affect adipocyte viability.
TABLE 1.
Effect of tunicamycin and thapsigargin on cell viability of adipocytes
MTT and lactate dehydrogenase assays were performed after adipocytes were incubated for 8 h with Thaps or tunicamycin (Tun). Cell viability data by MTT assay are given as percentage of control values. LDH leakage data are expressed as percentage of LDH activity in the culture media compared with total LDH activity both in the culture media and in whole adipocyte extracts. Results are means ± S.E. of three individual experiments performed in triplicate.
| MTT assay cell viability | LDH assay LDH leakage | |
|---|---|---|
| % control | % total | |
| Control | 100.0 ± 0.0 | 10.4 ± 0.7 |
| Thaps (0.5 μm) | 98.7 ± 1.5 | 9.6 ± 0.5 |
| Thaps (1 μm) | 95.0 ± 7.2 | 10.2 ± 0.3 |
| Tun (5 μg/ml) | 92.2 ± 6.0 | 12.7 ± 0.6 |
| Tun (10 μg/ml) | 82.6 ± 0.5a | 18.5 ± 0.3a |
a p < 0.01 versus control.
Lipolysis Response to ER Stress Is Mediated by Activation of cAMP/PKA Signaling
cAMP and PKA are the major signals for controlling lipolysis (2). We determined cAMP content in adipocytes by 125I radioimmunoassay (5, 9). Incubation with 1 μm thapsigargin for 1 h increased cAMP production by 68%, which persisted during the 8-h stimulation (Fig. 3A). As a positive control, the classical lipolytic agent isoproterenol caused a similar increase in cellular cAMP level (Fig. 3A). Furthermore, PKA activity was analyzed by immunoblotting with a primary antibody against a specific motif (RRX(S/T)) of PKA phosphosubstrates (9). Stimulation with 1 μm thapsigargin for various times significantly promoted PKA activity (Fig. 3B), although this effect was relatively weaker than that induced by isoproterenol (Fig. 3C). Preincubation with the PKA inhibitor H89 diminished PKA phosphosubstrate level (Fig. 3C) and greatly attenuated the lipolysis stimulation of thapsigargin (Fig. 3D) or isoproterenol (data not shown). These data suggest that lipolysis response to ER stress was mediated mainly by activating cAMP/PKA signals in adipocytes.
FIGURE 3.

ER stress triggers lipolysis by elevating cellular cAMP and activating PKA. A, 125I radioimmunoassay of cellular cAMP level in rat differentiated adipocytes incubated with 1 μm Thaps for 1 and 8 h or with 1 μm Iso (as positive control) for 0.5 h. B, determination of PKA activity. Adipocytes were stimulated with 1 μm Thaps for the indicated times, and then whole cell lysates underwent immunoblot (IB) analysis with a primary antibody against PKA phosphosubstrate motif (RRX(S/T)). C, stimulation with 1 μm Thaps for 1 h or with 1 μm isoproterenol for 20 min promoted the level of PKA phosphosubstrates in adipocytes, which was attenuated by PKA inhibition with 20 μm H89. D, glycerol assay. Lipolysis in adipocytes stimulated with 1 μm Thaps for 8 h was inhibited by 1-h preincubation with 20 μm H89. The blots are representative of three separate experiments, and the glycerol data are mean ± S.E. of at least three separate experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control; †††, p < 0.001 versus Thaps.
ERK1/2 Activation Accounts for ER Stress-induced Lipolysis
In addition to cAMP/PKA, ERK1/2 participates in lipolysis regulation (7, 11). Immunoblotting results showed that thapsigargin time-dependently promoted the phosphorylation of Raf-1 (Fig. 4A), an upstream Ser/Thr kinase in the MAPK cascade. In addition, phosphorylation of ERK1/2 was induced rapidly after a 0.5-h exposure to thapsigargin and was sustained at a high level during an 8-h stimulation (Fig. 4A). Tunicamycin and dithiothreitol, two other ER stress inducers, also stimulated significant phosphorylation of ERK1/2 in adipocytes (Fig. 4B). Preincubation with the ERK1/2 inhibitor U0126 eliminated ERK1/2 phosphorylation stimulated by thapsigargin or a control agent TNF-α (Fig. 4C) (6–8). The total level of ERK1/2 and Raf-1 protein was unchanged during ER stress.
FIGURE 4.

