

Background: Pkd2L1 is a calcium- and acid-activated channel likely involved in acid sensing, but how it is regulated remains unclear.
Results: Receptor for activated C kinase 1 (RACK1), known to regulate various receptors/channels, interacts with and inhibits the function of Pkd2L1.
Conclusion: Pkd2L1 is a novel target channel of RACK1.
Significance: Pkd2L1-RACK1 interaction may play important physiological roles through regulating channel activation by calcium or acid.
Keywords: Electrophysiology, Oocyte, Plasma Membrane, Protein-Protein Interactions, TRP Channels, Biotinylation, Blocking Peptide, Co-immunoprecipitation, Receptor for Activated C Kinase 1 (RACK1), Two-microelectrode Voltage Clamp
Abstract
Pkd2L1 (also called TRPP3) is a non-selective cation channel permeable to Ca2+, Na+, and K+ and is activated by Ca2+. It is also part of an acid-triggered off-response cation channel complex. We previously reported roles of the Pkd2L1 C-terminal fragments in its channel function, but the role of the N terminus remains unclear. Using a yeast two-hybrid screening, we found that the Pkd2L1 N terminus interacts with the receptor for activated C kinase 1 (RACK1), a scaffolding/anchoring protein implicated in various cellular functions. This interaction requires the last two Trp-Asp (WD) motifs of RACK1 and fragment Ala19–Pro45 of Pkd2L1. The interaction was confirmed by GST pulldown, blot overlay, and co-immunoprecipitation assays. By 45Ca tracer uptake and two-microelectrode voltage clamp electrophysiology, we found that in Xenopus oocytes with RACK1 overexpression Pkd2L1 channel activity is abolished or substantially reduced. Combining with oocyte surface biotinylation experiments, we demonstrated that RACK1 inhibits the function of Pkd2L1 channel on the plasma membrane in addition to reducing its total and plasma membrane expression. Overexpressing Pkd2L1 N- or C-terminal fragments as potential blocking peptides for the Pkd2L1-RACK1 interaction, we found that Pkd2L1 N-terminal fragment Met1–Pro45, but not Ile40–Ile97 or C-terminal fragments, abolishes the inhibition of Pkd2L1 channel by overexpressed and oocyte-native RACK1 likely through disrupting the Pkd2L1-RACK1 association. Taken together, our study demonstrated that RACK1 inhibits Pkd2L1 channel function through binding to domain Met1–Pro45 of Pkd2L1. Thus, Pkd2L1 is a novel target channel whose function is regulated by the versatile scaffolding protein RACK1.
Introduction
Pkd2L1 is a homologue of Pkd2 with 54% sequence identity (1). Both proteins are Ca2+-modulated cation channels permeable to Ca2+, Na+, and K+ (2–4). Pkd2 and Pkd2L1 share high similarity in membrane topology to the transient receptor potential (TRP)2 superfamily of cation channels and have been termed TRPP2 and TRPP3, respectively, as members of the TRP polycystin (TRPP) subfamily (5). Mutations in the Pkd1 or Pkd2 gene account for most cases of autosomal dominant polycystic kidney disease with an incidence of 0.1–0.2% worldwide. We previously reported that Pkd2L1 channel is activated by application of extracellular Ca2+ followed by an ensuing inactivation (3) and is inhibited by amiloride analogues and large monovalent cations such as tetrapentylammonium and tetrabutylammonium, which led to an estimation of its channel pore size of ∼7 Å (6, 7).
Pkd2L1 channel activity is modulated by membrane potential, pH, and cell volume (3, 8, 9). We also reported that Pkd2L1 physically interacts with α-actinin, an important component of the actin filament, in brain and other tissues and is functionally stimulated by α-actinin in vitro (10). Although overexpressed Pkd2L1 is targeted to the plasma membrane (PM) of Xenopus oocytes, it mostly localizes in intracellular membranes of mammalian cells when expressed alone (10–12). Pkd2L1 co-localizes with Pkd1 in the centrosome and may function in the cell cycle (13). Interestingly, co-expression of Pkd2L1 with Pkd1 in human embryonic kidney (HEK) 293 cells resulted in Pkd2L1 trafficking to the PM where Pkd2L1 seemed to mediate Ca2+ entry in the presence of a hypo-osmotic extracellular solution (11). Pkd1L3, a homologue of Pkd1, is critical in the PM trafficking of Pkd2L1 because knock-out of Pkd1L3 resulted in internalization of Pkd2L1 in mouse taste bud neurons (12). How Ca2+ triggers the activation remains unknown. So far it is known that EGTA preinjection abolishes this activation (3), indicating that an increase in the intracellular Ca2+ concentration is required for the activation and that deletion of the Pkd2L1 C-terminal domain Thr622–Ser805 does not abolish Ca2+-induced activation (14).
Pkd2L1 localizes to tongue taste receptor cells together with Pkd1L3, which is possibly involved in sour tasting (15–17), and to central canal neurons of the spinal cord, which is possibly involved in proton-dependent regulation of action potentials (15). Mouse Pkd2L1 and Pkd1L3 together mediate pH-dependent cation conductance (16), which was later described as the off-response (i.e. the channel is activated only after the low extracellular pH is removed) in a report proposing that the Pkd2L1-mediated off-response may work together with an on-response mediated by other acid receptors to account for acid sensing at large pH ranges (18). It was shown that Pkd2L1 together with a carbonic anhydrase is involved in sensing gaseous CO2 in the tongue (19). Interestingly, in the tongue of two patients with an acquired sour ageusia (i.e. unresponsive to sour stimuli), the mRNA and protein of Pkd2L1, Pkd1L3, and acid-sensing ion channels were undetectable (20). Indeed, mice with Pkd2L1 knock-out (KO) have reduced responses to sour stimuli (21). However, no anomaly in tasting was observed in Pkd1L3 KO mice (21, 22), and responses in sour taste cells are associated with acid-activated proton, but not Na+, currents (23), which challenges the implication in acid sensing of Pkd1L3 or Pkd2L1, a Na+-permeable channel. Chang et al. (23) also found that these proton currents are not affected by targeted deletion of Pkd1L3, a result that is incompatible with the study by Ishimaru et al. (12) about Pkd1L3-dependent plasma membrane trafficking of Pkd2L1, which would let one argue whether these proton currents are indeed mediated by Pkd2L1. Thus, whether and how Pkd1L3 or Pkd2L1 is involved in acid sensing and whether there is a link between Ca2+- and proton-induced channel activation remain subjects of further studies. Pkd2L1 is also present in ganglion cells and other retina neuron cells (24), several compartments of mouse brain (10), kidney, liver, and heart (14). The physiological role of Pkd2L1 in these tissues and whether it involves Ca2+ and/or acid dependence of the channel remain to be determined.
