

Abstract
Background
Chronic β-adrenoceptor antagonist (β-blocker) treatment in patients is associated with a potentially anti-arrhythmic prolongation of the atrial action potential duration (APD), which may involve remodelling of repolarising K+ currents.
Objective
To investigate the effects of chronic β-blockade on transient outward, sustained and inward rectifier K+ currents (ITO, IKSUS and IK1) in human atrial myocytes and on the expression of underlying ion channel subunits.
Methods
Ion currents were recorded from human right atrial isolated myocytes using the whole-cell-patch clamp technique. Tissue mRNA and protein levels were measured using real time RT-PCR and western blotting.
Results
Chronic β-blockade was associated with a 41% reduction in ITO density: 9.3±0.8 (30 myocytes, 15 patients) vs 15.7±1.1 pA/pF (32, 14), p<0.05; without affecting its voltage-, time- or rate-dependence. IK1 was reduced by 34% at −120 mV (p<0.05). Neither IKSUS, nor its increase by acute β-stimulation with isoprenaline, were affected by chronic β-blockade. Mathematical modelling suggested that the combination of ITO- and IK1-decrease could result in a 28% increase in APD90. Chronic β-blockade did not alter mRNA or protein expression of the ITO pore-forming subunit, Kv4.3, or mRNA expression of the accessory subunits KChIP2, KChAP, Kvβ1, Kvβ2 or frequenin. There was no reduction in mRNA expression of Kir2.1 or TWIK to account for the reduction in IK1.
Conclusion
A reduction in atrial ITO and IK1 associated with chronic β-blocker treatment in patients may contribute to the associated action potential prolongation, and this cannot be explained by a reduction in expression of associated ion channel subunits.
Keywords: Human, Atrial myocyte, Ion current, Ion channel, β-adrenoceptor, Action potential
Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia, and is associated with significant morbidity and mortality [3]. Beta-adrenoceptor antagonists (β-blockers) are used in the treatment of AF to control the ventricular rate, and they also exert atrial anti-arrhythmic effects [22]. The mechanisms underlying these atrial anti-arrhythmic effects are multifactorial [32, 36], but there is evidence to suggest that long-term treatment with β-blockers can alter atrial cellular electrophysiology by prolonging the action potential duration (APD) and effective refractory period (ERP) [29, 35] and, therefore, potentially inhibiting intra-atrial reentry.
This “pharmacological remodelling” [35] of atrial action potentials in patients treated chronically with β-blockers was initially demonstrated by our group to be associated with a reduction in the density of the atrial transient outward K+ current, ITO. However, the electrophysiological and molecular mechanisms of this ion current change are unknown. Thus, the effects of chronic β-blockade on characteristics of human atrial ITO other than its amplitude, such as voltage-dependency and kinetics, have not yet been investigated. Furthermore, the effects of chronic β-blockade on the expression of ion channel proteins which carry and modulate this current; proteins known to be associated with electrophysiological remodelling in AF and other cardiac pathologies [24], also have not yet been studied.
Preliminary data from our laboratory have suggested that chronic β-blockade may also reduce the human atrial inward rectifier K+ current, IK1, since the atrial cellular input resistance was reduced in patients taking β-blockers [35], although this current was not measured directly. Since there is increasing evidence to suggest that an increase in IKI may be a crucial determinant of the pro-arrhythmic atrial APD-shortening seen in AF-remodelling [28], it is important to establish the effects of chronic β-blockade on IK1 and on the expression of ion channel proteins associated with this current.
The aims of this study, therefore, were to investigate the effects of chronic β-blockade in patients on the time- and voltage-dependent characteristics of the main repolarising K+ currents in human atrium and on the expression of their underlying ion channel subunits, and also to investigate the likely effects of these ion current changes on the atrial action potential.
Methods
Human atrial myocyte isolation
Right atrial appendage tissues were obtained from adult patients undergoing cardiac surgery, who were in sinus rhythm with no history of AF, and either treated with a β-blocker for a minimum of 4 weeks prior to surgery, or not treated with a β-blocker. All procedures were approved by the institutional ethics committee (99MC002) and informed consent was obtained from all patients. The investigation conforms with the principles outlined in the Declaration of Helsinki. Atrial myocytes were isolated by enzymatic dissociation and mechanical disaggregation, as described in detail previously [33]. Briefly, tissue chunks were shaken for 45 min in a low [Ca2+] (50 μM) solution containing protease (4 U/ml Type XXIV, Sigma). Protease was then replaced by collagenase (400 U/ml Type 1, Worthington), which was exchanged 3 times, at 15 min intervals. At each exchange, cells were separated by filtration through nylon gauze and centrifugation at 40 g for 2 min, and washed of residual enzyme in a high [K+], low [Ca2+] solution. Cells were stored in a 0.2 mM Ca2+-containing physiological salt solution for up to 8 hr before electrical recording.
