

Background: Mutations in malin cause ∼40% cases of Lafora disease; however, the underlying mechanism of disease pathogenesis is poorly understood.
Results: Malin interacts with and promotes degradation of dishevelled2 leading to down-regulation of β-catenin-mediated transcriptional activity.
Conclusion: Malin negatively regulates Wnt signaling.
Significance: Aberrant Wnt signaling pathway might be implicated in Lafora disease pathogenesis.
Keywords: Autophagy, Glycogen Storage Disease, Proteasome, Ubiquitin Ligase, Wnt Signaling
Abstract
Using yeast-two hybrid screening followed by co-immunoprecipitation assay, we have found that the Lafora disease ubiquitin ligase malin interacts with dishevelled2, a key mediator of Wnt signaling pathway. Overexpression of malin enhances the degradation of dishevelled2 and inhibits Wnt signaling, which is evident from the down-regulation of β-catenin target genes and the decrease in β-catenin-mediated transcriptional activity. Partial knockdown of malin significantly increases the level of dishevelled2 and up-regulates Wnt signaling. Several malin mutants are found to be ineffective in degrading dishevelled2 and regulating the Wnt pathway. We have also found that malin enhances K48- and K63-linked ubiquitination of dishevelled2 that could lead to its degradation through both proteasome and autophagy. Altogether, our results indicate that malin regulates Wnt signaling pathway through the degradation of dishevelled2 and suggest possible deregulation of Wnt signaling in Lafora disease.
Introduction
Lafora disease (LD)3 is the most severe form of progressive myoclonus epilepsy and is inherited in an autosomal recessive manner. Severe myoclonic seizures, dementia, ataxia, muscle wasting, and respiratory failure typically characterize the disease (1–5). Onset of the disease is usually in the teen years, and the patient dies within 10 years of the first seizure. One of the common pathological hallmarks of LD is the intracellular accumulation of polyglucosan inclusions (commonly known as Lafora bodies) in many organs including brain, liver, heart, skeletal muscle, and skin (2, 3, 6). LD is caused by mutations in either the EPM2A or EPM2B gene (7, 8). The EPM2A gene encodes for a dual specificity phosphatase protein called laforin, whereas the EPM2B gene encodes malin, an E3 ubiquitin ligase having RING finger domain (7–10).
Several recent studies have demonstrated that malin in association with laforin regulates the turnover of a variety of proteins, mainly those that are involved in glycogen metabolism like glycogen synthase, glycogen debranching enzyme, protein targeting to glycogen, and neuronatin (9, 11–16). These findings could potentially explain the formation of Lafora bodies in LD, but how mutations in either malin or laforin cause disease pathogenesis and the involvement of Lafora bodies in the disease progression is poorly understood. There are reports suggesting that the formation of Lafora bodies and neuronal loss in LD might be two independent consequences (17, 18). This is further supported by some recent studies indicating the role of either laforin or malin in cell survival pathways (19, 20). Emerging evidence now points toward the defect in intracellular protein degradation pathways in LD pathogenesis (21–24). Because malin is an ubiquitin ligase, its loss of function could result in improper clearance and accumulation of its target substrates, leading to the pathogenesis of LD. Therefore, identification of novel cellular substrates of malin could provide further insight in understanding the disease pathogenesis.
Dishevelled (Dvl) is a central mediator of Wnt signaling pathway, which is involved in development, tissue self-renewal and tumorigenesis (25–29). It is a cytoplasmic phosphoprotein and exits as three isoforms in mammals (Dv1-1, Dv1-2, and Dv1-3). Dvl has three highly conserved domains: an amino-terminal DIX domain, a central PDZ domain, and a carboxyl-terminal DEP domain (29). In canonical Wnt pathway, Dvl regulates Wnt/β-catenin-mediated target gene expression by preventing the constitutive degradation of cytosolic β-catenin through ubiquitin-proteasome system (25, 26, 29). The binding of Wnt ligand to its membrane receptor frizzled causes polymerization and assembly of Dvl in cell membrane. This provides a platform for axin and glycogen synthase kinase (GSK)-3β, along with adenomatous polyposis coli protein and casin kinase-1, to relocate to the membrane leading to disassembly of the β-catenin destruction complex. β-Catenin then translocates to the nucleus and forms complexes with transcription factors of lymphoid enhancer-binding factor/T-cell factor family and mediates transcription of Wnt target genes (25–27). Dvl also plays an important role in noncanonical and calcium-dependent Wnt pathways (29).
The activity of Dvl is dynamically modulated by phosphorylation and ubiquitin-mediated degradation (29, 30). Several ubiquitin ligases are implicated in the selective ubiquitination and degradation of Dvl (31–33). Here we demonstrate for the first time that malin interacts with Dvl2 and promotes its degradation through both proteasome and autophagy. Malin also negatively regulates Wnt signaling pathway.
MATERIALS AND METHODS
Expression Plasmids, Antibodies, and Other Chemicals
The construction of full-length and delRING malin in pcDNA3.1 vector having V5 and His tags and the source of C26S malin were described elsewhere (34). Dvl2-FLAG construct was a kind gift from Dr. Mariann Bienz (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK). TOPflash luciferase vector (measures T-cell factor-dependent transcriptional activity) was a kind gift from Dr. Randall T. Moon (University of Washington, Seattle), and mutant ubiquitin constructs were a kind gift from Dr. Lim Kah Leong (National Neuroscience Institute, Singapore).
All of the cell culture reagents; MG132, rapamycin, bafilomycin A (BFA), and mouse monoclonal anti-FLAG were purchased from Sigma. Lipofectamine® 2000, optiMEM, and mouse monoclonal V5 antibody were purchased from Invitrogen. Mouse monoclonal anti-Myc, rabbit polyclonal anti-GAPDH, and anti-β-catenin were purchased from Santa Cruz Biotechnologies. Rabbit polyclonal anti-ubiquitin and mouse monoclonal anti-HA were procured from Dako and Roche Applied Science, respectively. Rabbit polyclonal anti-cyclinD1 was from Abcam. Human-specific malin and β-catenin siRNA (a pool of three target-specific 20–25-nucleotide siRNA) along with control (scrambled sequence) were purchased from Santa Cruz Biotechnologies. Alkaline phosphatase and fluorophore-conjugated secondary antibodies were purchased from Vector Laboratories. All other chemicals were purchased from Sigma unless otherwise mentioned.