The receptor for activated C kinase 1 (RACK1) is a guanine nucleotide-binding protein, β2-like 1 (also called GNB2L1) and a conserved scaffolding protein that regulates a range of cell activities, including cell growth, shape, and protein translation (25, 26). RACK1 was originally identified as an anchoring protein for activated protein kinase C and a scaffolding protein that recruits PKCs (and other proteins) into a signaling complex (27, 28). RACK1 is a 36-kDa protein composed of seven repeating units of Trp-Asp 40 (WD40) motifs forming an overall β-propeller structure (27). These WD40 motifs represent structural domains formed by four antiparallel β-strands and are thought to mediate protein-protein interaction. In the last decade, several of its new cellular functions have been discovered (29). In fact, RACK1 acts as a scaffolding/anchoring protein for or regulates the expression/function of a number of binding partners, ranging from signaling proteins such as protein kinase C (27), Src tyrosine kinase (30), protein-tyrosine phosphatase PTPμ (31), cAMP-specific phosphodiesterase PDE4D5 (32), and planar plan polarity protein Vangl2 (33) to receptor/channel proteins such as TRPM6 and TRPC3 (34, 35), integrin β subunit (36), type 1 IFN receptor (37), NMDA receptor (NMDAR) NR2B subunit (38), GABAA receptor β subunits (39), G protein-coupled receptor for thromboxane A2 (40), and inositol 1,4,5-trisphosphate receptors (41). In addition, recent data suggest that the binding of RACK1 plays a role in gene expression, translation, and ribosome assembly and activation (25, 26). RACK1 mediates the regulation of NMDAR by ethanol (42) and together with PKC is an integral part of the circadian feedback loop (43). RACK1 is ubiquitously expressed in the tissues of higher mammals and human, including brain, liver, and spleen, suggesting that it has an important functional role in most if not all cells (29, 38). Therefore, RACK1 is a versatile protein that regulates various biological functions.
In the present study, we first examined the physical interaction of Pkd2L1 with RACK1 using different protein-protein interaction approaches. We then investigated the effect of RACK1 on channel function and expression of Pkd2L1 and the importance of their physical interaction in the regulation of the channel by RACK1.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
We cloned a RACK1 cDNA from a human kidney cDNA library (Clontech) through a standard PCR approach with a pair of specific primers spanning the entire open reading frame: forward, 5′ CTCTAGAGGATCCATGACTGAGCAGATGACCC, and reverse, 5′ CCATATGCTAGCGTGTGCCAATGGTCACC. XbaI, BamHI, and NdeI sites (underlined) were engineered into the primers to facilitate subsequent cloning. The authenticity of all constructs was confirmed by sequencing. Human Pkd2L1 and RACK1 were subcloned into vectors pCDNA3.1 and pEGFPC2, respectively, for mammalian cell expression and vector pTLN2 (44) for Xenopus laevis oocyte expression. His-tagged Pkd2L1 N- and C-terminal fragments tagged with 5′ His were subcloned into vector pCHGF for oocyte expression. The pCHGF vector was generated by deleting the enhanced GFP fragment and part of the CMV promoter in vector pEGFP-C2 using restriction enzymes NheI and SmaI and then ligating in (by the same enzymes) the β-globin 5′ and 3′ untranslated regions generated by PCR from vector pTLN2.
Preparation of mRNAs and Microinjection into Oocytes
Pkd2L1 and RACK1 cDNAs in vector pCHGF were linearized with MluI followed by phenol/chloroform purification and ethanol precipitation. Linearized DNAs were used to in vitro synthesize capped mRNAs using the mMessageMachineTM kit (Ambion, Austin, TX). Stage V-VI oocytes were isolated from X. laevis. Defolliculation of oocytes was performed through incubation in Ca2+-free Barth's solution (14) containing collagenase (2 mg/ml) at room temperature (RT) for 2–2.5 h. Oocytes were then incubated at 18 °C in Barth's solution for at least 3 h before injection of 50 nl of H2O containing variable amounts of mRNAs. An equal volume of H2O was injected into control oocytes. Injected oocytes were incubated at 16–18 °C in Barth's solution supplemented with antibiotics for 2–4 days prior to experiments.
Cell Culture
HEK293, mouse inner medullary collecting duct-3 (IMCD3), and NIH 3T3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells of less than 25 passages were cultured to full confluence before collection.
Yeast Two-hybrid Analysis
Human Pkd2L1 N terminus (Pkd2L1N; amino acids Met1–Ile97) was generated by PCR and subcloned in-frame into the GAL4 DNA binding domain vector pGBKT7 (Clontech). A yeast two-hybrid assay using yeast strain AH109 and Pkd2L1N as bait to screen a human kidney cDNA library constructed in vector pACT2 (Clontech) was performed according to protocols reported previously (45, 46).
GST Pulldown
Precleared mammalian cell lysates containing HA-Pkd2L1N were incubated with 2 μg of purified GST-RACK1 protein in binding buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm CaCl2). The mixture was incubated at RT for 1 h with gentle shaking followed by another hour of incubation after addition of 100 μl of glutathione-agarose beads (Sigma-Aldrich). The beads were then washed several times with 140 mm NaCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, pH 7.5, and the remaining proteins were eluted using 10 mm glutathione, 50 mm Tris, pH 8.0. The protein samples were resolved by SDS-PAGE (10%) and transferred to a nitrocellulose membrane (Bio-Rad). The filters were then blocked with 3% skim milk powder, immunoblotted with HA antibody (Cell Signaling Technology, Danvers, MA), and visualized with enhanced chemiluminescence (Amersham Biosciences).
Blot Overlay Assay
Purified proteins were resolved by SDS-PAGE (10%) and transferred to a nitrocellulose membrane. The membrane was then incubated at 4 °C overnight with lysates of NIH 3T3 cells in blocking buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm CaCl2, 1% BSA), washed three times (10 min each) in blocking buffer containing 0.05% Tween 20, detected with a RACK1 antibody (1:1500; catalog number 610178, BD Biosciences) and a horseradish peroxidase-coupled secondary antibody (1:1500; Chemicon International, Temecula, CA), and visualized with enhanced chemiluminescence (Amersham Biosciences).
Co-immunoprecipitation (co-IP)
Co-IP experiments using Xenopus oocytes co-expressing Pkd2L1 and RACK1 were performed using a modified protocol (47). Briefly, each sample contained 10 healthy oocytes that were injected with Pkd2L1 and/or RACK1 mRNAs and harvested in 500 μl of ice-cold CelLytic M lysis buffer (Sigma-Aldrich) 3 days after injection. Total proteins from the postnuclear supernatant were incubated on ice for 2 h either with anti-Pkd2L1 antibody PR71 or anti-RACK1 antibody (B-3, catalog number sc-17754, Santa Cruz Biotechnology for mammalian cells or BD Biosciences for oocytes) followed by another 2-h incubation with gentle shaking upon addition of 100 μl of protein A (for the anti-Pkd2L1 sample; Sigma-Aldrich) or protein L (for the anti-RACK1 sample; Pierce)-Sepharose. The immunoprecipitates absorbed to protein A/L-Sepharose were resuspended in 40 μl of Laemmli sample buffer, and a 20-μl aliquot of the extract was subjected to SDS-PAGE followed by immunoblotting. Co-IP using native tissues and mammalian cultured cells was conducted as described recently (48).
45Ca Uptake Measurements
45CaCl2 (Amersham Biosciences) at 30 μm was added to uptake solution (100 mm N-methyl-d-glucamine, 2 mm KCl, 1 mm MgCl2, 10 mm Hepes, pH 7.5) plus 1 mm non-radioactive CaCl2. Ten oocytes of each sample were incubated in 0.5 ml of uptake solution for 30 min, and the incubation was terminated by washing oocytes through six wells containing an ice-cold NaCl-containing solution (6). Individual oocytes were then dissolved in 250 μl of 10% SDS and mixed with 2.5 ml of scintillation mixture prior to scintillation counting. Data were compared using the paired or unpaired Student's t test and expressed as means ± S.E. p < 0.05 and p < 0.01 were considered statistically significant and very significant, respectively.