Electrical recording
Ion currents were recorded from human atrial myocytes using the whole-cell-patch clamp technique with an Axopatch-1D amplifier (Axon Instruments) in voltage-clamp mode in conjunction with WinWCP software (J Dempster, Strathclyde University). Myocytes were superfused at 37 °C with a solution containing (mM): NaCl (130), KCl (4.0), MgCl2 (1), CaCl2 (2), CdCl2 (0.2), glucose (10) and HEPES (10); pH 7.35. The pipette solution contained (mM): K-aspartate (110.0), KCl (20.0), MgCl2 (1.0), EGTA (0.15), Na2ATP (4.0), Na2GTP (0.4) and HEPES (5.0); pH 7.25. A liquid junction potential of −7 mV was compensated for prior to recordings, as were capacitative transients and series resistances (64-72% compensation). Voltage-dependent activation of ITO was measured by stimulating cells at 0.3 Hz from a holding potential of −50 mV, with voltage pulses of 100 ms duration increasing in 10 mV steps from −40 to +60 mV. ITO amplitude was calculated as the peak outward current minus end-pulse current. IKSUS, considered to reflect mainly the ultra-rapid delayed rectifier K+ current, IKur [25], was measured as the end-pulse current. Voltage-dependent inactivation of ITO was measured using a 1 s pre-pulse which was increased from −90 mV to +60 mV in 10 mV steps, followed by a second, 100 ms pulse, to +60 mV. ITO reactivation was measured using a standard S1-S2 pulse protocol with an initial inter-pulse interval of 500 ms, which was reduced in 50 ms intervals to 150 ms, and then by 10 ms intervals to 10 ms. ITO rate-dependence was measured using 6 trains of 8 pulses at 75-500 beats/minute. Barium (0.5 mM)-sensitive IK1 voltage-dependent activation was determined using 500 ms voltage pulses increasing from −120 mV to +60 mV in 10 mV steps. In a subset of cells, the effect of acute β-adrenoceptor stimulation with isoprenaline (ISO) on IK1 (in the absence of Ba2+), ITO and/or IKSUS, was assessed. ISO was used at 1 μM; close to Emax concentration for human atrial IKur [20].
Mathematical modelling
To investigate the potential contribution of changes in ion currents to the action potential (AP) configuration, such changes were simulated using a well established mathematical model of the human atrial AP; described in Courtemanche et al [6] and implemented using the CESE Pro 1.4.8 modelling software (Simulogic Inc., Halifax, Canada). Since the typical AP configuration recorded in human atrial cells from our laboratory [35] is Type 3 (or “no dome” [8]), and the Courtemanche model typically produces Type 1 APs (or “spike and dome”) [8], we modified the model to produce “no dome” APs that mimicked those recorded in our laboratory, as follows. The L-type Ca2+ current (ICaL) and IKSUS are the largest two currents that contribute to the plateau (“dome”) phase of the action potential in the Courtemanche model: see Figure 2 of [32] and Figure 14 of [6]. The peak conductance of ICaL in the Courtemanche model (0.124 nS/pF) is larger than that typically recorded in our laboratory (0.099 nS/pF; [35]), and the peak conductance of IKur in the Courtemanche model (0.054 nS/pF) is smaller than that recorded in cells in the present study (0.075 nS/pF). Therefore, ICaL and IKur conductances were decreased and increased, respectively, in steps of 10%, until the resulting action potential closely resembled that from a representative cell obtained from a patient not treated with a β-blocker: Figure 1A of [35]. This was achieved with a 50% scaling of ICaL and 150% IKur, i.e., peak conductances of 0.062 nS/pF and 0.081 nS/pF, respectively.
Quantitative real-time PCR
Human atrial tissue samples were stored at −20°C in RNAlater solution (Quiagen), before cryosectioning at 20 μm. Total RNA was extracted using RNeasy Mini Kit (Quiagen) and reverse transcribed with Superscript III reverse transcriptase (Invitrogen), using random hexamer priming and a PCR thermo-cycler (Thermohybaid PCR Express). The relative abundance of cDNA was determined using an ABI 7900HT Fast Real-Time PCR system (Applied Biosystems), Power SYBR Green PCR MasterMix (Applied Biosystems) and primer assays (10× QuantiTect, Quiagen or Applied Biosystems) (Table 1). Samples were tested in triplicate for each primer with a 40-cycle PCR reaction followed by melting curves. Negative controls consisted of molecular grade water. Mean threshold cycles (Ct) values were determined and ΔCt derived relative to one tissue sample used as an internal calibrator for each primer reaction. cDNA abundance for each tissue sample (a) was expressed relative to mean abundance of 28S and GAPDH (b) for that sample, using the equation: relative abundance=EaΔCta/EbΔCtb. E=efficiency of the PCR reaction and was determined from √Fn/F-2n, where F=fluorescence at the end of each elongation step and n=cycle number.
Table 1.