Yeast Two-hybrid Screening
Wild type human malin was cloned into the pGBKT7 plasmid and used as bait to screen a human fetal brain cDNA library (Clontech, Palo Alto, CA) as described earlier (16). Colonies were grown on selection medium lacking amino acids (Leu, Trp, and His), and positive clones were screened using the auxotrophic marker genes and the α-galactosidase assay. Plasmids isolated from yeast were transformed in Escherichia coli, amplified, and sequenced. The interactions were confirmed by retransforming the identified pACT-cDNA plasmids together with the bait (pGBKT7-malin) into the yeast.
Cell Culture, Transfection, and RT-PCR Analysis
HEK293 and HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum and antibiotics penicillin/streptomycin. For routine experiments, the cells were plated onto six-well tissue culture plates at subconfluent density. After 24 h of plating, the cells were transfected with various plasmids using Lipofactamine® 2000 according to the manufacturer's instructions (transfection efficiency was ∼50–60%), and then the cells were processed for co-immunoprecipitation, immunoblotting analysis, or immunofluorescence studies. The RT-PCR analysis of malin was described earlier (16).
Co-immunoprecipitation and Immunoblotting Experiment
HEK293 cells were transiently transfected with wild type or mutant malin plasmids in 6-well tissue culture plates. After 36 h, the cells were washed with ice-cold PBS, collected by centrifugation, and lysed on ice for 30 min with Nonidet P-40 lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, and complete protease inhibitor mixture). The cell lysates were briefly sonicated and centrifuged for 10 min at 15 000 × g at 4 °C, and the supernatants were used for immunoprecipitation as described earlier (35). Bound proteins were eluted from the beads with 1× SDS sample buffer, vortexed, boiled for 6 min, and analyzed by immunoblotting. The total cell lysate and the immunoprecipitated proteins were separated through SDS-polyacrylamide gel electrophoresis and proceeded for immunoblot analysis as previously described (35). Dvl2, β-catenin, cyclin D1, GAPDH, and FLAG antibodies were used in 1:1000 dilutions, and V5 and HA antibodies were used in 1:5000 dilutions.
Immunofluorescence Techniques
HEK293 cells grown in chamber slides were transiently transfected with appropriate plasmids. Thirty-six hours post-transfection, the cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.5% Triton X-100 in PBS for 5 min, washed four or five times, and then blocked with 4% nonfat dried milk in PBS for 2 h. The cells were then incubated with primary antibody overnight at 4 °C. After multiple washings with PBS, the cells were incubated with appropriate fluorescent-labeled secondary antibody (1:500 dilutions) for 1 h, washed several times, and then mounted. The samples were observed in a fluorescence microscope (Apotome, Zeiss, Germany), and digital images were assembled using Adobe Photoshop. The anti-Dvl2, anti-V5, and anti-Myc were used in 1:1000 dilutions, and anti-HA was used in 1:250 dilutions.
Luciferase Reporter Assay
HEK293 cells were transfected onto 6-well tissue cultured plates with a mixture of TOPflash reporter and PRL-SV40 (express Renilla luciferase) plasmids along with either various malin or Dvl2 constructs as indicated in the respective figure legends. Thirty-six hours after transfection, the cells were harvested and subjected to dual luciferase assay according to the manufacturer's instructions. PRL-SV40 used for co-transfection to normalize the data (ratio of firefly to Renilla values) and transfected at a very low concentration (500-fold lower than TOPflash luciferase plasmid). The data are represented as relative luciferase activity.
In Vitro Ubiquitination Assay
The in vitro ubiquitination assay was performed as described earlier (36). Dvl2-FLAG protein was immunoprecipitated with FLAG antibody from the HEK293 cell transfected with Dvl2-FLAG. The immunoprecipitated protein was washed several times and then used for in vitro ubiquitination assay. Briefly, equal amounts of immunoprecipitated Dvl2-FLAG were incubated in a reaction volume of 50 μl of 50 mm of Tris-HCl, pH 7.4, containing 50 ng of E1, 500 ng of E2 (UBCH5c), 2 μg of wild type or deRING malin, 6 μg of ubiquitin, 1 mm of DTT, 2 mm of MgCl2, and 4 mm of ATP. The incubation was carried out at 30 °C for 2 h. The reaction was terminated by the addition of SDS sample buffer, boiled, and separated through 10% SDS-polyacrylamide gel electrophoresis. The blot was probed with ubiquitin antibody.
Statistical Analysis
Statistical analysis was performed out using SigmaStat software. The values are presented as the means ± S.D. Intergroup comparisons were analyzed by two-tailed Student's t test. p < 0.05 was considered statistical significant.