ERK1/2 activation accounts for lipolysis response to ER stress. A and B, immunoblot analysis of phosphorylation of Raf-1 and ERK1/2 in rat differentiated adipocytes incubated with or without 1 μm Thaps, 5 μg/ml tunicamycin (Tun), or 1 mm dithiothreitol (DTT). C, preincubation with 10 μm U0126 inhibited ERK1/2 phosphorylation induced by 1 μm Thaps or 50 ng/ml TNF-α as a test control. D–F, lipolysis assay. Adipocytes were preincubated with 10 μm U0126 (D), 50 μm PD98059 (E), or U0126 plus H89 each at 10 μm (F) and then stimulated for 8 h with 1 μm Thaps or 50 ng/ml TNF-α. Glycerol release in the medium was determined. Data are mean ± S.E. of three separate experiments performed in triplicate. **, p < 0.01 versus control; †, p < 0.05; ††, p < 0.01 versus Thaps. ***, p < 0.001 versus control.
Next, we examined the effects of the kinase inhibitors on the lipolysis response. After preincubation for 1 h with the ERK1/2 kinase inhibitor PD98059 or U0126, the lipolysis of thapsigargin was inhibited by 50 and 42%, respectively (Fig. 4, D and E). By contrast, U0126 decreased the lipolysis of TNF-α by 78% (Fig. 4D). These data indicate that inhibition of the ERK signal partially suppressed the ER stress-responsive lipolysis. However, when ERK and PKA were simultaneously inhibited by U0126 plus H89, the lipolysis of thapsigargin was fully abrogated (Fig. 4F), which suggests that PKA and ERK1/2 cooperate to regulate ER stress-induced lipolysis.
c-Jun N-terminal Kinase (JNK) and p38 MAPK and PKC Do Not Regulate ER Stress-stimulated Lipolysis
JNK (7) and PKC (12) regulate lipolysis action of isoproterenol or TNF-α. JNK is activated by ER stress in adipocytes (29, 38). Therefore, we investigated whether JNK and PKC are involved in the lipolysis response to ER stress. Immunoblot analysis revealed that JNK phosphorylation was elevated 2 h after thapsigargin stimulation (Fig. 5A), but phosphorylation of p38 MAPK was undetectable (data not shown). However, preincubation for 1 h with the JNK inhibitor SP600125 did not alter glycerol release stimulated by thapsigargin (Fig. 5B). Also, the lipolysis activation was not affected by the p38 inhibitor SB203580 (Fig. 5C) or PKC inhibitor Ro-31-8220 (Fig. 5D). Therefore, ERK1/2 but not JNK, p38 MAPK, or PKC is involved in lipolytic regulation in ER-stressed adipocytes.
FIGURE 5.

JNK, p38 MAPK, and PKC do not regulate ER stress-stimulated lipolysis. A, immunoblot analysis of JNK phosphorylation in adipocytes stimulated with 1 μm Thaps. B–D, glycerol assay of rat differentiated adipocytes preincubated for 1 h with or without JNK inhibitor SP600125 at 12.5 μm (B), p38 inhibitor SB203580 at 10 μm (C), or PKC inhibitor Ro-31-8220 at 1 μm (D) and then stimulated with 1 μm Thaps for 8 h. Data are mean ± S.E. of three separate experiments in triplicate. ***, p < 0.001 versus control.
Enhanced Phosphorylation of Perilipin on ER Stress
Perilipins are the most abundant proteins located at the surface of lipid droplets in adipocytes (16, 36). Phosphorylation (13–15, 17) or down-regulation (9) of perilipins facilitate lipolysis. Because a specific antibody against phosphorylated perilipin is not available, we performed two protocols for immunodetection of perilipin phosphorylation. First, adipocyte extracts were separated on low-bis SDS-polyacrylamide gels (10:0.07% acrylamide/bisacrylamide), because the gel system allows better resolution of the 67-kDa phosphorylated perilipin A (9, 15, 16). Immunoblot analysis with an anti-perilipin antibody revealed that thapsigargin at 1 μm caused time-dependent shifts of the perilipin A from the 65-kDa native form to the 67-kDa phosphorylated species, which indicates that phosphorylation of perilipin A occurs during ER stress (Fig. 6A). Such a migration shift was due to increased incorporation of phosphate in perilipin A proteins, as confirmed previously with 32P labeling of adipocytes and autoradiography (15, 16, 36).
FIGURE 6.