Primer details for real time RT-PCR
|
Target transcript |
Primer Assay Number |
|---|---|
| hGAPDH | QT01192646 |
| hKChIP2 | Hs00752497s1 |
| hKChAP | QT00053158 |
| hKvβ1 | QT00031136 |
| hKvβ2 | QT00051961 |
| hFrequenin | QT00038598 |
|
Target transcript |
Accession number |
Primer Sequence 5′-3′ |
Fragment length (bp) |
|---|---|---|---|
| 28S | AF460236 | GTTGTTGCCATGGTAAT CCTGCTCAGTACGTCTG ACTTAGAGGCGTTCAGT CATAATCCC |
133 |
| Kv4.3 | NM004980 | TGGCCTTCTACGGCATCC TGCTCGGCGTTCTCCCTCT |
84 |
Western blotting
Human atrial tissue samples were homogenised for 90 s with an Ultraturrax T8 homogeniser (VWR) in 1 ml of homogenisation buffer containing: 15 μl protease inhibitor (Sigma P8340), NaCl 150 mM, KCl 5.4 mM, MgCl2 1.2 mM, NaHEPES 5 mM, Glucose 10 mM, CaCl2 1.0 mM; pH 7.4, and stored at −80°C. Kv4.3 and GAPDH protein levels were measured using sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), immunoblotting and optical densitometry. Tissue samples were blinded with respect to β-blocker status and randomised so that 2 samples from β-blocked and 2 from non-β-blocked patients were loaded onto each gel, at concentrations which we first established to result in a linear relationship with optical density (total protein load of 5-10 μg per lane). Electrophoresis and immunoblotting were performed using the NuPAGE system (Invitrogen) with 4-12% gradient Bis-Tris NuPAGE gels, and molecular weight marker Magic Mark Western Standard. Protein immunodetection was performed using the Invitrogen Western Breeze kit unless otherwise stated, and analysed using Quantity-One software (BIORAD). Blots were incubated overnight with monoclonal anti-Kv4.3 (Neuromab) at 1:133 or anti-GAPDH (mAbcam 9484) at 1:20000. Western blotting controls were performed using rabbit and rat brain or lung tissues. All procedures conformed with the Institutional Guidelines for the Care and Use of Animals, and with the Guidance on the Operation of the Animals (Scientific Procedures) Act 1986. A New Zealand White rabbit was killed with an intravenous injection of 100 mg/kg pentobarbital sodium (UK Project Licence No. PPL60/3538), and a Wistar rat was killed by cervical dislocation in accordance with Schedule 1. Tissue homogenates were used to compare bands generated by the monoclonal primary antibody (Neuromab anti-Kv4.3) plus secondary antibodies with secondary antibody alone, and also to compare a polyclonal anti-Kv4.3 (Alomone) at 1:1600 with a solution of this antibody pre-incubated with Kv4.3 antigenic peptide (Alomone) at 1:10.
Statistical analysis
Comparisons between patient groups for current and ion channel expression were made using Student’s t-tests, with Graphpad Prism software. Multiple linear regression was performed using Stata Version 10, Stata Corp., College Station, Texas, USA. Statistical significance was taken as p<0.05.
Results
Patient characteristics
Table 2 shows the patients’ clinical characteristics, and a comparison between treatment and non-treatment with a β-blocker. The majority of patients underwent coronary artery bypass graft surgery (89%) and had angina (88%). LV function was normal in 74% of patients. Of the patients treated with a β-blocker, 95% received a cardiac selective β1-adrenoceptor antagonist (atenolol or bisoprolol), and 2 patients received carvedilol, which additionally antagonises β2- and α1-receptors. No patient was given sotalol (which has additional class III activity). Patients treated and not treated with a β-blocker were generally of similar age, male-female ratio and disease history (Table 2). Those receiving a β-blocker had a lower incidence of calcium channel blocker use, and a lower heart rate (Table 2).
Table 2.
Patient characteristics. Values are numbers of patients [n; (% of total within group)] with specified clinical characteristics, except for age and heart rate (mean±SEM), in groups of patients not treated, or treated chronically, with a beta-adrenoceptor antagonist (β-blocker: no, and b-blocker: yes, respectively). Age and heart rate were compared between groups by unpaired Student’s t-test; categorical data by χ2 with Yates’ correction.