Electrophysiology
Current and voltage signals were measured using the conventional two-microelectrode voltage clamp technique with a commercial amplifier (TEV-200A, Dagan, Minneapolis, MN). Electrodes were fabricated from borosilicate glass (Warner Instruments, Hamden, CT) by a micropipette puller (P-87, Sutter Instruments, Novato, CA) and filled with 3 m KCl with a typical tip resistance of 0.5–3 megaohms. The Digidata 1320A converter and pClamp 8 (Axon Instruments, Union City, CA) were used for data acquisition and analysis, respectively. SigmaPlot 11 or 12 (Jandel Scientific Software, San Rafael, CA) was used for further data analysis, including data plotting, t test, analysis of variance, and regressions. In experiments using a ramp or gap-free protocol (3), current/voltage signals were digitized at 0.2 or 200 ms/sample.
Immunofluorescence
Xenopus oocytes expressing Pkd2L1 alone or Pkd2L1 plus RACK1 were embedded in a cryostat medium and shock frozen, and 10-μm sections were sliced on a cryostat and mounted onto Superfrost Plus microscope slides (Fisher). Sections were dried for 30 min at RT and then fixed in phosphate-buffered saline (PBS) plus 2% paraformaldehyde for 10 min. After rinses in PBS, sections were blocked in 2% normal rabbit serum and 0.1% Triton X-100 in PBS for 5 min and incubated at RT for 1 h with anti-Pkd2L1 rabbit polyclonal antibody PR71. Sections were then rinsed and incubated with a secondary FITC-conjugated anti-rabbit antibody (1:100; Chemicon International) for 1 h. After washing, sections were mounted in Vectashield (Vector Laboratories, Burlington, Ontario, Canada) for fluorescence detection with a Zeiss 510 confocal laser-scanning microscope.
RESULTS
Interaction between Pkd2L1 and RACK1 Revealed by Yeast Two-hybrid Analysis
Our previous study showed that the C-terminal EF-hand motif Glu637–Leu665 acts as a negative regulator of Ca2+-induced Pkd2L1 channel activation and that truncation mutant T622X still exhibits channel function, indicating that the domain C-terminal of Thr622 is not essential for channel function (14). In the present study, we utilized the N terminus (Pkd2L1N; Met1–Ile97) as bait to screen for interacting partner proteins by the yeast two-hybrid approach. For this, Pkd2L1N was fused into vector pGBKT7 as bait to screen a kidney cDNA library in yeast strain AH109. The C terminus of RACK1 (Lys139–Arg317; named RACK1C) was identified from the library as a Pkd2L1N binding partner (Fig. 1A). The physical association between the two protein fragments was confirmed by an individual pair test in yeast. To further delineate the corresponding regions within Pkd2L1N and RACK1 that mediate the association, we constructed a series of truncated cDNA fragments that encode peptides expressed as fusion proteins in yeast strain Y187 using vector pGBKT7. Upon examination for interaction with Pkd2L1N, we found that at least two domains of RACK1, including Lys139–Glu223 and Met217–Arg317, interact with Pkd2L1N (Fig. 1A) and that on the other hand fragment Ala19–Ile97 of Pkd2L1N interacts with RACK1C (Fig. 1B). Furthermore, our yeast two-hybrid assays found that RACK1 also interacts with the Pkd2L1 C terminus (Pkd2L1C; Glu566–Ser805), indicating that more than one domain in both RACK1 and Pkd2L1 are involved in their interaction.
FIGURE 1.

Interaction between Pkd2L1 N terminus and RACK1 identified by yeast two-hybrid assay. The start and end amino acid residue numbers for each domain are indicated. “++”, “+,” and “−” indicate the presence of a strong interaction, positive interaction, and absence of interaction, respectively. A, RACK1 was identified from a human kidney library by yeast two-hybrid screening using the Pkd2L1 N-terminal segment Met1–Ile97 (Pkd2L1N) as bait. RACK1C and several truncated fragments of RACK1 were assessed for their interaction with Pkd2L1N. B, Pkd2L1 N-terminal fragments were examined for interaction with RACK1C. Domain Met1–Pro45 exhibited self-activation (SA) and was not used to assess the interaction with RACK1.
Interaction between Pkd2L1 and RACK1 under in Vivo and in Vitro Conditions
We performed other assays to further document the interaction between Pkd2L1 and RACK1. By GST pulldown, we showed that HA-Pkd2L1N expressed in HEK293 cells is able to interact with GST-RACK1 purified from Escherichia coli (Fig. 2A) with full-length human RACK1 generated by PCR. We also used a blot overlay assay to verify this interaction in vitro. Purified fusion proteins, including GST-Pkd2L1N, GST-Met1–Pro45, and GST-Ala19–Pro45, were resolved using SDS-PAGE and transferred to a nitrocellulose membrane followed by incubation with NIH 3T3 cell lysates. Immunoreactive protein bands from probing with a RACK1 antibody were observed only in the presence of GST-Pkd2L1N, GST-Met1–Pro45, or GST-Ala19–Pro45 but not with GST alone or bovine albumin (Fig. 2B), which is indicative of specific binding between Pkd2L1N and RACK1.
FIGURE 2.
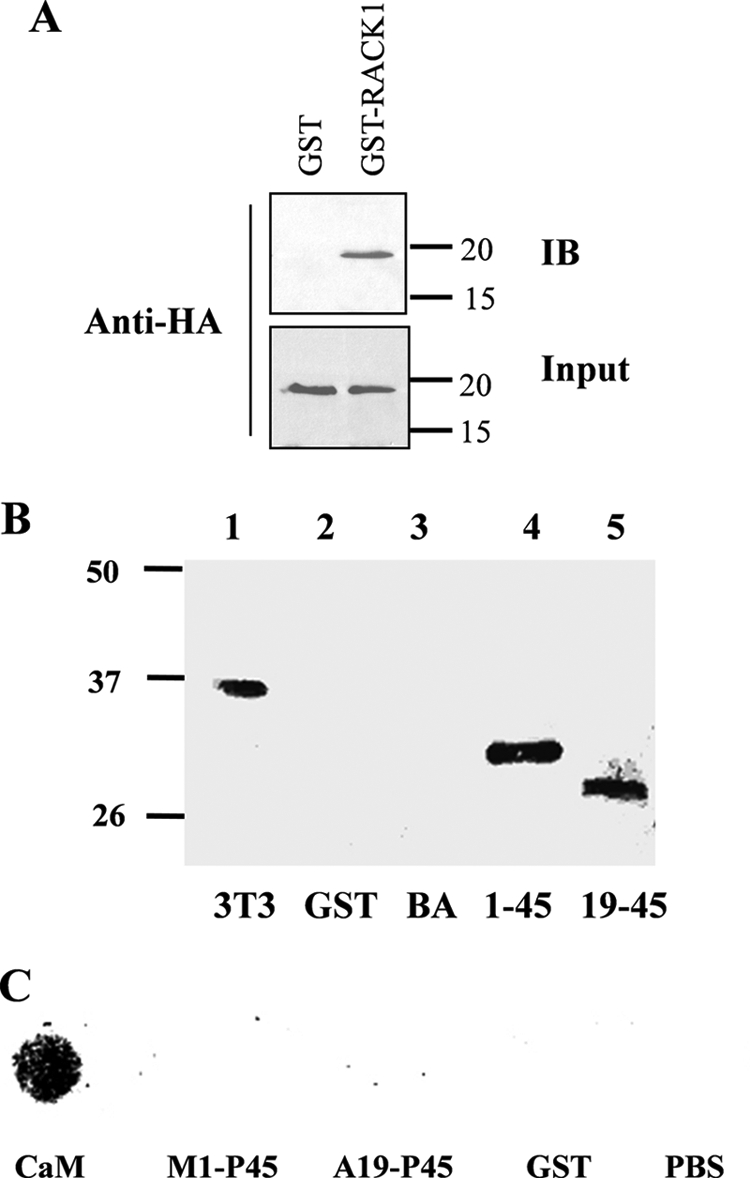
Interaction between Pkd2L1N and RACK1 by GST pulldown and blot overlay. A, GST pulldown. Precleared HEK293 cell lysates containing HA-Pkd2L1N were incubated with purified GST-RACK1 protein (2 μg) in binding buffer. B, blot overlay for interaction between a Pkd2L1 N-terminal fragment and endogenous RACK1. A nitrocellulose membrane containing purified GST-fused Pkd2L1 N-terminal fragments was incubated with total protein lysates of NIH 3T3 cells and immunoblotted (IB) by the anti-RACK1 antibody. NIH 3T3 cell lysates were used as a positive control (lane 1). C, Ca2+ binding of Pkd2L1 N-terminal domains Met1–Pro45 and Ala19–Pro45. GST fusion polypeptides purified from E. coli were spotted on the nitrocellulose membrane strips, overlaid with 45Ca in PBS buffer, and autoradiographed. Calmodulin (CaM) served as a positive control, whereas GST alone and bovine albumin (BA) served as negative controls.