ER stress causes phosphorylation of perilipin. A, rat differentiated adipocytes were treated with 1 μm Thaps and then lysates underwent SDS-PAGE and immunoblot analysis with the anti-perilipin antibody. Electrophoresis was run for 7 h on a medium sized gel especially prepared with a low-bis concentration (10:0.07% acrylamide/bisacrylamide); these conditions allow for maximal resolution of perilipin proteins but may cause band distortion. The migration shift from 65 to 67 kDa indicates hyperphosphorylation of full-length perilipin A. The 46-kDa bands are the truncated perilipin B isoforms. B, top image, a control test for accessibility of phosphorylated perilipin with the anti-phospho-Ser/Thr antibody. The extracts of primary adipocytes stimulated with Iso were immunoprecipitated (IP) (with a monoclonal antibody against phospho-Ser/Thr PKA substrates), then immunoblotted (IB), and recognized by anti-perilipin antibodies. Bottom images, phosphorylation of perilipin A induced by Thaps was immunodetected with the anti-phospho-Ser/Thr antibody. Then the blot was stripped and reprobed with anti-perilipin antibodies to detect the native perilipins. C, adipocytes were stimulated with 1 μm Thaps, 5 μg/ml tunicamycin (Tun), or 1 mm DTT, and lysates underwent immunoblot analysis with antibodies against phospho-Ser/Thr or perilipin, respectively. D, H89 attenuated perilipin phosphorylation in adipocytes stimulated with 1 μm Thaps or Iso as a positive control for PKA activation. The blots represent data from three separate experiments. peri, perilipin; p-peri, phosphorylated perilipin.
As an alternative, adipocyte extracts were run on traditional polyacrylamide gels and underwent immunoblotting with an antibody against phospho-Ser/Thr PKA substrates, which was previously used to recognize phosphorylated perilipins (5, 8). The control test showed that the immunoprecipitants of primary adipocyte with anti-phospho-Ser/Thr antibodies were recognized by anti-perilipin antibodies to raise two concrete bands of phosphorylated perilipin A together with its truncated 46-kDa isoform perilipin B induced by isoproterenol (Fig. 6B, top image). Furthermore, immunoblot analysis confirmed that phosphorylation of perilipin A was effectively induced in adipocytes stimulated with thapsigargin, tunicamycin, or dithiothreitol (Fig. 6, B, top image, and C). Phosphorylation of the perilipin B isoform was also moderately promoted. However, the protein level of native perilipins was not altered (Fig. 6, A and bottom image in B). In the presence of the PKA inhibitor H89, phosphorylation of perilipin A (Fig. 6D) and the accompanying lipolysis action (data not shown) were reduced in adipocytes stimulated with thapsigargin or the positive lipolytic agent isoproterenol.
Regulation of Lipase Activity on ER Stress
HSL and ATGL are two major lipases in adipocytes. We assayed the hydrolysis rate of triolein substrates in the reaction with adipocyte extracts, which served as an index of lipase activity. Thapsigargin increased lipase activity by 44%, as compared with 80% with isoproterenol at 30 min (Fig. 7A). Protein levels of HSL and ATGL were not altered after thapsigargin treatment (Fig. 7B). However, thapsigargin promoted HSL phosphorylation at Ser-563 and Ser-660 residues (Fig. 7C), two critical phosphoserine sites for controlling HSL activity (19). Translocation of HSL from cytosol to lipid droplets is crucial for conferring a full lipolysis reaction (14, 18, 39). Immunofluorescent staining indicated that thapsigargin moderately increased HSL translocation to the lipid droplet surface, whereas isoproterenol significantly promoted this process (Fig. 7D).
FIGURE 7.

Regulation of lipase activity in ER-stressed adipocytes. A, lipase activity was determined by measuring glycerol release from the hydrolysis of emulsified triolein substrates in the reaction containing extracts of primary adipocytes stimulated with 1 μm Thaps for 8 h. Stimulation with 1 μm Iso for 30 min served as a positive control. Data are mean ± S.E. of at least three separate experiments performed in triplicate. *, p < 0.05; **, p < 0.01 versus control. B, immunoblot analysis of protein levels of HSL and ATGL in differentiated adipocytes stimulated with 1 μm Thaps. C, HSL phosphorylation was detected with primary antibodies against the phospho-Ser-563 or -Ser-660 motif of HSL. D, immunofluorescence. Rat differentiated adipocytes were incubated with 1 μm Thaps for 8 h or with 1 μm Iso (as a positive control) for 30 min and then immunostained for HSL. The dark circles inside adipocytes indicate lipid droplets, and enhanced fluorescence at the lipid droplet surface indicates the translocation of HSL. Images are representative of three separate experiments.