|
β-blocker: No n (%) |
β-blocker: Yes n (%) |
p | |
|---|---|---|---|
| Total patients | 35 | 38 | - |
| Male/female | 28/7 | 26/12 | 0.390 |
| Age (years) | 63.7±1.7 | 64.9±1.4 | 0.606 |
| Heart rate (beats.min−1) | 74±2 | 57±2 | <0.0001* |
| CABG | 30 (86) | 35 (92) | 0.618 |
| AVR | 7 (20) | 3 (8) | 0.245 |
| MVR | 1 (3) | 0 (0) | 0.967 |
| β-blocker (any): | 0 (0) | 38 (100) | - |
| atenolol | 0 (0) | 30 (79) | - |
| bisoprolol | 0 (0) | 6 (16) | - |
| carvedilol | 0 (0) | 2 (5) | - |
| ACE inhibitor | 15 (43) | 21 (55) | 0.409 |
| CCB | 19 (54) | 8 (21) | 0.007* |
| Statin | 31 (89) | 38 (100) | 0.103 |
| Nicorandil | 19 (54) | 15 (39) | 0.302 |
| Digoxin | 1 (3) | 0 (0) | 0.967 |
| No LVSD | 24 (69) | 30 (79) | 0.458 |
| Mild/moderate LVSD | 11 (31) | 7 (18) | 0.310 |
| Severe LVSD | 0 (0) | 1 (3) | 0.967 |
| Previous MI | 12 (34) | 13 (34) | 0.810 |
| Angina | 30 (86) | 34 (89) | 0.895 |
| Hypertension | 17 (49) | 24 (63) | 0.308 |
| Diabetes | 10 (29) | 5 (13) | 0.181 |
=p<0.05.
CABG=coronary artery bypass graft, AVR=aortic valve replacement, MVR=mitral valve replacement, CCB=calcium channel blocker, LVSD=left ventricular systolic dysfunction, MI=myocardial infarction.
Chronic β-blockade reduced the density of human atrial ITO and IK1, but not IKSUS
Figure 1A shows representative current traces of ITO and IKSUS voltage-dependent activation in myocytes of similar capacitance from a non-β-blocked patient and a β-blocked patient. There was a significant reduction in mean ITO density at all voltages from +10 to +60 mV associated with chronic β-blockade (Figure 1B). Peak ITO density at +60 mV was 41% lower in myocytes from patients treated with a β-blocker compared to non-β-blocked patients (Figure 1C). There was no significant difference in mean capacity of these cells between patients treated and not treated with a β-blocker (67±4 vs 76±3 pF; p>0.05). Single linear regression analysis of the effects of chronic β-blockade on ITO demonstrated a change in current density of −4.14 pA/pF (95% CI −7.50 to −0.77, p<0.05). There was a significant reduction in ITO density of −4.19 pA/pF (95% CI −7.88 to −0.49, p<0.05) in association with chronic β-blockade after adjusting for calcium channel blocker use. In a separate model, adjusting for heart rate, chronic β-blockade was not statistically significantly associated with ITO density, −1.61 pA/pF (95% CI −6.35 to 3.14, p=0.49). There was no difference in peak IKSUS density, recorded in the same cells, between β-blocked and non-β-blocked patients (Figure 1C). Figure 2 shows mean current-voltage relationships for Ba2+-sensitive IK1, recorded in 21 cells from β-blocked patients and 31 cells from non-β-blocked patients; several of which (6 and 9 cells, respectively) were used to record ITO. There was a significant, 34% reduction in peak IK1, at −120 mV, in myocytes from β-blocked patients, and no significant difference in IK1 at the less negative potentials at which the small outward portion of the current occurred. There was no significant difference in mean capacity of cells in which IK1 was recorded, between β-blocked and non-β-blocked patients (85±5 vs 87±4 pF; p>0.05).
Figure 1.

Chronic β-blockade is associated with a reduction in the density of human atrial ITO, but not IKSUS. (A) Representative current traces of ITO and IKSUS in isolated atrial myocytes from a patient treated (●) or not treated (○) with a β-blocker. (B) Mean±SEM current-voltage-relationship for ITO density (●: n=30 myocytes, 15 patients, ○: n=32 myocytes, 14 patients) (C) Mean±SEM peak ITO and IKSUS densities at +60 mV. *=p<0.05; NS= not significant (p>0.05).
Figure 2.
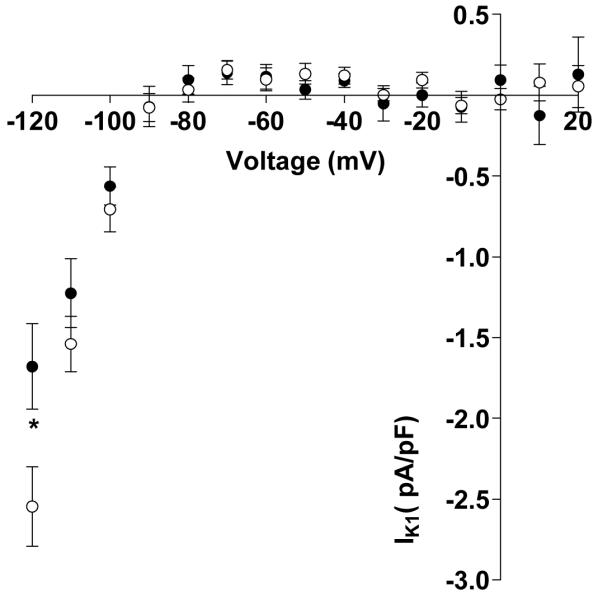
Chronic β-blockade is associated with a reduction in the inward component of IK1 (at −120 mV). Values are means±SEM current-voltage-relationship for Ba2+-sensitive IK1, in myocytes from patients treated (●: n=21 myocytes, 11 patients) or not treated (○: n=31 myocytes, 12 patients) with a β-blocker. *=p<0.05.