RESULTS
Malin Interacts with Dvl2
To identify the novel interacting partner of malin, we employed yeast two-hybrid screening of human fetal brain cDNA library using wild type malin as bait. DNA sequencing analysis revealed few of the clones as Dvl2 (Fig. 1A). Interaction between Dvl2 and malin was further confirmed by co-immunoprecipitation assay. HEK293 cells were transiently transfected with malin or its RING domain-deleted mutant (delRING), and then cell lysates were processed for co-immunoprecipitation using Dvl2 antibody. The blots were detected with Dvl2 and V5 antibodies (that detects full-length and delRING malin). We found that both wild type and delRING malin interact with Dvl2 (Fig. 1B). Fig. 1C depicts the domain structure of wild type malin and Dvl2 along with the position of various mutants that were used in the experiment. The C26S mutant of malin is detected in subsets of LD patients, and it is functionally inactive. The delRING deletion mutant is also functionally inactive because the RING domain is necessary for ubiquitin ligase function. We next performed double immunofluorescence staining of malin and Dvl2 to study their co-localization. HEK293 cells were transiently transfected with plasmids encoding wild type and mutant malin along with Dvl2, and 36 h later, the cells were subjected to immunofluorescence staining. As shown in Fig. 2, ectopic expression of Dvl2 formed cytoplasmic puncta as was demonstrated earlier (37, 38). Interestingly, wild type malin as well as two of its mutants (delRING and C26S mutant) were co-localized with cytoplasmic puncta of Dvl2. These findings further suggest that wild type malin or its two mutants (delRING and C26S) also associate with Dvl2. It is also important to note that wild type malin, as well as its mutant, showed both cytoplasmic and nuclear distribution, as has been shown earlier (34). Because of the unavailability of good malin antibody, we were unable to check the endogenous co-localization of malin with Dvl2. However, we studied the co-localization of very low levels of overexpressed malin with endogenous Dvl2 in HEK293 cells. The endogenous Dvl2 showed diffuse cytoplasmic staining with a few puncate structures where malin was co-localized. We have also noticed a decrease in the numbers of cytoplasmic puncta of Dvl2 in cells overexpressing malin (supplemental Fig. S1).
FIGURE 1.

Malin interacts with Dvl2. A, identification of Dvl2 as an interacting partner of malin in yeast two-hybrid screening. The wild type malin was used as bait to screen human fetal cDNA library as described under “Experimental Procedures.” Identified Dvl2 clone (in GAL4-BD vector) was retransformed into yeast cells along with malin or lamin C (negative control) and grown on plates lacking amino acids Leu, Trp, and His in culture media. In positive control, yeast cells were transformed with pGADT7-RecT (express SV40 large T-antigen as a GAL4-AD fusion) along with pGBKT7–53 (express p53 as a GAL4-BD fusion). B, co-immunoprecipitation of Dvl2 with malin. HEK293 cells were transiently transfected with plasmids encoding LacZ, wild type, and delRING malin (all having V5 tag), and 48 h later the cells were collected, and lysates were made and subjected to co-immunoprecipitation using Dvl2 antibody. The blots were probed with antibodies against Dvl2 and V5 (to detect LacZ, wild type, and delRING malin). WCL, whole cell lysate; IP, immunoprecipitation; IB, immunoblot. C, domain structure of malin and Dvl2 including positions of specific mutations.
FIGURE 2.

Malin and its mutants co-localize with Dvl2 vesicles. HEK293 cells were transfected with plasmids encoding wild type malin or its two mutants along with Dvl2-FLAG (250 ng of each plasmid/well in a 2-well chamber slide). Thirty-six hours later, the cells were processed for double immunofluorescence staining using antibodies against Dvl2/V5 (to detect wild type and delRING malin) and Dvl2/Myc (to detect C26S). FITC-conjugated secondary antibody was used to recognize wild type or mutant malins, and Texas Red-conjugated secondary antibody was used to label Dvl2. The nuclei were counterstained with DAPI. Scale bar, 20 μm.
Malin Promotes Degradation of Dvl2 and Negatively Regulates Wnt Signaling Pathway
Because malin interacts and co-localizes with Dvl2, we further investigated the effect of malin on Dvl2 degradation and regulation of Wnt signaling pathway. HEK293 cells were transfected with different concentrations of plasmid encoding malin, and 36 h later, the cells were collected and subjected to immunoblot analysis using antibodies against Dvl2 and other markers of Wnt pathway. As shown in Fig. 3 (A and B), the steady-state level of endogenous Dvl2 was decreased upon overexpression of malin in a concentration-dependent manner. The endogenous level of β-catenin and its two downstream target genes (c-Myc and cyclin D1) were also significantly reduced. Malin, however, did not have any direct effect on β-catenin, because there was no interaction between these two proteins (supplemental Fig. S2). Malin also degraded ectopically expressed Dvl2 (Fig. 3, C and D). We further studied the effect of malin on Wnt/β-catenin-mediated transcriptional activity using TOPflash reporter gene assay. HEK293 and HeLa cells were transfected with different concentrations of malin plasmid along with a mixture of TOPflash reporter and PRL-SV40 plasmids, and 36 h post-transfection, the cells were collected and subjected to dual luciferase reporter assay. In one set of experiment, the cells were treated with Wnt3a to stimulate the Wnt signaling pathway. Fig. 4 (A and B) showed that malin significantly reduced the TOPflash reporter activity in a concentration-dependent way in both uninduced and induced HEK293 and HeLa cells. Fig. 4 (C and D) demonstrated the suppressive effect of malin on TOPflash reporter activity when HEK293 or HeLa cells were treated with different concentrations of Wnt3a. To further confirm our findings, we performed siRNA-mediated knockdown of malin and then studied the level of Dvl2 and TOPflash activity under unstimulated and stimulated conditions. Partial knockdown of malin significantly increased the level of Dvl2 and β-catenin and concomitantly stimulated (significantly over control) Wnt signaling as evident from the increase in TOPflash reporter activity (Fig. 5). Partial knockdown of β-catenin prevented the Wnt3a-induced TOPflash activity (Fig. 5C). We also studied the effect of two malin mutants (delRING and C26S) on Dvl2 degradation and TOPflash reporter activity and observed that none of the mutants had any significant effect on Dvl2 degradation and TOPflash reporter activity (Fig. 6).
FIGURE 3.

Malin decreases Dvl2 level and downstream targets of Wnt/β-catenin pathway in the HEK293 cell. A, cells were transfected with different concentrations of wild type malin, and 48 h post-transfection, the cell lysates were made and subjected to immunoblot analysis using antibodies against Dvl2, β-catenin, c-Myc, cyclin D1, GAPDH, and V5 (to detect malin). B, band intensities of the blot shown above were quantified using NIH image analysis software, normalized with GAPDH, and expressed as percentages of change. The values are the means ± S.D. of three independent experiments. *, p < 0.05 in comparison with control. C, cells were transfected with different concentrations of plasmid encoding malin along with fixed amounts of Dvl2 for 48 h. The collected cells were then subjected to immunoblot analysis using FLAG, GAPDH, and V5 antibodies. D, quantitation of the Dvl2 blot shown in C. The band intensities were normalized with GAPDH and expressed as percentages of change. *, p < 0.01 in comparison with control.