DISCUSSION
In this study, we demonstrated that ER stress stimuli induced lipolysis by activating cAMP/PKA and ERK1/2 signaling in adipocytes. This lipolytic activation is probably an adaptive response that regulates energy homeostasis but, with sustained ER stress, may contribute to lipotoxicity and impair insulin sensitivity because of persistently accelerated FFA efflux from adipocytes to the bloodstream and various tissues.
Significant ER stress occurs in adipocytes in obesity and diabetes (28–30), yet its origins are unknown. The agents that disrupt ER homeostasis allow for exploring ER stress responses. Thapsigargin, tunicamycin, and brefeldin A trigger ER stress by inhibiting the ER Ca2+ pump and protein glycosylation or interfering with ER-Golgi protein trafficking, respectively (23, 38). With thapsigargin stimulation, we observed a rapid increase in GRP78 protein level and an induction of eIF2α and PERK phosphorylation, thus confirming that ER stress was evoked in rat adipocytes. Thapsigargin, tunicamycin, and brefeldin A, which elicit ER stress by different pathways, effectively activated significant lipolysis in rat adipocytes.
The mechanisms by which ER stress stimulates lipolysis may be multifactorial. cAMP and PKA are the major early signals that control lipolysis with catecholamine hormone stimulation (2). Our radioimmunoassay results revealed that cellular cAMP production increased by 68%, along with a significant elevation of PKA phosphosubstrates in thapsigargin-treated adipocytes. PKA inhibition with H89 decreased PKA phosphosubstrates and inhibited the lipolysis stimulation of thapsigargin. Recent reports show that the survival of INS-1 cells and rat pancreatic β-cells under thapsigargin-induced ER stress can be improved by the PKA activator forskolin or by the glucagon-like peptide-1 receptor agonist that may elevate cAMP. This protective effect was attenuated on PKA inhibition, which implicates an involvement of cAMP/PKA signals in modulating the unfolded protein response during ER stress (25, 40). Increased cAMP production might occur as an adaptive response to ER stress challenge, which then activates PKA to modulate several components in the ER stress cascade. However, to date, direct evaluation of cellular cAMP and PKA activation during ER stress is lacking. Our data reveal an activation of cAMP/PKA at least in ER-stressed adipocytes stimulated with thapsigargin. Although the mechanism needs to be clarified, the activation of cAMP/PKA is a major signaling event triggering the lipolytic cascade in ER-stressed adipocytes.
In addition to PKA, ERK1/2 participates in regulating chronic lipolysis stimulation of TNF-α or lipopolysaccharides (6–9). JNK (7) and PKC (12) may also modulate lipolysis. Recent studies suggest that ERK1/2 and JNK are readily activated with ER stress. ERK1/2 activation protects cells against ER stress-induced death (41), but JNK activation impairs insulin action (38). We observed that thapsigargin, tunicamycin, and dithiothreitol promoted phosphorylation of Raf-1, an upstream kinase in the MAPK cascade, accompanied by rapid phosphorylation of both ERK1/2 and JNK. Nevertheless, inhibition of JNK and p38 MAPK or PKC failed to inhibit the lipolysis response to ER stress. Instead, preincubation with ERK1/2 kinase inhibitors abrogated ERK1/2 phosphorylation and moderately attenuated the lipolysis in ER-stressed adipocytes. Furthermore, when PKA and ERK1/2 were simultaneously inhibited with H89 plus U0126, lipolysis was completely abolished. Of note, PKA-independent effects of H89 may occur at high concentrations, although H89 is more selective for PKA inhibition. Davies et al. (42) reported that H89 at the concentration of ∼10 μm, used in this study, can inhibit several other kinases such as AMPK, ERK2, and PKC by 81, 13, and 21%, respectively. Because these kinases may modulate lipolysis with different stimulations, the inhibitory effects of H89 on the lipolytic signaling in ER-stressed adipocytes should be discussed. AMPK was proposed to activate lipolysis (43), but several studies debated that AMPK has anti-lipolytic activity (44, 45). The latter view is supported by our previous work that antidiabetic biguanide metformin, which induces AMPK (44), can inhibit the lipolysis action of isoproterenol and TNF-α in primary rat adipocytes (46, 47). Thus, by preferring an antilipolytic action of AMPK, we speculate that AMPK inactivation of H89, although significant, may not account for the lipolytic inhibition of H89 in ER-stressed adipocytes. However, the slight suppression of H89 on ERK2 but not PKC could contribute in part to the inhibitory effect of H89 on ER stress-mediated lipolytic signaling. Therefore, PKA and ERK1/2 may be the key signals that cooperatively regulate lipolysis in ER-stressed adipocytes. PKA may govern a rapid lipolytic action, and ERK1/2 may act in the chronic phase of the lipolysis response to ER stress.