Furthermore, we in vitro transcribed complementary RNAs of Pkd2L1 and RACK1 and injected them into Xenopus oocytes. After 3 days, total protein extracts were used to perform co-IP assays between the two proteins. Precipitation with anti-Pkd2L1 antibody PR71 resulted in the pulldown of RACK1, and reciprocally, anti-RACK1 antibody was also able to co-precipitate Pkd2L1 in oocytes (Fig. 3A), demonstrating that the two proteins are in the same complex in oocytes. We further substantiated the interaction between Pkd2L1 and RACK1 by co-IP using native HEK293 and IMCD3 cells and mouse kidney and heart tissues (Fig. 3B), indicating that the two proteins interact with each other in vivo. It is well known that RACK1 is abundantly expressed in brain and interacts with brain proteins such as NMDA and GABA receptors (38, 39, 49). On the other hand, Pkd2L1 was also recently found in several brain compartments such as hippocampus, thalamus, and spinal cord, although its physiological roles there remain unclear (10, 15). We thus tested their interaction in mouse brain tissues by co-IP. Indeed, we found that Pkd2L1 and RACK1 precipitate with each other and reciprocally (Fig. 3C). Taken together, our data demonstrated the interaction between Pkd2L1 and RACK1 under in vitro and in vivo conditions.
FIGURE 3.

Interaction between Pkd2L1 and RACK1 by co-IP. A, Xenopus oocytes overexpressing Pkd2L1 + RACK1 or RACK1 and control (Ctrl) oocytes were used for reciprocal co-IP using anti-Pkd2L1 (PR71) or anti-RACK1 antibody for IP or immunoblotting (IB) as indicated. B, left panel, cell lysates from native HEK293 cells were precipitated with PR71, non-immune rabbit IgG serum (RIgG; as control), or no antibody (None; as control). Immunoprecipitated proteins were analyzed by WB using the anti-RACK1 antibody (BD Biosciences). Right panel, representative data obtained from similar co-IP assays using native IMCD3 cells and mouse kidney and heart tissues with anti-Pkd2L1 and -RACK1 antibodies for IP and immunoblotting, respectively. C, interaction between Pkd2L1 and RACK1 in mouse brain by reciprocal co-IP. Left panel, cell lysates from mouse brain were precipitated with PR71 or rabbit IgG serum (RIgG). Immunoprecipitated proteins were analyzed by WB using the anti-RACK1 antibody. Right panel, reciprocal co-IP experiments were performed using RACK1 antibody B-3 or non-immune mouse IgG serum (mIgG) for IP and PR71 for immunoblotting.
Inhibition of Pkd2L1 Channel Activity by RACK1
We next tested whether RACK1 has any effect on Pkd2L1 channel function. By co-expressing Pkd2L1 and RACK1 in Xenopus oocytes, we found that overexpression of RACK1 abolishes the Ca2+ transport ability of Pkd2L1 (Fig. 4A). Under voltage clamp conditions, RACK1 abolished both the basal conductance and Ca2+-induced channel activation of Pkd2L1 (Fig. 4, B and C). On average, RACK1 inhibited 65 and 87% of basal and Ca2+-activated currents, respectively (Fig. 4D). Because Xenopus and human RACK1 are 95% identical and 99% similar, we assume that endogenous RACK1, if present, is detectable by our antibody and inhibits part of Pkd2L1 channel activity. Indeed, our Western blotting (WB) assays detected the endogenous RACK1 and showed that the total Pkd2L1 expression is reduced by overexpressed RACK1 (to 68 ± 3%; averaged from five experiments) and increased by endogenous RACK1 knockdown by siRNA (Fig. 5A) with sequences 5′-CAAUCAAACUGUGGAACACTT-3′ and 5′-GUGUUCCACAGUUUGAUUGTT-3′ synthesized by Shanghai GenePharma Co. (Shanghai, China). Consistently, RACK1 knockdown on average increased Ca2+-induced channel activity of Pkd2L1 by 79% (n = 4) (Fig. 5B). We then performed biotinylation assays to examine whether abolished or substantially reduced Pkd2L1 activity by RACK1 could be accounted for by a decrease in the Pkd2L1 PM expression. We found that Pkd2L1 PM expression in the presence of RACK1 overexpression was decreased to 64 ± 7% (averaged from four experiments) (Fig. 5C). Thus, RACK1 reduces similarly the total and PM expression of Pkd2L1 to about two-thirds, indicating that it does not alter the Pkd2L1 trafficking. This reduction in the PM expression does not seem to be sufficient to account for the decrease in the Ca2+-induced current (to 13%), indicating that RACK1 inhibits Pkd2L1 channel function. Immunofluorescence data are also in agreement with the conclusion that RACK1 overexpression and knockdown, respectively, reduce and increase the Pkd2L1 surface membrane expression (Fig. 5D). To provide further documentation, we injected oocytes with decreasing amounts of Pkd2L1 mRNA to identify an amount that generates channel activity comparable with that produced by 50 ng of Pkd2L1 + 25 ng of RACK1. We found that the current obtained from injection of 50 ng of Pkd2L1 + 25 ng of RACK1 is comparable with that from the injection of 6.2 ng of Pkd2L1 alone, but the latter has much less PM expression than the former (Fig. 6). In fact, although the Pkd2L1 PM expression is comparable between injection of 50 ng of Pkd2L1 + 25 ng of RACK1 and that of 25 ng of Pkd2L1 alone, the latter produced far larger channel currents than the former. In this biotinylation assay, we used Na,K-ATPase as a PM marker control and calreticulin and β-actin as non-PM marker controls. These data demonstrated that RACK1 substantially inhibits Pkd2L1 channel function.
FIGURE 4.