FIGURE 4.

Malin inhibits Wnt signaling pathway. A and B, malin inhibits β-catenin-dependent luciferase activity (TOPflash activity) in a concentration-dependent manner. HEK293 (A) or HeLa (B) cells were plated onto 6-well tissue cultured plate, and on the following day the cells were transfected with increasing concentrations of plasmid encoding malin along with a mixture of TOPflash (500 ng) and Renilla luciferase (1 ng) vectors. Twenty-four hours post-transfection, the cells were left untreated or treated with Wnt3a (100 ng/ml) for another 24 h. The cells were then collected and subjected to dual luciferase assay. The values are the means ± S.D. of three independent experiments each performed in triplicate. *, p < 0.01 in comparison with the respective control group. C and D, HEK293 or HeLa cells were transfected with either empty pcDNA3.1 or malin plasmid (2 μg/well) along with a mixture of TOPflash and Renilla reporter vectors, and 24 h post-transfection, the cells were treated with different concentrations of Wnt3a for another 24 h. The collected cells were then processed for dual luciferase assay. The values are the means ± S.D. of two independent experiments each performed triplicate. *, p < 0.01 in comparison with respective control group.
FIGURE 5.

Knockdown of malin increases the level of Dvl2 and activates Wnt signaling pathway. A, HEK293 cells were transfected with scrambled siRNA (control) or human malin specific siRNA (each 50 pmol/well of 6-well tissue culture plate) for 48 h. The cells were then collected and either processed for RNA extraction followed by RT-PCR analysis of malin and β-actin or subjected to immunoblot analysis using antibodies against Dvl2, β-catenin, and GAPDH. B, quantitation of the level of Dvl2 and β-catenin in the knockdown experiment described above. The values were normalized against GAPDH and expressed as percentages of change. The values represent the means ± S.D. of three independent experiments. *, p < 0.05 in comparison with respective control group. C, HEK293 cells were transfected with scrambled siRNA (control, Con.), β-catenin (β cat.) siRNA, or malin siRNA (each 50 pmol/well of 6-well tissue culture plate) along with TOPflash and Renilla luciferase vector for 24 h. The cells were then treated with Wnt3a (200 ng/ml) for another 24 h, and the collected cells were subjected to dual luciferase assay. The values are the means ± S.D. of three independent experiments each performed in triplicate. *, p < 0.001 in comparison with control siRNA-transfected + Wnt3a-treated group.
FIGURE 6.
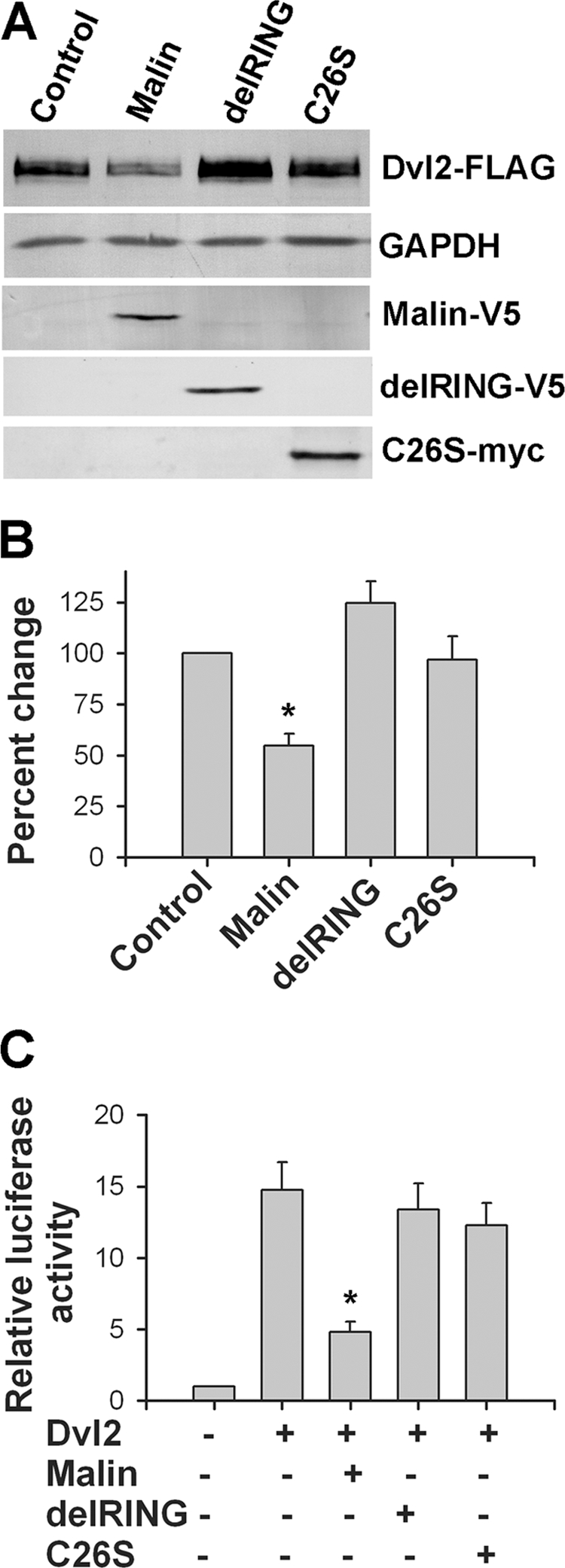
Effect of two malin mutants on Dvl2 level and TOPflash activity. A, HEK293 cells were transfected with plasmids encoding wild type or mutants of malin (delRING and C26S) along with Dvl2-FLAG (each plasmid 2 μg/well of 6-well tissue culture plate). Thirty-six hours later, the cells were processed for immunoblot analysis using antibodies against FLAG (to detect Dvl2), V5 (to detect wild type and delRING malin), Myc (to detect C26S), and GAPDH. B, Dvl2 blot collected from three independent experiments were quantified using NIH Image analysis software, normalized against GAPDH, and expressed as percentages of change. The values are the means ± S.D. of three independent experiments. *, p < 0.001 in comparison with the control group. C, HEK293 cells were transiently transfected with plasmid encoding wild type and mutant malins (along with Dvl2-FLAG and mixture of TOPflash and Renilla luciferase vector. Thirty-six hours later, the cells were processed for luciferase assay. The values are the means ± S.D. of three independent experiments each performed triplicate. *, p < 0.001 in comparison with only the Dvl2-transfected group.