In the downstream lipolytic cascade, perilipin is one of the most abundant PKA phosphoproteins in adipocytes (16, 36). Unlike its less abundant splicing variant (∼46-kDa perilipin B), the full-length perilipin A (simply termed perilipin in this study) is the major isoform, and its function on lipolytic regulation has been firmly established. Perilipins coat lipid droplets as a barrier to prevent triglyceride hydrolysis by lipases (13). Phosphorylation of perilipin in at least three of the six PKA consensus site serines may impair its barrier function and enhance HSL translocation to the lipid droplet, hence facilitating lipolysis (13–18). We found that ER stress did not alter the level of perilipin proteins but increased the phosphorylation. When perilipin phosphorylation was attenuated on PKA inhibition, the lipolysis response ceased. HSL and ATGL are two major lipases that control ∼95% of the triglyceride hydrolase activity in adipocytes (20, 21). HSL hydrolyzes both triglycerides and diglycerides, but the affinity for the latter is 10-fold higher (48). PKA-mediated phosphorylation of HSL at Ser-660, Ser-659, and Ser-563 is crucial for controlling lipase activity (19). The translocation of HSL from the cytosol to lipid droplets is an important step for PKA-regulated lipolysis (14, 18, 39), which requires the phosphorylation of both HSL (49) and perilipin (14, 15). Thapsigargin increased phosphorylation of HSL at Ser-660 and Ser-563 and moderately promoted its translocation to the lipid droplet surfaces, which indicates an activation of HSL during ER stress. Unlike HSL, ATGL predominantly hydrolyzes triglyceride but is not phosphorylated by PKA (20). A recent model proposes an indirect control of ATGL activity by PKA dependent on perilipin phosphorylation (50, 51). Perilipin binds the ATGL co-activator Abhd5/CGI-58 and thereby suppresses the interaction of the lipase with Abhd5. Phosphorylation of perilipin on Ser-492 or Ser-517 results in a release of sequestered Abhd5, hence allowing Abhd5 to interact with and activate ATGL (50, 51). In adipocytes stimulated with thapsigargin, protein levels of HSL and ATGL were unchanged, but total lipase activity was increased by 44%, which was associated with PKA activation and strong perilipin phosphorylation. Therefore, perilipin phosphorylation occurring during ER stress may indirectly promote ATGL activity by regulating accessibility to its coactivator Abhd5, although this speculation requires further investigation. Activated HSL and ATGL may cooperatively manipulate the hydrolysis of triglycerides and diglycerides in ER-stressed adipocytes.
In conclusion, we reveal a novel pathway of lipolysis response to ER stress in adipocytes. Obesity and diabetes mellitus are associated with increased adipocyte lipolysis and a high level of circulating FFA (3, 4), accompanied by significant ER stress in adipose cells (28–30). The lipolysis response to ER stress promotes FFA efflux from adipocytes to the plasma, which could be a cellular basis of lipotoxicity, dyslipidemia, and insulin resistance in these pathologies. Moreover, because fatty acids themselves can trigger ER stress in pancreatic β-cells, for example (26, 27), accelerated lipolysis and FFA efflux from already stressed adipocytes may produce a feed-forward machinery to further stimulate or worsen ER stress in other tissues. This phenomenon implicates a central role of adipose cells in the development of ER stress-related pathologies in various tissues in obesity and diabetes.
Acknowledgment
We thank Lingzi Xu from Peking University Health Science Center for the kind help with editing the manuscript.
This work was supported by National Basic Research Program of China Grants 2009CB941603 and 2012CB517505 and National Natural Science Foundation of China Grants 81070114 and 30890042.