Chronic β-blockade did not affect voltage-, time- or rate-dependent properties of human atrial ITO
The voltage-dependences of ITO activation and inactivation were not significantly different between myocytes from β-blocked and non-β-blocked patients (Figure 3A). The voltage of half maximal activation (V0.5) in β-blocked patients was 25.1±0.7 mV (29 myocytes, 15 patients) vs 23.8±0.5 mV (32 myocytes, 14 patients) in the non-β-blocked group (p>0.05). Inactivation V0.5 in cells from β-blocked and non-β-blocked patients was −19.0±1.0 mV (29, 13) vs −19.5±0.6 mV (32, 15), p>0.05. ITO time-dependent inactivation was bi-exponential in 93% of β-blocked patients and in 100% of non-β-blocked patients, and the resulting curve fits are shown in Figure 3B. The mean fast rate constants (inverse of time constants) were 0.16±0.01 for β-blocked patients (22 myocytes, 13 patients) and 0.15±0.01 for non-β-blocked patients (27 myocytes, 15 patients), p>0.05. The corresponding slow rate constants were 0.03±0.01 and 0.04±0.01, p>0.05, respectively. The contribution of each exponential to ITO inactivation in these cells, i.e., the amplitude of the fast and slow inactivation phases, was also not significantly different (p>0.05 for each phase) between the two patient groups (Figure 3B inset). ITO reactivation is shown in Figure 3C. Mean S2 ITO current densities were normalised to the 1st post-rest pulse of the protocol. Initially, at long inter-pulse intervals, ITO density remained unchanged, in keeping with the rapid recovery of ITO from inactivation [11]. As the inter-pulse interval was shortened to <150 ms, ITO reduced in myocytes from both patient groups. ITO reactivation in 80% of myocytes from β-blocked patients (24 myocytes, 12 patients) and in 93% from non-β-blocked patients (28 myocytes, 13 patients) best fitted a mono-exponential curve (Figure 3C). The time-course of ITO reactivation for each patient group was virtually superimposable, with mean time constants of reactivation of 33.5±2.6 ms and 31.3±1.9 ms, respectively; p>0.05. ITO density reduced significantly with increasing stimulation rate; reversible and not different between myocytes from β-blocked and non-β-blocked patients (Figure 3D). The reduction in ITO density associated with chronic β-blockade was maintained even at rapid rates.
Figure 3.

Lack of effect of chronic β-blockade on voltage- or time-dependent characteristics of ITO. Open symbols are data (means±SEM) from non-β-blocked patients (n=27-32 cells, 11-15 patients); filled symbols from β-blocked patients (17-29 cells, 6-15 patients). (A) Boltzmann curves-fits for voltage-dependent activation (_) and inactivation (---). (B) Bi-exponential curve-fits showing time-dependent ITO inactivation at +60 mV. Inset shows amplitudes of fast and slow inactivation phases, respectively. (C) Mono-exponential curve-fits showing ITO reactivation. Inset is an enlarged version of main graph at short interpulse intervals of the 2-pulse activation protocol. (D) ITO rate-dependence: normalised steady-state (8th pulse) ITO density vs stimulation rate.
Acute β-adrenergic stimulation increased atrial IKSUS; an effect not modified by β-blocker therapy
In atrial cells from a group of patients not treated with a β-blocker, acute superfusion with ISO (1 μM) significantly increased IKSUS, and had no significant effect on IK1 or ITO (Figure 4A). A similar increase in IKSUS and lack of change in IK1 or ITO was found in response to ISO in cells from a group of patients who were treated chronically with a β-blocker (Figure 4B).
Figure 4.

Effects of acute β-adrenoceptor stimulation on K+ currents in human atrial cells, and lack of effect of chronic β-blocker therapy. (A) Patients not treated with a β-blocker (n=14-15 cells, 3 patients). (B) Patients treated with a β-blocker (n=11-14 cells, 3-4 patients). CON=control; ISO=superfusion with isoprenaline 1 μM for 90 s. IK1 was measured as inward current at −120 mV. ITO and IKSUS were measured at +60 mV. Values are means±SEM. *=p<0.05 (paired t-test); NS=not significant, versus respective controls.