Malin Mediates Degradation of Dvl2 via Proteasome and Autophagy
Dvl2 has been shown to be degraded through both proteasome and autophagy (30). To explore the pathway by which malin degrades Dvl2, we first checked the effect of proteasome inhibitor MG132 on Dvl2 level and found that proteasome inhibition partially recovered the malin-induced down-regulation of Dvl2 (Fig. 7, A and B). Because Dvl2 also degraded through autophagy, we further checked the effect of autophagy inducers on Dvl2 degradation. HEK293 cells were transfected with plasmids encoding malin along with Dvl2, and then the cells were starved by serum deprivation to induce autophagy. In some experiments, the cells were treated with autophagy inhibitor BFA before collecting the cell. Serum starvation decreased the level of Dvl2, and the treatment of BFA partially recovered its level (Fig. 7, C and D). Serum starvation also augmented malin-induced decrease in the level of Dvl2, which can be rescued by BFA (Fig. 7, C and D). The malin-induced degradation of Dvl2 was also induced upon treatment of the autophagy inducer rapamycin (supplemental Fig. S3).
FIGURE 7.

Malin mediates Dvl2 degradation via proteasomal and autophagic pathways. A, HEK293 cells were transfected with Dvl2-FLAG plasmid along with either empty pcDNA3.1 (control) or malin plasmid (each 2 μg/well of 6-well plate) for 36 h. In some experiments, the cells were treated with 10 μm of MG132 for 8 h before cell collection. The cell lysates were then subjected to immunoblot analysis using FLAG, V5, and GAPDH antibodies. B, quantitation of the level of Dvl2 in the experiment described in A. The values were normalized against GAPDH. The values represent the means ± S.D. of three independent experiments. *, p < 0.01 in comparison with control. C, cells were transfected with different plasmids as above and then starved with serum-deficient media for 6 h before collection. In some experiment, the cells were treated with BFA along with serum starvation. Collected cells were then subjected to immunoblot analysis using FLAG (to detect Dvl2) and GAPDH antibodies. D, quantitation of the level of Dvl2 in the experiment described in C. The values were normalized against GAPDH. The values represent the means ± S.D. of three independent experiments. Starv., starvation. *, p < 0.05.
Ubiquitination of Dvl2 by Malin
To study the effect of malin on Dvl2 ubiquitination, we performed both in vivo and in vitro ubiquitination assays. With in vivo ubiquitination assays, HEK293 cells were transfected with plasmids encoding malin and two of its mutants along with Dvl2 and ubiquitin-HA. The cells were collected and subjected to co-immunoprecipitation using FLAG antibody (to pull down Dvl2), and blots were probed with FLAG and HA antibodies. Fig. 8A showed that overexpression of malin induced the ubiquitination of Dvl2. The delRING and C26S malin had no effect on Dvl2 ubiquitination. Malin (but not its mutant) also induced ubiquitination of Dvl2 in in vitro ubiquitination assay (Fig. 8B). It was shown that K48-linked ubiquitination targets a substrate for proteasomal degradation, whereas K63-linked ubiquitination for autophagy (39–41). Because malin promotes degradation of Dvl2 through both proteasome and autophagy, we checked the involvement of malin in K48- and K63-linked ubiquitination of Dvl2. Fig. 8C showed that malin induced K48- and K63-linked ubiquitination of Dvl2 in in vivo ubiquitination assay. Lysine-less ubiquitin (K0) was used as control, which was not attached to Dvl2. Double immunofluorescence staining confirmed that Dvl2 puncta co-localized with both K48 and K63 ubiquitin, suggesting further that Dvl2 is ubiquitinated by both K48- and K63-linked ubiquitin chain (Fig. 8D).
FIGURE 8.

Malin ubiquitinates Dvl2 in vivo and in vitro. A, HEK293 cells were transiently transfected onto 6-well tissue cultured plate with plasmids encoding wild type malin or its mutants (2 μg/well) along with Dvl2-FLAG (2 μg/well) and ubiquitin-HA (1 μg/well). Forty-eight hours later, the cells were collected and processed for co-immunoprecipitation using FLAG antibody, and blots were probed with either FLAG or HA antibody. B, Malin induces ubiquitination of Dvl2 in in vitro ubiquitination assay. Equal amounts of immunoprecipitated Dvl2 were incubated with malin, delRING malin along with ubiquitin, E1, and E2 (UBCH5c) as described under “Experimental Procedures.” The blot was detected with ubiquitin antibody. C, cells were transfected with plasmids encoding Dvl2, wild type malin as in A along with wild type or various mutant forms of ubiquitin-HA (each 1 μg/well) constructs. Forty-eight hours later the cells were processed for co-immunoprecipitation using FLAG antibody, and blot was detected with HA antibody (to detect ubiquitinated Dvl2). D, HEK293 cells were transfected with Dvl2-FLAG along with K48- or K63-ubiquitin-HA constructs in chamber slides. Thirty-six hours later, the cells were processed for double immunofluorescence staining using antibodies against Dvl2/HA (to detect ubiquitin). FITC-conjugated secondary antibody was used to detect ubiquitin, and Texas Red-conjugated secondary antibody was used to label Dvl2. The nuclei were counterstained with DAPI. IP, immunoprecipitation; Con, control. Scale bar, 20 μm.