Functional modulation of Pkd2L1 channel by RACK1. A, radiolabeled 45Ca uptake in oocytes expressing Pkd2L1 + RACK1 or Pkd2L1 alone and H2O-injected control oocytes. Radiolabeled 45Ca uptake was performed using uptake solution 3 days following RNA injection. Data were averaged from three experiments. The effect of RACK1 was analyzed by unpaired t test. B, representative whole-cell current tracings under voltage clamp (Vm = −50 mV) using the two-microelectrode voltage clamp technique. Different scales of current were used as indicated. Currents were measured using solution “Na” (the standard Na+-containing solution (100 mm NaCl, 2 mm KCl, 1 mm MgCl2, 10 mm Hepes, pH 7.5) or “Na+Ca” (standard Na+-containing solution + 5 mm CaCl2) by the two-microelectrode voltage-clamp technique with a voltage ramp protocol. C, representative current-voltage curves in an oocyte expressing Pkd2L1 alone or Pkd2L1 + RACK1 in the presence of the standard Na+-containing solution with (+Ca) or without (−Ca) 5 mm CaCl2 as indicated. D, averaged currents obtained in oocytes expressing Pkd2L1 + RACK1 (n = 17) or Pkd2L1 alone (n = 17) and H2O-injected control oocytes (n = 6) in the presence of Na+-containing solution with (Na+Ca) or without (Na) addition of 5 mm CaCl2. Inhibition of currents by RACK1 was very significant both with and without Ca2+ with p < 0.001 and p = 0.006 (unpaired t test), respectively. Two-way analysis of variance found that the inhibitory effect of RACK1 is very significant (p < 0.001), that the stimulatory effect of Ca2+ is very significant (p < 0.001), and that the effect of RACK1 is significantly different (p < 0.001) whether Ca2+ is present or not in the solution, consistent with t test analysis. Error bars indicate S.E.
FIGURE 5.

Effects of RACK1 on channel activity and total and PM expression of Pkd2L1 expressed in Xenopus oocytes. A and B, oocytes were injected with Pkd2L1 + RACK1, Pkd2L1 + si-RACK1, Pkd2L1 alone, or water (negative control). A, representative data on expression of Pkd2L1 and RACK1 by WB. B, Ca2+-activated currents were measured at −50 mV. Shown are averaged values from four oocytes per group with p values from a paired t test. Error bars indicate S.E. C, left panel, representative data on the PM expression of Pkd2L1 by biotinylation. Na,K-ATPase (PM marker) and calreticulin and β-actin (non-PM markers) were used as controls of the biotinylation assay. Middle and right panels, flow-through and WB input data, respectively, from the same experiment. D, representative data on subcellular distribution of Pkd2L1 and RACK1 by immunofluorescence.
FIGURE 6.

Inhibition of Pkd2L1 channel function by RACK1. Top, Xenopus oocytes were injected with various amounts of Pkd2L1 mRNA with or without co-injection of RACK1. Shown are averaged Ca2+-activated currents measured at −50 mV from different numbers of oocytes as indicated. p values obtained from an unpaired t test are indicated. Furthermore, linear and Michaelis-Menten regression analyses (current versus mRNA amount) indicate that current very significantly (p < 0.001) increases with the amount of injected mRNA. Bottom, the same batches of oocytes as in the top panel were used for biotinylation assays. Shown are representative data on the PM expression of Pkd2L1 with β-actin as control.
Effects of Mutations in RACK1 and Pkd2L1 on Pkd2L1 Channel Function
Although both fragments Met1–Ser205 and Met217–Arg317 of RACK1 bind Pkd2L1 (Fig. 1A), whether they are sufficient to exhibit inhibition of Pkd2L1 channel function remains unknown. To test this, we co-expressed each of various fragments of RACK1 with Pkd2L1 in oocytes and performed the two-electrode voltage clamp experiments. We found that, similar to the full-length RACK1, fragments Met1–Asp220 and Met1–Ile258 significantly reduce Pkd2L1 channel activity, whereas Met217–Arg317 has no significant effect (Fig. 7A). This indicates that, although Met217–Arg317 binds Pkd2L1N, the binding is not essential for the channel inhibition. We also found that two missense mutants of RACK1, Y228F and Y246F, have inhibitory effects similar to those of WT RACK1, suggesting that the phosphorylation of RACK1 at Tyr228 or Tyr246 by Src tyrosine kinase (50) is not involved in its inhibition of Pkd2L1 channel function. We also conducted Pkd2L1 transport activity by means of 45Ca radiotracer uptake. The results are in agreement with our data obtained from electrophysiology experiments (Fig. 7B).
FIGURE 7.

Effects of mutations in RACK1 and Pkd2L1. A and B, PKd2L1 was expressed in oocytes with or without co-expression of a RACK1 mutant. A, averaged Ca2+-activated currents measured at −50 mV with each group from 5 to 16 different oocytes. ** and *** indicate p < 0.01 and p < 0.001, respectively. B, averaged 45Ca tracer uptake from three independent experiments using uptake solution. Error bars indicate S.E. C, effect of RACK1 on the function of Pkd2L1 mutants. Pkd2L1 missense mutants were expressed in oocytes with or without co-expression of RACK1. Shown are averaged Ca2+-activated currents measured at −50 mV with each group from 7 to 11 different oocytes.
Because our yeast two-hybrid experiments revealed that RACK1 binds fragment Ala19–Pro45 of Pkd2L1 and RACK1 was reported to modulate the function of target channels through regulating their phosphorylation (34, 38), we made point mutations at potential phosphorylation sites within this fragment to determine whether these sites are important for channel function or inhibition by RACK1. The NetPhosK online program predicted that Thr34, Thr39, Ser41, and Ser42 in Pkd2L1N are candidate phosphorylation sites. We thus changed these residues to alanine and found that the resulting mutants still retain channel function comparable with that of WT Pkd2L1 (Fig. 7C). Furthermore, RACK1 overexpression inhibited the channel function of these mutants as well (Fig. 7C). This result suggests that phosphorylation within Ala19–Pro45 is not critical to Pkd2L1 channel function or to its inhibition by RACK1.
Role of Pkd2L1 Fragment Met1–Pro45 in Mediating Pkd2L1 Channel Inhibition by RACK1
To determine whether the physical interaction between Pkd2L1 and RACK1 through the Met1–Pro45 fragment of Pkd2L1 is important for the inhibition of Pkd2L1 channel function by RACK1, we co-expressed Pkd2L1 and RACK1 with His-tagged Met1–Pro45 peptide that should serve as a blocking peptide through competing with Pkd2L1 for binding RACK1. Indeed, expression of His-Met1–Pro45 rescued the channel function of Pkd2L1 otherwise inhibited by RACK1 (Fig. 8A). In contrast, the remaining part of the N terminus (His-Ile40–Ile97) and C-terminal fragments (His-tagged Glu566–Thr622 and Gln617–Ser805) had no rescuing effects (Fig. 8B). By WB, we verified that the functional rescue by Met1–Pro45 is not through decreasing RACK1 overexpression (Fig. 8C). Consistently, we found by co-IP that Met1–Pro45 indeed substantially decreases the Pkd2L1-RACK1 interaction (Fig. 8D). In negative control experiments, we verified that Met1–Pro45 expressed alone has no channel function (data not shown). Taken together, our data support that binding between Pkd2L1 and RACK1 through its N-terminal fragment Met1–Pro45, but not Pkd2L1C or the remaining part of Pkd2L1N, is required for inhibition of Pkd2L1 channel function by RACK1.