- FFA
- free fatty acid
- ATGL
- adipose triglyceride lipase
- AMPK
- AMP-activated protein kinase
- ER
- endoplasmic reticulum
- GRP78
- glucose-regulated protein 78
- HSL
- hormone-sensitive lipase
- Iso
- isoproterenol
- PERK
- RNA-dependent protein kinase-like eIF2α kinase
- Thaps
- thapsigargin
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Matthews C. K., van Holde K. E. (eds) (1996) Biochemistry, pp. 819–859, Benjamin-Cummings, Menlo Park, CA [Google Scholar]
- 2. Londos C., Brasaemle D. L., Schultz C. J., Adler-Wailes D. C., Levin D. M., Kimmel A. R., Rondinone C. M. (1999) On the control of lipolysis in adipocytes. Ann. N.Y. Acad. Sci. 892, 155–168 [DOI] [PubMed] [Google Scholar]
- 3. Arner P. (2002) Insulin resistance in type 2 diabetes. Role of fatty acids. Diabetes Metab. Res. Rev. 18, S5–S9 [DOI] [PubMed] [Google Scholar]
- 4. Jensen M. D. (2007) Adipose tissue metabolism. An aspect we should not neglect? Horm. Metab. Res. 39, 722–725 [DOI] [PubMed] [Google Scholar]
- 5. Xu C., He J., Jiang H., Zu L., Zhai W., Pu S., Xu G. (2009) Direct effect of glucocorticoids on lipolysis in adipocytes. Mol. Endocrinol. 23, 1161–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang H. H., Halbleib M., Ahmad F., Manganiello V. C., Greenberg A. S. (2002) Tumor necrosis factor-α stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes 51, 2929–2935 [DOI] [PubMed] [Google Scholar]
- 7. Ryden M., Dicker A., van Harmelen V., Hauner H., Brunnberg M., Perbeck L., Lonnqvist F., Arner P. (2002) Mapping of early signaling events in tumor necrosis factor-α-mediated lipolysis in human fat cells. J. Biol. Chem. 277, 1085–1091 [DOI] [PubMed] [Google Scholar]
- 8. Zu L., Jiang H., He J., Xu C., Pu S., Liu M., Xu G. (2008) Salicylate blocks lipolytic actions of tumor necrosis factor-α in primary rat adipocytes. Mol. Pharmacol. 73, 215–223 [DOI] [PubMed] [Google Scholar]
- 9. Zu L., He J., Jiang H., Xu C., Pu S., Xu G. (2009) Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. J. Biol. Chem. 284, 5915–5926 [DOI] [PubMed] [Google Scholar]
- 10. Honnor R. C., Dhillon G. S., Londos C. (1985) cAMP-dependent protein kinase and lipolysis in rat adipocytes. II. Definition of steady-state relationship with lipolytic and antilipolytic modulators. J. Biol. Chem. 260, 15130–15138 [PubMed] [Google Scholar]
- 11. Greenberg A. S., Shen W. J., Muliro K., Patel S., Souza S. C., Roth R. A., Kraemer F. B. (2001) Stimulation of lipolysis and hormone-sensitive lipase via the extracellular signal-regulated kinase pathway. J. Biol. Chem. 276, 45456–45461 [DOI] [PubMed] [Google Scholar]
- 12. Fricke K., Heitland A., Maronde E. (2004) Cooperative activation of lipolysis by protein kinase A and protein kinase C pathways in 3T3-L1 adipocytes. Endocrinology 145, 4940–4947 [DOI] [PubMed] [Google Scholar]
- 13. Brasaemle D. L. (2007) Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 48, 2547–2559 [DOI] [PubMed] [Google Scholar]
- 14. Sztalryd C., Xu G., Dorward H., Tansey J. T., Contreras J. A., Kimmel A. R., Londos C. (2003) Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J. Cell Biol. 161, 1093–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He J., Jiang H., Tansey J. T., Tang C., Pu S., Xu G. (2006) Calyculin and okadaic acid promote perilipin phosphorylation and increase lipolysis in primary rat adipocytes. Biochim. Biophys. Acta 1761, 247–255 [DOI] [PubMed] [Google Scholar]
- 16. Greenberg A. S., Egan J. J., Wek S. A., Garty N. B., Blanchette-Mackie E. J., Londos C. (1991) Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 266, 11341–11346 [PubMed] [Google Scholar]
- 17. Zhang H. H., Souza S. C., Muliro K. V., Kraemer F. B., Obin M. S., Greenberg A. S. (2003) Lipase-selective functional domains of perilipin A differentially regulate constitutive and protein kinase A-stimulated lipolysis. J. Biol. Chem. 278, 51535–51542 [DOI] [PubMed] [Google Scholar]