DISCUSSION
Here we provide evidence that malin promotes degradation of Dvl2 and thereby negatively regulates Wnt signaling pathway. First, we have shown that malin interacts with Dvl2 in a yeast two-hybrid system followed by co-immunoprecipitation assay. Overexpression of malin promotes the degradation of Dvl2 and consequently inhibits Wnt signaling. Partial knockdown of malin increases the steady-state level of Dvl2 and stimulates Wnt pathway. Catalytically inactive malin mutants are ineffective in degrading Dvl2 and regulating Wnt signaling. We have also demonstrated that malin promotes K48- and K63-linked ubiquitination of Dvl2, which could lead to its degradation through both proteasome and autophagy.
Dvl is a key component of Wnt signaling and transmits Wnt signals from membrane receptors to downstream effectors (25, 26, 28, 29). The current model of the canonical Wnt pathway suggests that in the absence of Wnt signals, β-catenin is phosphorylated and degraded by the ubiquitin proteasome system. The phosphorylation of β-catenin occurs as part of the multi-protein complex that includes GSK-3β, axin, casin kinase-1, and adenomatous polyposis coli. Wnt ligand initiates binding of Dvl to frizzled and axin to LDL-receptor-related protein-5/6, which results in disassembly of the β-catenin destruction complex and consequently accumulation of β-catenin in the nucleus. In the nucleus, β-catenin forms a complex with the T-cell factor/lymphoid-enhancer binding factor family of transcription factors and activates the transcription of target genes like c-Myc, cyclin D1, etc. Ectopic expression of Dvl also causes activation of canonical Wnt signaling pathway (32). Our findings indicate that malin could be one of the important regulators of canonical as well as noncanonical and calcium-dependent Wnt signaling by promoting the degradation of Dvl2.
Several ubiquitin ligases have been demonstrated to promote the ubiquitination and proteasome-mediated degradation of Dvl including NEDL1, Kelch-like-12-Cullin-3, and prickle-1 (31–33). Overexpression of these ligases negatively regulates the Wnt pathway. A recent report demonstrates the degradation of Dvl2 through autophagy under metabolic stress and suggests that autophagy could potentially regulate Wnt signaling pathway (30). Our findings show that malin promotes the degradation Dvl2 through both proteasome and autophagic degradation pathways. Malin also promotes both K48- and K63-linked ubiquitination of Dvl2, indicating further that malin uses both proteasomal and autophagic pathways for the clearance of Dvl2. Increasing evidence now suggests that K48-linked ubiquitination targets the substrate for proteasomal degradation, whereas K63-linked ubiquitination targets the substrate for autophagy (39–41).
Interestingly, laforin (a protein phosphatase), another protein implicated in LD, has been shown to down-regulate the Wnt signaling pathway by activating GSK-3β (20). Inactivation of laforin results in increased Wnt signaling and tumorigenesis (20). Laforin is an important interacting partner of malin, and these two proteins have been suggested to regulate some common physiological pathways, because LD patients with mutations in either malin or laforin are phenotypically indistinguishable (5, 42). In fact, laforin-malin complex has been shown to regulate the level and activity of many key enzymes that are involved in glycogen metabolism (11–15). These findings could provide a mechanistic basis in the formation of Lafora bodies in each and every LD patient. Our present findings, along with others (20), suggest that the Wnt signaling pathway could be another common pathway that is regulated by both malin and laforin (Fig. 9).
FIGURE 9.
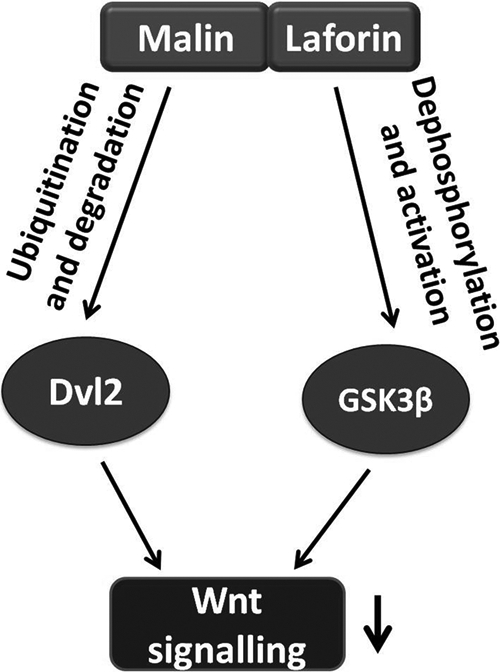
Schematic representation of the regulation of Wnt pathway by malin and laforin. Laforin is shown to function as a phosphatase for GSK-3β. It stimulates the degradation of β-catenin and down-regulates Wnt signaling by activating GSK-3β (20). Our finding shows that malin down-regulates Wnt pathway by promoting degradation of Dvl2. Therefore, the loss of function of either malin or laforin could result in aberrant Wnt signaling, which might be one of the possible cause of LD pathogenesis.
In addition to its well known involvement in several developmental processes including synaptic differentiation, the Wnt signaling pathway has also been implicated in regulating synaptic plasticity and neurogenesis in adults (28, 43). Loss of function of either malin or laforin could potentially increase Wnt signaling in developing or adult brain, leading to abnormal synaptic differentiation, synaptic plasticity, or other abnormalities. The teen age onset of the disease also indicates possible defects in the developmental program in LD. At present there is no evidence of altered Wnt signaling in LD. Interestingly, mutation in prickle-1, another ubiquitin ligase involved in the down-regulation of Wnt signaling through degradation of Dvl, also causes autosomal recessive progressive myoclonus epilepsy-ataxia syndrome (33, 44). These findings, together with ours, suggest a possible role for altered Wnt signaling in progressive myoclonus epilepsy including LD. Further studies in malin and laforin knock-out mice could provide insight in understanding the role of altered Wnt signaling in LD pathogenesis.
Laforin has also been implicated in the regulation of autophagy, and laforin knock-out mice exhibit defects in the autophagic degradation pathway (23, 24). Our findings of malin-dependent K63-linked ubiquitination and autophagic degradation of Dvl2 suggest that the laforin-malin complex might be involved in the autophagic degradation of not only Dvl2 but also other autophagic substrates.