FIGURE 8.

Effects of Pkd2L1 N- and C-terminal fragments on inhibition of Pkd2L1 by RACK1 and on Pkd2L1-RACK1 interaction. A, representative recordings of currents measured at −50 mV in oocytes expressing Pkd2L1 and RACK1 with or without co-expression of Met1–Pro45 in the presence of Na+-containing solution ±5 mm Ca2+ as indicated. B, averaged Ca2+-induced currents measured at −50 mV in oocytes expressing Pkd2L1 alone (n = 15), Pkd2L1 + RACK1 (n = 12), or Pkd2L1 + RACK1 + a blocking peptide (Met1–Pro45, n = 14; Ile40–Ile97, n = 11; Glu566–Thr622, n = 9; and Gln617–Ser805, n = 6). p values obtained from comparison with Pkd2L1 alone (by unpaired t test) are indicated. C, expression of RACK1 and Pkd2L1 with or without co-expression of Met1–Pro45 in oocytes assessed by WB. D, the Pkd2L1-RACK1 interaction assessed by co-IP with or without (control (Ctrl)) co-expression of Met1–Pro45. Pkd2L1, GFP-RACK1, and/or His-Met1–Pro45 were expressed in oocytes. Shown are representative data in which GFP antibody was used for IP and Pkd2L1 and GFP antibodies were used for immunoblotting (IB) as indicated. E, effect of overexpression of Met1–Pro45 on Pkd2L1 activity. Shown are averaged Ca2+-induced currents from oocytes expressing Pkd2L1 (n = 24) or Pkd2L1 plus Met1–Pro45 (n = 20, p = 0.005; **, unpaired t test). F, representative data of Pkd2L1 expression by WB in individual oocytes expressing either Pkd2L1 (lanes 1–6) or Pkd2L1 plus Met1–Pro45 (lanes 7–10). These oocytes are part of the oocytes used in generating currents in E. Error bars indicate S.E.
Because oocytes possess significant endogenous RACK1 levels, we reasoned that Met1–Pro45 should block the inhibition of Pkd2L1 channel function by endogenous RACK1. Indeed, we found that overexpression of blocking peptide Met1–Pro45 roughly doubles the Ca2+-induced current (at −50 mV) from an average of 2.5 ± 0.3 μA (n = 24) in oocytes expressing Pkd2L1 to 4.8 ± 0.6 μA (n = 20, p = 0.005) in those expressing Pkd2L1 + Met1–Pro45. Furthermore, in the presence of Met1–Pro45, the Pkd2L1 expression assessed by WB was slightly decreased (Fig. 8F) possibly due to limited expression capability of oocytes. These data indicate that Met1–Pro45 blocks the inhibition of Pkd2L1 by endogenous RACK1 in agreement with the data obtained from using overexpressed RACK1 (Fig. 8, A and B).
DISCUSSION
The mechanism of Ca2+- or acid-induced Pkd2L1 channel activation has remained obscure. The C terminus of Pkd2L1 contains a number of motifs, including a putative Ca2+-binding EF-hand and a coiled coil domain, but when these motifs were removed as in the truncation mutant T622X or when the EF-hand was deleted, Ca2+-induced activation was still present and even increased (14). In an effort to examine the role of the Pkd2L1 N terminus in channel activation, we found in the present study that Pkd2L1N associates with RACK1 and that RACK1 inhibits Pkd2L1 channel function through binding to Pkd2L1N. RACK1 was initially identified as a receptor for activated PKC, but recent studies showed that it is also involved in many other cellular functions. For example, RACK1 acts as a novel inhibitory scaffolding protein in complex with tyrosine kinase Fyn and its substrate, the NR2B subunit of NMDAR, and this association prevents the phosphorylation of NR2B by Fyn and inhibits NMDAR channel activity (38, 42). The presence of RACK1 in CA1 hippocampal slices resulted in a 50% decrease in NMDAR-mediated current (38).
Because RACK1 reduced both the total and PM expression of Pkd2L1, it remained to be determined whether RACK1-inhibited whole-cell Pkd2L1 channel activity is solely due to the reduced PM level of Pkd2L1 or also to the inhibited activity of individual channel molecules. First, although co-expression of RACK1 reduced the Pkd2L1 PM level by 36%, it inhibited 87% of the Ca2+-activated whole-cell current, indicating that RACK1 represses the activity of each individual Pkd2L1 channel. Then we found that although the Pkd2L1 surface membrane expression from injection of 50 ng of Pkd2L1 and 25 ng of RACK1 mRNAs, is comparable with that from injection of 25 ng of Pkd2L1 mRNA alone, the whole-cell current associated with injection of 50 ng of Pkd2L1 + 25 ng of RACK1 is only comparable with that associated with injection of 6.2 ng of Pkd2L1 alone (Fig. 6). Thus, these data together showed that RACK1 substantially inhibits Pkd2L1 channel function in addition to reducing its total and surface membrane expressions.
We found that two RACK1 fragments bind Pkd2L1 and that RACK1 binds both the N and C termini of Pkd2L1. It was thus unclear whether all physical bindings are important for inhibition of Pkd2L1 function by RACK1. Although we could utilize truncation/deletion mutations in Pkd2L1 and/or RACK1 that disrupt the Pkd2L1-RACk1 interaction to examine whether a mutant RACK1 still inhibits WT channel function or whether WT RACK1 inhibits the function of a mutant Pkd2L1, the information we obtain would not be sufficient to make a conclusion because the normal function of RACK1 or Pkd2L1 may readily be affected by these mutations. In view of this, we utilized co-expression of Pkd2L1 N- and C-terminal fragments as potential blocking peptides to break down the physical association between WT Pkd2L1 and RACK1. Our data demonstrated that fragment Met1–Pro45 is able to substantially reduce the Pkd2L1-RACK1 binding and abolish inhibition by RACK1, indicating that their binding through domain Met1–Pro45 is strongest and critical for RACK1 to exert functional inhibition. In contrast, although Pkd2L1C also binds RACK1, overexpression of Pkd2L1C was unable to significantly affect the Pkd2L1-RACK1 binding, suggesting that their binding through Pkd2L1C is not as strong as through Met1–Pro45 and is not important for functional regulation. It is interesting to note that similarly, although both Met1–Asp220 and Met217–Arg317 of RACK1 associate with Pkd2L1, only Met1–Asp220 inhibited Pkd2L1 channel activity, indicating that physical interaction of Met217–Arg317 with Pkd2L1 is not essential for the channel inhibition.
How the binding of RACK to domain Met1–Pro45 of Pkd2L1 inhibits the channel function remains unclear. Because RACK1 was known as a chaperone of protein kinases to promote the phosphorylation of target proteins, it is possible that binding of RACK1 to domain Met1–Pro45 prevents phosphorylation of a residue within the domain. Therefore, we mutated candidate phosphorylation residues to alanine: T34A, T39A, S41A, and S42A. However, all these mutants still possessed channel function comparable with that of WT channel and were inhibitable by RACK1, indicating that these potential phosphorylation sites are not important for channel function or binding with RACK1. On the other hand, if Ca2+ could bind to Met1–Pro45 of Pkd2L1 then RACK1 may prevent the Ca2+ binding, thereby inhibiting channel activation. However, our dot blot calcium overlay assays for testing whether Met1–Pro45 can directly bind Ca2+ ions using GST-fused peptides Met1–Pro45 and Ala19–Pro45 purified from E. coli revealed that GST-tagged Met1–Pro45 and Ala19–Pro45 cannot bind Ca2+ (Fig. 2C). Thus, it is possible that Ca2+-induced channel activation is mediated by an unknown protein that binds Met1–Pro45 and that RACK1 competitively disrupts their interaction, thereby inhibiting Pkd2L1 channel function. Further studies to identify and characterize such an unknown protein would shed light on the mechanism underlying Ca2+-induced Pkd2L1 activation.