Mathematical modelling of human atrial ion current changes associated with chronic β-blockade
Action potentials (AP) from the Courtemanche model featured a prominent plateau (phase 2) and a marked “spike and dome” (Type-1) morphology (trace i in Figure 5A). We progressively reduced the plateau amplitude by sequentially scaling ICaL and IKsus, until the AP configuration and approximate APD at 90% repolarisation (APD90) as recorded previously in our laboratory in atrial myocytes from non-β-blocked patients [35], was attained: trace ii in Figure 5A (APD90=186 ms). Repeating this simulated AP (trace ii) with IK1 reduced by the same magnitude as observed at −120 mV in the myocytes from β-blocked patients, 34% (Figure 2), resulted in a 26% increase in APD90 (Figure 5B). A simulated voltage-ramp-clamp of IK1 indicated that this AP change was due to a reduction in outward IK1 (Figure 5C). Reducing ITO independently by 41%, i.e. as associated with β-blocker therapy (Figure 1C), also prolonged APD90 (Figure 5E), but by a lesser degree, 9%, than with IK1 reduction. Simultaneous reduction in IK1 and ITO resulted in a 28% increase in APD90 (Figure 5F), versus a 21% increase previously associated with β-blocker therapy [35]. By contrast, using the Type-1 AP (non-representative of APs recorded from our laboratory; trace i of Figure 5A), the same reductions in IK1, ITO, and their combination, produced negligible changes in APD90: 3% increase; 2% decrease; and 2% increase, respectively.
Figure 5.

Mathematical modelling of ion current changes associated with β-blocker therapy. (A) Superimposed simulated APs (last APs of 1 Hz trains of 8 APs) resulting from progressive (in 10% steps) simultaneous ICaL-decrease and IKsus-increase. Trace i=100% ICaL+100% IKsus; ii=50% ICaL+150% IKsus; iii=0% ICaL+200% IKsus. (B) Effect of reducing IK1 on AP shape. Control AP=trace ii of panel A. (C) Effect of reducing IK1 on simulated quasi-steady-state IK1-voltage relationship obtained with the voltage-ramp in D. (E) Effect of independent reduction in ITO on AP shape. (F) Effect of simultaneous reduction in IK1 and ITO on AP shape.
Chronic β-blockade did not reduce mRNA expression of human atrial K+ channel subunits
Figure 6A-F shows the relative mean abundance of mRNA for Kv4.3, the pore-forming ion channel α-subunit responsible for ITO, and its accessory protein subunits KChIP2, Kvβ1, Kvβ2, KChAP and frequenin [27]. The expression of mRNA for these subunits was not different in the tissue samples from β-blocked patients compared to those from non-β-blocked patients. There was also no change in the expression of Kv1.5, the pore-forming ion channel α-subunit carrying IKSUS [26] (Figure 6G). Kir2.1, one of the pore-forming subunits carrying IK1 tended to increase in the tissue from β-blocked patients (p=0.05; Figure 6H), in contrast to the reduction in current density in this patient group. Kir2.2 was not detectable in either group of tissue samples, and there was no change in TWIK-1, the pore-forming subunit of a weak inwardly-rectifying current that may contribute to IK1 [26, 19] (Figure 6I). Expression of pore-forming subunits for ICaL and the Na+/Ca2+-exchanger (Cav1.2 and NCX-1, respectively) also did not differ between patient groups.
Figure 6.

Mean±SEM abundance of mRNA relative to 28S and GAPDH for ion channel subunits associated with the ion currents ITO, IKSUS and IK1, in atrial tissues from β-blocked (■; n=8) and non-β-blocked (□; n=6) patients (NS=p>0.05). 28S and GAPDH expression did not differ significantly between patient groups.
Chronic β-blockade did not affect Kv4.3 protein expression in human atrial tissue
Kv4.3 was detected in western blots using two primary antibodies: a monoclonal antibody, and a polyclonal antibody which was supplied with the corresponding antigen for use in control experiments. The polyclonal anti-Kv4.3 detected Kv4.3 at 65 kDa in rat and rabbit brain tissue, in which Kv4.3 is highly expressed, and this band was markedly reduced when the antibody was pre-incubated with the Kv4.3 antigen (Figure 7A). This polyclonal primary antibody did not detect any similar band in human atrial tissues, presumably because of lower levels of Kv4.3 expression, and hence the monoclonal antibody was then used for increased sensitivity. This monoclonal anti-Kv4.3 also detected Kv4.3 at 65 kDa in the rodent brain tissue positive control, and this band was markedly reduced in rodent lung tissue, in which Kv4.3 expression is minimal [17] (Figure 7B). Using this antibody, a band at 65 kDa was also detected in human atrial tissue (Figure 7B). The specificity of this antibody was tested by excluding non-specific secondary antibody binding (Figure 7B). No antigenic peptide was available for control testing with this monoclonal antibody. Figure 7C shows a western blot of continuously varying concentrations of total protein from tissue from two β-blocked and two non-β-blocked patients. The blot was cut horizontally just above the 40 kDa marker (dashed horizontal line in Figure 7C) prior to incubation with primary antibody. The top half was treated with the monoclonal anti-Kv4.3 and the bottom with a monoclonal anti-GAPDH that detected GAPDH at 38 kDa. There was no significant difference in the mean optical density (OD) of either GAPDH (9.56±0.70 vs 10.66±1.61 ODu/mm2, p>0.05) or Kv4.3 (11.44±1.03 vs 11.57±1.31 ODu/mm2, p>0.05) between tissues from β-blocked (n=8) and non-β-blocked (n=10) patients. All ODs were compared at total protein load 7.5 μg. The ratios of Kv4.3:GAPDH were 1.24±0.13 in samples from β-blocked, and 1.30±0.27 in samples from non-β-blocked, patients, p>0.05.