In conclusion, our studies identified Dvl2 as a new substrate of malin. Malin negatively regulates Wnt signaling through degradation of Dvl2. Our findings suggest a possible involvement of altered Wnt signaling in LD pathogenesis.
Supplementary Material
Acknowledgments
We are thankful to Dr. Mariann Bienz (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK) for providing the Dvl2-FLAG construct. TOPflash luciferase vector was a kind gift from Dr. Randall T. Moon (University of Washington, Seattle), and mutant ubiquitin constructs were a kind gift from Dr. Lim Kah Leong (National Neuroscience Institute, Singapore).
This work was supported by the Department of Biotechnology of the Government of India.

This article contains supplemental Figs. S1–S3.
- LD
- Lafora disease
- GSK
- glycogen synthase kinase
- BFA
- bafilomycin A
- Dvl.
- dishevelled.
REFERENCES
- 1. Ganesh S., Puri R., Singh S., Mittal S., Dubey D. (2006) Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J. Hum. Genet. 51, 1–8 [DOI] [PubMed] [Google Scholar]
- 2. Acharya J. N., Satishchandra P., Asha T., Shankar S. K. (1993) Lafora's disease in south India. A clinical, electrophysiologic, and pathologic study. Epilepsia 34, 476–487 [DOI] [PubMed] [Google Scholar]
- 3. Delgado-Escueta A. V. (2007) Advances in lafora progressive myoclonus epilepsy. Curr. Neurol. Neurosci. Rep. 7, 428–433 [DOI] [PubMed] [Google Scholar]
- 4. Minassian B. A. (2001) Lafora's disease. Towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 25, 21–29 [DOI] [PubMed] [Google Scholar]
- 5. Singh S., Ganesh S. (2009) Lafora progressive myoclonus epilepsy. A meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum. Mutat. 30, 715–723 [DOI] [PubMed] [Google Scholar]
- 6. Carpenter S., Karpati G. (1981) Ultrastructural findings in Lafora disease. Ann. Neurol. 10, 63–64 [DOI] [PubMed] [Google Scholar]
- 7. Minassian B. A., Lee J. R., Herbrick J. A., Huizenga J., Soder S., Mungall A. J., Dunham I., Gardner R., Fong C. Y., Carpenter S., Jardim L., Satishchandra P., Andermann E., Snead O. C., 3rd, Lopes-Cendes I., Tsui L. C., Delgado-Escueta A. V., Rouleau G. A., Scherer S. W. (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 20, 171–174 [DOI] [PubMed] [Google Scholar]
- 8. Chan E. M., Young E. J., Ianzano L., Munteanu I., Zhao X., Christopoulos C. C., Avanzini G., Elia M., Ackerley C. A., Jovic N. J., Bohlega S., Andermann E., Rouleau G. A., Delgado-Escueta A. V., Minassian B. A., Scherer S. W. (2003) Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 35, 125–127 [DOI] [PubMed] [Google Scholar]
- 9. Gentry M. S., Worby C. A., Dixon J. E. (2005) Insights into Lafora disease. Malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. U.S.A. 102, 8501–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ganesh S., Agarwala K. L., Ueda K., Akagi T., Shoda K., Usui T., Hashikawa T., Osada H., Delgado-Escueta A. V., Yamakawa K. (2000) Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum. Mol. Genet. 9, 2251–2261 [DOI] [PubMed] [Google Scholar]
- 11. Cheng A., Zhang M., Gentry M. S., Worby C. A., Dixon J. E., Saltiel A. R. (2007) A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori's disease. Genes Dev. 21, 2399–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lohi H., Ianzano L., Zhao X. C., Chan E. M., Turnbull J., Scherer S. W., Ackerley C. A., Minassian B. A. (2005) Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 14, 2727–2736 [DOI] [PubMed] [Google Scholar]
- 13. Solaz-Fuster M. C., Gimeno-Alcañiz J. V., Ros S., Fernandez-Sanchez M. E., Garcia-Fojeda B., Criado Garcia O., Vilchez D., Dominguez J., Garcia-Rocha M., Sanchez-Piris M., Aguado C., Knecht E., Serratosa J., Guinovart J. J., Sanz P., Rodriguez de Córdoba S. (2008) Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum. Mol. Genet. 17, 667–678 [DOI] [PubMed] [Google Scholar]
- 14. Vilchez D., Ros S., Cifuentes D., Pujadas L., Vallès J., García-Fojeda B., Criado-García O., Fernández-Sánchez E., Medraño-Fernández I., Domínguez J., García-Rocha M., Soriano E., Rodríguez de Córdoba S., Guinovart J. J. (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10, 1407–1413 [DOI] [PubMed] [Google Scholar]
- 15. Worby C. A., Gentry M. S., Dixon J. E. (2008) Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 283, 4069–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharma J., Rao S. N., Shankar S. K., Satishchandra P., Jana N. R. (2011) Lafora disease ubiquitin ligase malin promotes proteasomal degradation of neuronatin and regulates glycogen synthesis. Neurobiol. Dis. 44, 133–141 [DOI] [PubMed] [Google Scholar]
- 17. Ganesh S., Delgado-Escueta A. V., Sakamoto T., Avila M. R., Machado-Salas J., Hoshii Y., Akagi T., Gomi H., Suzuki T., Amano K., Agarwala K. L., Hasegawa Y., Bai D. S., Ishihara T., Hashikawa T., Itohara S., Cornford E. M., Niki H., Yamakawa K. (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 11, 1251–1262 [DOI] [PubMed] [Google Scholar]
- 18. Chan E. M., Ackerley C. A., Lohi H., Ianzano L., Cortez M. A., Shannon P., Scherer S. W., Minassian B. A. (2004) Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum. Mol. Genet. 13, 1117–1129 [DOI] [PubMed] [Google Scholar]
- 19. Garyali P., Siwach P., Singh P. K., Puri R., Mittal S., Sengupta S., Parihar R., Ganesh S. (2009) The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum. Mol. Genet. 18, 688–700 [DOI] [PubMed] [Google Scholar]