Acknowledgment
We thank Yi Shi for assistance in data analysis.
This work was supported by the Canadian Institutes of Health Research, the Alberta Innovates Health Solutions, and the Kidney Foundation of Canada (to X. -Z. C.).
- TRP
- transient receptor potential
- TRPP
- TRP polycystin
- PM
- plasma membrane
- RACK1
- receptor for activated C kinase 1
- NMDAR
- NMDA receptor
- IP
- immunoprecipitation
- WB
- Western blotting.
REFERENCES
- 1. Nomura H., Turco A. E., Pei Y., Kalaydjieva L., Schiavello T., Weremowicz S., Ji W., Morton C. C., Meisler M., Reeders S. T., Zhou J. (1998) Identification of PKDL, a novel polycystic kidney disease 2-like gene whose murine homologue is deleted in mice with kidney and retinal defects. J. Biol. Chem. 273, 25967–25973 [DOI] [PubMed] [Google Scholar]
- 2. Koulen P., Cai Y., Geng L., Maeda Y., Nishimura S., Witzgall R., Ehrlich B. E., Somlo S. (2002) Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 4, 191–197 [DOI] [PubMed] [Google Scholar]
- 3. Chen X. Z., Vassilev P. M., Basora N., Peng J. B., Nomura H., Segal Y., Brown E. M., Reeders S. T., Hediger M. A., Zhou J. (1999) Polycystin-L is a calcium-regulated cation channel permeable to calcium ions. Nature 401, 383–386 [DOI] [PubMed] [Google Scholar]
- 4. Vassilev P. M., Guo L., Chen X. Z., Segal Y., Peng J. B., Basora N., Babakhanlou H., Cruger G., Kanazirska M., Ye C. p., Brown E. M., Hediger M. A., Zhou J. (2001) Polycystin-2 is a novel cation channel implicated in defective intracellular Ca2+ homeostasis in polycystic kidney disease. Biochem. Biophys. Res. Commun. 282, 341–350 [DOI] [PubMed] [Google Scholar]
- 5. Montell C. (2005) The TRP superfamily of cation channels. Sci. STKE 2005, re3. [DOI] [PubMed] [Google Scholar]
- 6. Dai X. Q., Ramji A., Liu Y., Li Q., Karpinski E., Chen X. Z. (2007) Inhibition of TRPP3 channel by amiloride and analogs. Mol. Pharmacol. 72, 1576–1585 [DOI] [PubMed] [Google Scholar]
- 7. Dai X. Q., Karpinski E., Chen X. Z. (2006) Permeation and inhibition of polycystin-L channel by monovalent organic cations. Biochim. Biophys. Acta 1758, 197–205 [DOI] [PubMed] [Google Scholar]
- 8. Liu Y., Li Q., Tan M., Zhang Y. Y., Karpinski E., Zhou J., Chen X. Z. (2002) Modulation of the human polycystin-L channel by voltage and divalent cations. FEBS Lett. 525, 71–76 [DOI] [PubMed] [Google Scholar]
- 9. Shimizu T., Janssens A., Voets T., Nilius B. (2009) Regulation of the murine TRPP3 channel by voltage, pH, and changes in cell volume. Pflugers Arch. 457, 795–807 [DOI] [PubMed] [Google Scholar]
- 10. Li Q., Dai X. Q., Shen P. Y., Wu Y., Long W., Chen C. X., Hussain Z., Wang S., Chen X. Z. (2007) Direct binding of α-actinin enhances TRPP3 channel activity. J. Neurochem. 103, 2391–2400 [DOI] [PubMed] [Google Scholar]
- 11. Murakami M., Ohba T., Xu F., Shida S., Satoh E., Ono K., Miyoshi I., Watanabe H., Ito H., Iijima T. (2005) Genomic organization and functional analysis of murine PKD2L1. J. Biol. Chem. 280, 5626–5635 [DOI] [PubMed] [Google Scholar]
- 12. Ishimaru Y., Katano Y., Yamamoto K., Akiba M., Misaka T., Roberts R. W., Asakura T., Matsunami H., Abe K. (2010) Interaction between PKD1L3 and PKD2L1 through their transmembrane domains is required for localization of PKD2L1 at taste pores in taste cells of circumvallate and foliate papillae. FASEB J. 24, 4058–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bui-Xuan E. F., Li Q., Chen X. Z., Boucher C. A., Sandford R., Zhou J., Basora N. (2006) More than colocalizing with polycystin-1, polycystin-L is in the centrosome. Am. J. Physiol. Renal Physiol. 291, F395–F406 [DOI] [PubMed] [Google Scholar]
- 14. Li Q., Liu Y., Zhao W., Chen X. Z. (2002) The calcium-binding EF-hand in polycystin-L is not a domain for channel activation and ensuing inactivation. FEBS Lett. 516, 270–278 [DOI] [PubMed] [Google Scholar]
- 15. Huang A. L., Chen X., Hoon M. A., Chandrashekar J., Guo W., Tränkner D., Ryba N. J., Zuker C. S. (2006) The cells and logic for mammalian sour taste detection. Nature 442, 934–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ishimaru Y., Inada H., Kubota M., Zhuang H., Tominaga M., Matsunami H. (2006) Transient receptor potential family members PKD1L3 and PKD2L1 form a candidate sour taste receptor. Proc. Natl. Acad. Sci. U.S.A. 103, 12569–12574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. LopezJimenez N. D., Cavenagh M. M., Sainz E., Cruz-Ithier M. A., Battey J. F., Sullivan S. L. (2006) Two members of the TRPP family of ion channels, Pkd1l3 and Pkd2l1, are co-expressed in a subset of taste receptor cells. J. Neurochem. 98, 68–77 [DOI] [PubMed] [Google Scholar]
- 18. Inada H., Kawabata F., Ishimaru Y., Fushiki T., Matsunami H., Tominaga M. (2008) Off-response property of an acid-activated cation channel complex PKD1L3-PKD2L1. EMBO Rep. 9, 690–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chandrashekar J., Yarmolinsky D., von Buchholtz L., Oka Y., Sly W., Ryba N. J., Zuker C. S. (2009) The taste of carbonation. Science 326, 443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huque T., Cowart B. J., Dankulich-Nagrudny L., Pribitkin E. A., Bayley D. L., Spielman A. I., Feldman R. S., Mackler S. A., Brand J. G. (2009) Sour ageusia in two individuals implicates ion channels of the ASIC and PKD families in human sour taste perception at the anterior tongue. PLoS One 4, e7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horio N., Yoshida R., Yasumatsu K., Yanagawa Y., Ishimaru Y., Matsunami H., Ninomiya Y. (2011) Sour taste responses in mice lacking PKD channels. PLoS One 6, e20007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nelson T. M., Lopezjimenez N. D., Tessarollo L., Inoue M., Bachmanov A. A., Sullivan S. L. (2010) Taste function in mice with a targeted mutation of the pkd1l3 gene. Chem. Senses 35, 565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang R. B., Waters H., Liman E. R. (2010) A proton current drives action potentials in genetically identified sour taste cells. Proc. Natl. Acad. Sci. U.S.A. 107, 22320–22325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Basora N., Nomura H., Berger U. V., Stayner C., Guo L., Shen X., Zhou J. (2002) Tissue and cellular localization of a novel polycystic kidney disease-like gene product, polycystin-L. J. Am. Soc. Nephrol. 13, 293–301 [DOI] [PubMed] [Google Scholar]