- 18. Brasaemle D. L., Levin D. M., Adler-Wailes D. C., Londos C. (2000) The lipolytic stimulation of 3T3-L1 adipocytes promotes the translocation of hormone-sensitive lipase to the surfaces of lipid storage droplets. Biochim. Biophys. Acta 1483, 251–262 [DOI] [PubMed] [Google Scholar]
- 19. Anthonsen M. W., Rönnstrand L., Wernstedt C., Degerman E., Holm C. (1998) Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 273, 215–221 [DOI] [PubMed] [Google Scholar]
- 20. Zimmermann R., Strauss J. G., Haemmerle G., Schoiswohl G., Birner-Gruenberger R., Riederer M., Lass A., Neuberger G., Eisenhaber F., Hermetter A., Zechner R. (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306, 1383–1386 [DOI] [PubMed] [Google Scholar]
- 21. Schweiger M., Schreiber R., Haemmerle G., Lass A., Fledelius C., Jacobsen P., Tornqvist H., Zechner R., Zimmermann R. (2006) Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J. Biol. Chem. 281, 40236–40241 [DOI] [PubMed] [Google Scholar]
- 22. Rydén M., Jocken J., van Harmelen V., Dicker A., Hoffstedt J., Wirén M., Blomqvist L., Mairal A., Langin D., Blaak E., Arner P. (2007) Comparative studies of the role of hormone-sensitive lipase and adipose triglyceride lipase in human fat cell lipolysis. Am. J. Physiol. Endocrinol. Metab. 292, E1847–E1855 [DOI] [PubMed] [Google Scholar]
- 23. Kaufman R. J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13, 1211–1233 [DOI] [PubMed] [Google Scholar]
- 24. Harding H. P., Calfon M., Urano F., Novoa I., Ron D. (2002) Transcriptional and translational control in the mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 18, 575–599 [DOI] [PubMed] [Google Scholar]
- 25. Yusta B., Baggio L. L., Estall J. L., Koehler J. A., Holland D. P., Li H., Pipeleers D., Ling Z., Drucker D. J. (2006) GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 4, 391–406 [DOI] [PubMed] [Google Scholar]
- 26. Cunha D. A., Hekerman P., Ladrière L., Bazarra-Castro A., Ortis F., Wakeham M. C., Moore F., Rasschaert J., Cardozo A. K., Bellomo E., Overbergh L., Mathieu C., Lupi R., Hai T., Herchuelz A., Marchetti P., Rutter G. A., Eizirik D. L., Cnop M. (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 121, 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kharroubi I., Ladrière L., Cardozo A. K., Dogusan Z., Cnop M., Eizirik D. L. (2004) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology 145, 5087–5096 [DOI] [PubMed] [Google Scholar]
- 28. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 29. Sharma N. K., Das S. K., Mondal A. K., Hackney O. G., Chu W. S., Kern P. A., Rasouli N., Spencer H. J., Yao-Borengasser A., Elbein S. C. (2008) Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J. Clin. Endocrinol. Metab. 93, 4532–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gregor M. F., Hotamisligil G. S. (2007) Thematic review series. Adipocyte Biology. Adipocyte stress. The endoplasmic reticulum and metabolic disease. J. Lipid Res. 48, 1905–1914 [DOI] [PubMed] [Google Scholar]
- 31. Ohsaki Y., Cheng J., Suzuki M., Fujita A., Fujimoto T. (2008) Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J. Cell Sci. 121, 2415–2422 [DOI] [PubMed] [Google Scholar]
- 32. Robenek H., Hofnagel O., Buers I., Robenek M. J., Troyer D., Severs N. J. (2006) Adipophilin-enriched domains in the ER membrane are sites of lipid droplet biogenesis. J. Cell Sci. 119, 4215–4224 [DOI] [PubMed] [Google Scholar]
- 33. Blanchette-Mackie E. J., Dwyer N. K., Barber T., Coxey R. A., Takeda T., Rondinone C. M., Theodorakis J. L., Greenberg A. S., Londos C. (1995) Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J. Lipid Res. 36, 1211–1226 [PubMed] [Google Scholar]
- 34. Ozeki S., Cheng J., Tauchi-Sato K., Hatano N., Taniguchi H., Fujimoto T. (2005) Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 118, 2601–2611 [DOI] [PubMed] [Google Scholar]