- 20. Wang Y., Liu Y., Wu C., Zhang H., Zheng X., Zheng Z., Geiger T. L., Nuovo G. J., Liu Y., Zheng P. (2006) Epm2a suppresses tumor growth in an immunocompromised host by inhibiting Wnt signaling. Cancer Cell 10, 179–190 [DOI] [PubMed] [Google Scholar]
- 21. Rao S. N., Maity R., Sharma J., Dey P., Shankar S. K., Satishchandra P., Jana N. R. (2010) Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum. Mol. Genet. 19, 4726–4734 [DOI] [PubMed] [Google Scholar]
- 22. Vernia S., Rubio T., Heredia M., Rodríguez de Córdoba S., Sanz P. (2009) Increased endoplasmic reticulum stress and decreased proteasomal function in Lafora disease models lacking the phosphatase laforin. PLoS One 4, e5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puri R., Suzuki T., Yamakawa K., Ganesh S. (2012) Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum. Mol. Genet. 21, 175–184 [DOI] [PubMed] [Google Scholar]
- 24. Aguado C., Sarkar S., Korolchuk V. I., Criado O., Vernia S., Boya P., Sanz P., de Córdoba S. R., Knecht E., Rubinsztein D. C. (2010) Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 19, 2867–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moon R. T., Kohn A. D., De Ferrari G. V., Kaykas A. (2004) WNT and β-catenin signalling. Diseases and therapies. Nat. Rev. Genet. 5, 691–701 [DOI] [PubMed] [Google Scholar]
- 26. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 27. Logan C. Y., Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 28. Inestrosa N. C., Arenas E. (2010) Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 11, 77–86 [DOI] [PubMed] [Google Scholar]
- 29. Gao C., Chen Y. G. (2010) Dishevelled: The hub of Wnt signaling. Cell Signal. 22, 717–727 [DOI] [PubMed] [Google Scholar]
- 30. Gao C., Cao W., Bao L., Zuo W., Xie G., Cai T., Fu W., Zhang J., Wu W., Zhang X., Chen Y. G. (2010) Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 12, 781–790 [DOI] [PubMed] [Google Scholar]
- 31. Miyazaki K., Fujita T., Ozaki T., Kato C., Kurose Y., Sakamoto M., Kato S., Goto T., Itoyama Y., Aoki M., Nakagawara A. (2004) NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J. Biol. Chem. 279, 11327–11335 [DOI] [PubMed] [Google Scholar]
- 32. Angers S., Thorpe C. J., Biechele T. L., Goldenberg S. J., Zheng N., MacCoss M. J., Moon R. T. (2006) The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-β-catenin pathway by targeting Dishevelled for degradation. Nat. Cell Biol. 8, 348–357 [DOI] [PubMed] [Google Scholar]
- 33. Chan D. W., Chan C. Y., Yam J. W., Ching Y. P., Ng I. O. (2006) Prickle-1 negatively regulates Wnt/β-catenin pathway by promoting Dishevelled ubiquitination/degradation in liver cancer. Gastroenterology 131, 1218–1227 [DOI] [PubMed] [Google Scholar]
- 34. Rao S. N., Sharma J., Maity R., Jana N. R. (2010) Co-chaperone ChIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease. J. Biol. Chem. 285, 1404–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mishra A., Dikshit P., Purkayastha S., Sharma J., Nukina N., Jana N. R. (2008) E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J. Biol. Chem. 283, 7648–7656 [DOI] [PubMed] [Google Scholar]
- 36. Mishra A., Godavarthi S. K., Maheshwari M., Goswami A., Jana N. R. (2009) The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 284, 10537–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yanagawa S., van Leeuwen F., Wodarz A., Klingensmith J., Nusse R. (1995) The dishevelled protein is modified by wingless signaling in Drosophila. Genes Dev. 9, 1087–1097 [DOI] [PubMed] [Google Scholar]
- 38. Yang-Snyder J., Miller J. R., Brown J. D., Lai C. J., Moon R. T. (1996) A frizzled homolog functions in a vertebrate Wnt signaling pathway. Curr. Biol. 6, 1302–1306 [DOI] [PubMed] [Google Scholar]
- 39. Tan J. M., Wong E. S., Kirkpatrick D. S., Pletnikova O., Ko H. S., Tay S. P., Ho M. W., Troncoso J., Gygi S. P., Lee M. K., Dawson V. L., Dawson T. M., Lim K. L. (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439 [DOI] [PubMed] [Google Scholar]
- 40. Shi C. S., Kehrl J. H. (2010) TRAF6 and A20 regulate lysine 63-linked ubiquitination of beclin-1 to control TLR4-induced autophagy. Science Signal. 3, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lim K. L., Lim G. G. (2011) K63-linked ubiquitination and neurodegeneration. Neurobiol. Dis. 43, 9–16 [DOI] [PubMed] [Google Scholar]
- 42. Singh S., Sethi I., Francheschetti S., Riggio C., Avanzini G., Yamakawa K., Delgado-Escueta A. V., Ganesh S. (2006) Novel NHLRC1 mutations and genotype-phenotype correlations in patients with Lafora's progressive myoclonic epilepsy. J. Med. Genet. 43, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De Ferrari G. V., Moon R. T. (2006) The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene 25, 7545–7553 [DOI] [PubMed] [Google Scholar]
- 44. Bassuk A. G., Wallace R. H., Buhr A., Buller A. R., Afawi Z., Shimojo M., Miyata S., Chen S., Gonzalez-Alegre P., Griesbach H. L., Wu S., Nashelsky M., Vladar E. K., Antic D., Ferguson P. J., Cirak S., Voit T., Scott M. P., Axelrod J. D., Gurnett C., Daoud A. S., Kivity S., Neufeld M. Y., Mazarib A., Straussberg R., Walid S., Korczyn A. D., Slusarski D. C., Berkovic S. F., El-Shanti H. I. (2008) A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am. J. Hum. Genet. 83, 572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.