- 25. Ceci M., Gaviraghi C., Gorrini C., Sala L. A., Offenhäuser N., Marchisio P. C., Biffo S. (2003) Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature 426, 579–584 [DOI] [PubMed] [Google Scholar]
- 26. Nilsson J., Sengupta J., Frank J., Nissen P. (2004) Regulation of eukaryotic translation by the RACK1 protein: a platform for signalling molecules on the ribosome. EMBO Rep. 5, 1137–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ron D., Chen C. H., Caldwell J., Jamieson L., Orr E., Mochly-Rosen D. (1994) Cloning of an intracellular receptor for protein kinase C: a homolog of the β subunit of G proteins. Proc. Natl. Acad. Sci. U.S.A. 91, 839–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schechtman D., Mochly-Rosen D. (2001) Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene 20, 6339–6347 [DOI] [PubMed] [Google Scholar]
- 29. McCahill A., Warwicker J., Bolger G. B., Houslay M. D., Yarwood S. J. (2002) The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol. Pharmacol. 62, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 30. Chang B. Y., Conroy K. B., Machleder E. M., Cartwright C. A. (1998) RACK1, a receptor for activated C kinase and a homolog of the β subunit of G proteins, inhibits activity of Src tyrosine kinases and growth of NIH 3T3 cells. Mol. Cell. Biol. 18, 3245–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mourton T., Hellberg C. B., Burden-Gulley S. M., Hinman J., Rhee A., Brady-Kalnay S. M. (2001) The PTPμ protein-tyrosine phosphatase binds and recruits the scaffolding protein RACK1 to cell-cell contacts. J. Biol. Chem. 276, 14896–14901 [DOI] [PubMed] [Google Scholar]
- 32. Yarwood S. J., Steele M. R., Scotland G., Houslay M. D., Bolger G. B. (1999) The RACK1 signaling scaffold protein selectively interacts with the cAMP-specific phosphodiesterase PDE4D5 isoform. J. Biol. Chem. 274, 14909–14917 [DOI] [PubMed] [Google Scholar]
- 33. Li S., Esterberg R., Lachance V., Ren D., Radde-Gallwitz K., Chi F., Parent J. L., Fritz A., Chen P. (2011) Rack1 is required for Vangl2 membrane localization and planar cell polarity signaling while attenuating canonical Wnt activity. Proc. Natl. Acad. Sci. U.S.A. 108, 2264–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cao G., Hoenderop J. G., Bindels R. J. (2008) Insight into the molecular regulation of the epithelial magnesium channel TRPM6. Curr. Opin. Nephrol. Hypertens. 17, 373–378 [DOI] [PubMed] [Google Scholar]
- 35. Bandyopadhyay B. C., Ong H. L., Lockwich T. P., Liu X., Paria B. C., Singh B. B., Ambudkar I. S. (2008) TRPC3 controls agonist-stimulated intracellular Ca2+ release by mediating the interaction between inositol 1,4,5-trisphosphate receptor and RACK1. J. Biol. Chem. 283, 32821–32830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liliental J., Chang D. D. (1998) Rack1, a receptor for activated protein kinase C, interacts with integrin β subunit. J. Biol. Chem. 273, 2379–2383 [DOI] [PubMed] [Google Scholar]
- 37. Usacheva A., Smith R., Minshall R., Baida G., Seng S., Croze E., Colamonici O. (2001) The WD motif-containing protein receptor for activated protein kinase C (RACK1) is required for recruitment and activation of signal transducer and activator of transcription 1 through the type I interferon receptor. J. Biol. Chem. 276, 22948–22953 [DOI] [PubMed] [Google Scholar]
- 38. Yaka R., Thornton C., Vagts A. J., Phamluong K., Bonci A., Ron D. (2002) NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc. Natl. Acad. Sci. U.S.A. 99, 5710–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brandon N. J., Jovanovic J. N., Smart T. G., Moss S. J. (2002) Receptor for activated C kinase-1 facilitates protein kinase C-dependent phosphorylation and functional modulation of GABAA receptors with the activation of G-protein-coupled receptors. J. Neurosci. 22, 6353–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parent A., Laroche G., Hamelin E., Parent J. L. (2008) RACK1 regulates the cell surface expression of the G protein-coupled receptor for thromboxane A2. Traffic 9, 394–407 [DOI] [PubMed] [Google Scholar]
- 41. Patterson R. L., van Rossum D. B., Barrow R. K., Snyder S. H. (2004) RACK1 binds to inositol 1,4,5-trisphosphate receptors and mediates Ca2+ release. Proc. Natl. Acad. Sci. U.S.A. 101, 2328–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ron D. (2004) Signaling cascades regulating NMDA receptor sensitivity to ethanol. Neuroscientist 10, 325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robles M. S., Boyault C., Knutti D., Padmanabhan K., Weitz C. J. (2010) Identification of RACK1 and protein kinase Cα as integral components of the mammalian circadian clock. Science 327, 463–466 [DOI] [PubMed] [Google Scholar]
- 44. Lorenz C., Pusch M., Jentsch T. J. (1996) Heteromultimeric CLC chloride channels with novel properties. Proc. Natl. Acad. Sci. U.S.A. 93, 13362–13366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Q., Dai Y., Guo L., Liu Y., Hao C., Wu G., Basora N., Michalak M., Chen X. Z. (2003) Polycystin-2 associates with tropomyosin-1, an actin microfilament component. J. Mol. Biol. 325, 949–962 [DOI] [PubMed] [Google Scholar]
- 46. Li Q., Shen P. Y., Wu G., Chen X. Z. (2003) Polycystin-2 interacts with troponin I, an angiogenesis inhibitor. Biochemistry 42, 450–457 [DOI] [PubMed] [Google Scholar]
- 47. Li Q., Liu Y., Shen P. Y., Dai X. Q., Wang S., Smillie L. B., Sandford R., Chen X. Z. (2003) Troponin I binds polycystin-L and inhibits its calcium-induced channel activation. Biochemistry 42, 7618–7625 [DOI] [PubMed] [Google Scholar]
- 48. Li Q., Montalbetti N., Shen P. Y., Dai X. Q., Cheeseman C. I., Karpinski E., Wu G., Cantiello H. F., Chen X. Z. (2005) α-Actinin associates with polycystin-2 and regulates its channel activity. Hum. Mol. Genet. 14, 1587–1603 [DOI] [PubMed] [Google Scholar]
- 49. Sklan E. H., Podoly E., Soreq H. (2006) RACK1 has the nerve to act: structure meets function in the nervous system. Prog. Neurobiol. 78, 117–134 [DOI] [PubMed] [Google Scholar]
- 50. Chang B. Y., Harte R. A., Cartwright C. A. (2002) RACK1: a novel substrate for the Src protein-tyrosine kinase. Oncogene 21, 7619–7629 [DOI] [PubMed] [Google Scholar]