Figure 7.

Western blots of Kv4.3. Molecular weight markers are indicated in kDa. (A) Kv4.3 was detected at 65 kDa in rat brain (rB) and rabbit brain (RB) using polyclonal α-Kv4.3 primary antibody (1°Ab) (left section of membrane), and was markedly reduced when treated with 1°Ab pre-incubated with antigen, Ag (right). (B) Kv4.3 was also detected at the same molecular weight using a monoclonal 1°Ab in both rodent brain and human atrium (HA), but was markedly reduced in negative control rabbit lung (RL), and absent with secondary antibody (2°Ab) only. (C) Increasing protein concentrations (5-10 μg) of whole human atrial homogenate from two β-blocked (BBYes) and two non-β-blocked (BBNo) patients treated with monoclonal α-Kv4.3 (upper section) or monoclonal α-GAPDH (lower section).
Discussion
This is the first study, to our knowledge, to show that the chronic treatment of patients in sinus rhythm with β-blockers is associated with a significant reduction in both atrial ITO and IK1; that the reduction in ITO occurred without any changes in its time-, rate- or voltage-dependence; and that neither ion current change resulted from any changes in expression of underlying ion channel subunits. Furthermore, we suggested, using mathematical modelling, that the combination of the reduction in ITO and IK1 may contribute to the established prolongation of atrial action potential repolarisation in patients treated with β-blockers.
The magnitude of ITO reduction was similar to that found in a previously reported smaller study by our group [35], in which other properties of this current were not measured. Further supporting these findings, chronic treatment of rabbits with the β-blocker carvedilol reduced atrial ITO [4], but no other studies examining the effects on human atrial ITO could be found. However, in catecholamine-depleted rats, and also in rats with chronically reduced cardiac sympathetic innervation, ventricular ITO was reduced in conjunction with prolongation of the APD [2, 21]. Given the rapid inactivation kinetics of ITO, the effect of changes in this current on late repolarisation is presently unclear and under debate [7, 14, 10]. We established that chronic β-blockade in patients was also associated with a reduction in atrial IK1, measured as the inwardly-rectifying Ba2+-sensitive current, and this might be expected to prolong action potential terminal repolarisation [12]. Our mathematical modelling of changes in ITO and IK1 suggested that the reduction in each current alone could result in APD-prolongation, and also that when combined, these ion current changes could prolong the APD as much as was reported with chronic β-blockade [35]. Of the two currents, IK1 might be expected to contribute more to the APD-prolongation. Upregulation of IK1 in chronic AF is considered to be a crucial factor in stabilising the high frequency rotors that help to sustain fibrillation [28], and it is conceivable that the present reduction of IK1 seen in patients treated with β-blockers might contribute to their anti-arrhythmic actions by opposing such rotor stabilisation.
We found that pharmacological remodelling by chronic β-blockade was not associated with any change in the expression of the ITO ion channel pore, Kv4.3, either at protein or mRNA level. There was also no change in the expression of mRNA for its related ion channel subunits, although the protein expression of these subunits was not measured. While some accessory subunits, such as KChAP and KChIP2 are involved in trafficking of Kv4.3 to the cell membrane and, therefore, directly influence current density, many subunits modify multiple current characteristics of ITO additional to its density [27]. Therefore, the observed absence of change in the expression of ITO accessory subunits is consistent with the observed absence of change in ITO voltage- and time-dependent characteristics. The reduction in IK1 density was not the result of any significant reduction in mRNA expression of potential pore-forming components, Kir2.1 or TWIK, although neither was examined at protein level as part of this study.