- 35. Jiang H., He J., Pu S., Tang C., Xu G. (2007) Heat shock protein 70 is translocated to lipid droplets in rat adipocytes upon heat stimulation. Biochim. Biophys. Acta 1771, 66–74 [DOI] [PubMed] [Google Scholar]
- 36. Egan J. J., Greenberg A. S., Chang M. K., Londos C. (1990) Control of endogenous phosphorylation of the major cAMP-dependent protein kinase substrate in adipocytes by insulin and β-adrenergic stimulation. J. Biol. Chem. 265, 18769–18775 [PubMed] [Google Scholar]
- 37. Peled N., Krenz M. C. (1981) A new assay of microbial lipases with emulsified trioleoyl glycerol. Anal. Biochem. 112, 219–222 [DOI] [PubMed] [Google Scholar]
- 38. Hotamisligil G. S. (2005) Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 54, S73–S78 [DOI] [PubMed] [Google Scholar]
- 39. Egan J. J., Greenberg A. S., Chang M. K., Wek S. A., Moos M. C., Jr., Londos C. (1992) Mechanism of hormone-stimulated lipolysis in adipocytes: translocation of hormone-sensitive lipase to the lipid storage droplet. Proc. Natl. Acad. Sci. U.S.A. 89, 8537–8541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cunha D. A., Ladrière L., Ortis F., Igoillo-Esteve M., Gurzov E. N., Lupi R., Marchetti P., Eizirik D. L., Cnop M. (2009) Glucagon-like peptide-1 agonists protect pancreatic beta-cells from lipotoxic endoplasmic reticulum stress through up-regulation of BiP and JunB. Diabetes 58, 2851–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu P., Han Z., Couvillon A. D., Exton J. H. (2004) Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 279, 49420–49429 [DOI] [PubMed] [Google Scholar]
- 42. Davies S. P., Reddy H., Caivano M., Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yin W., Mu J., Birnbaum M. J. (2003) Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis In 3T3-L1 adipocytes. J. Biol. Chem. 278, 43074–43080 [DOI] [PubMed] [Google Scholar]
- 44. Daval M., Diot-Dupuy F., Bazin R., Hainault I., Viollet B., Vaulont S., Hajduch E., Ferré P., Foufelle F. (2005) Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J. Biol. Chem. 280, 25250–25257 [DOI] [PubMed] [Google Scholar]
- 45. Djouder N., Tuerk R. D., Suter M., Salvioni P., Thali R. F., Scholz R., Vaahtomeri K., Auchli Y., Rechsteiner H., Brunisholz R. A., Viollet B., Mäkelä T. P., Wallimann T., Neumann D., Krek W. (2010) PKA phosphorylates and inactivates AMPKα to promote efficient lipolysis. EMBO J. 29, 469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ren T., He J., Jiang H., Zu L., Pu S., Guo X., Xu G. (2006) Metformin reduces lipolysis in primary rat adipocytes stimulated by tumor necrosis factor-α or isoproterenol. J. Mol. Endocrinol. 37, 175–183 [DOI] [PubMed] [Google Scholar]
- 47. Zhang T., He J., Xu C., Zu L., Jiang H., Pu S., Guo X., Xu G. (2009) Mechanisms of metformin inhibiting lipolytic response to isoproterenol in primary rat adipocytes. J. Mol. Endocrinol. 42, 57–66 [DOI] [PubMed] [Google Scholar]
- 48. Fredrikson G., Strålfors P., Nilsson N. O., Belfrage P. (1981) Hormone-sensitive lipase of rat adipose tissue. Purification and some properties. J. Biol. Chem. 256, 6311–6320 [PubMed] [Google Scholar]
- 49. Su C. L., Sztalryd C., Contreras J. A., Holm C., Kimmel A. R., Londos C. (2003) Mutational analysis of the hormone-sensitive lipase translocation reaction in adipocytes. J. Biol. Chem. 278, 43615–43619 [DOI] [PubMed] [Google Scholar]
- 50. Granneman J. G., Moore H. P., Granneman R. L., Greenberg A. S., Obin M. S., Zhu Z. (2007) Analysis of lipolytic protein trafficking and interactions in adipocytes. J. Biol. Chem. 282, 5726–5735 [DOI] [PubMed] [Google Scholar]
- 51. Granneman J. G., Moore H. P., Krishnamoorthy R., Rathod M. (2009) Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 284, 34538–34544 [DOI] [PMC free article] [PubMed] [Google Scholar]