We show here ion current changes, by pharmacological remodelling, that did not require changes in the expression of associated ion channel pore-forming or accessory subunits. This appears to contrast with some of the ion current changes which occur in chronic AF [34] that have been associated with changes in expression of corresponding ion channels [9]. However, there are exceptions, e.g., with dramatic changes in ICaL in chronic AF without corresponding changes in protein expression [5, 13, 31, 15]. Perhaps the most likely explanation underlying ion current reduction in the present study is a chronic β-blockade-induced, post-translation alteration in ion channel structure or function. This could result in changes to trafficking of ion channel components to the cell membrane, or to their turnover or recycling from the cell membrane, or to their interaction with other channel subunits and signalling molecules. The potential of chronic β-blockade to alter phosphorylation-dependent changes in channel function is one possible post-translation modification. Adrenoceptor-signalling is complex and varies with chronicity of stimulation. However, cyclic AMP-dependent protein kinase A (PKA), PKC and Ca2+-calmodulin-dependent protein kinase II (CaMKII) are all implicated in multiple downstream signalling pathways [37]. In addition, β-adrenoceptors localise to cardiac cell membrane lipid microdomains or caveolae, where they may interact or recruit ion channel subunits [23]. CaMKII has been shown to associate with Kv4.3 and can alter channel function and trafficking to the cell membrane by phosphorylation, as well as affecting gene transcription [27]. Kv4.3 has potential sites for phosphorylation by PKA and PKC, both of which can affect channel function [27]. PKA-dependent phosphorylation has been implicated in localisation of ITO channel complexes in the cell membrane to lipid microdomains via interaction with caveolin-3 [1]. In addition, human cardiac IK1 is also inhibited by both PKC and PKA-dependent signalling pathways [18, 16]. In the present study, acute β-stimulation increased IKSUS and had no effect on ITO or IK1, consistent with previous reports in human atrium (recently reviewed [32]). Furthermore, β-blocker therapy did not alter the ISO-response of these currents, consistent with a reported lack of effect of β-blocker therapy on ICaL increase by ISO [30]. However, remodelling by β-blocker therapy of other ion current or biochemical responses to β-, or α-, stimulation, should not be excluded.
Study limitations
We studied cells from patients in sinus rhythm, and it is unknown whether the ability of β-blocker therapy to alter ion currents and action potentials is preserved in cells from patients with chronic AF. It should be noted, however, that β-blockers are used in the treatment of a variety of cardiac pathologies that predispose to AF, including ischaemia, heart failure and hypertension, all of which may cause potentially pro-arrhythmic electrophysiological remodelling [24, 36]. The effects of the potentially anti-arrhythmic remodelling of ITO, IK1 and APD by chronic β-blockade may, therefore, be expected to participate in addition to anti-adrenergic, anti-ischaemic, and structural remodelling effects of β-blockers. We focussed on ITO, IKSUS and IK1. ICaL is unaffected by β-blocker therapy [32], but other currents remain to be studied, e.g., IKACh, IKr, IKs and INa/Ca, as well as intracellular Ca2+ homeostasis. In the mathematical modelling study, we reduced IK1 by 34%, as was observed at −120 mV, although no change in IK1 was detected at more positive potentials. Inward IK1 is large and easily quantifiable at −120 mV, but it is recognised that outward IK1 at more positive potentials is small, and that the “small expression levels of IK1 indicate that the ‘natural variability’ between cells may make it difficult to determine how much IK1 is present at plateau potentials” [12]. Any effect of chronic β-blockade to reduce IK1 at such potentials may, therefore, be difficult to discern under voltage-clamp conditions. Since cardiac myocytes have a high resistance at the end of the plateau, the APD at this phase is very sensitive to intrinsic net current changes. We therefore considered it important to model a reduction in outward IK1 to investigate potential ionic mechanisms of effects of β-blockade on APD, and reasonable to assume a reduction of 34%. However, we do not exclude the possibility that β-blockade decreases IK1 at negative potentials without affecting outward IK1, through some modification of the voltage-sensitivity of IK1 channel gating, and that ion current changes additional to the observed reduction in ITO and IK1 are responsible for the increase in APD.
In conclusion, this study improves our understanding of electrophysiological and molecular mechanisms of the potentially anti-arrhythmic prolongation of human atrial action potentials associated with the chronic treatment of patients with β-blockers.
Acknowledgements
Glasgow Royal Infirmary cardiac surgical teams for providing atrial tissue. Viktoria Szuts, University of Szeged, for helpful discussion about the Kv4.3 monoclonal antibody.
Funding: British Heart Foundation (BHF) Clinical PhD Studentship (FS/04/087), and BHF Basic Science Lectureship Renewal (BS/06/003).
Footnotes
The experiments comply with the current laws of the country in which they were performed. The authors declare that they have no conflict of interest.
References
- 1.Alday A, Urrutia J, Gallego M, Casis O. α1-adrenoceptors regulate only the caveolae-located subpopulation of cardiac KV4 channels. Channels. 2010;4:168–178. doi: 10.4161/chan.4.3.11479. [DOI] [PubMed] [Google Scholar]
- 2.Bru-Mercier G, Deroubaix E, Rousseau D, Coulombe A, Renaud J-F. Depressed transient outward potassium current density in catecholamine-depleted rat ventricular myocytes. Am J Physiol. 2002;282:H1237–H1247. doi: 10.1152/ajpheart.00180.2001. [DOI] [PubMed] [Google Scholar]
- 3.Camm AJ, Kirchof P, Lip GYH, Schotten U, Savelieva I, Ernst S, Van Gelder IC, Al-Attar N, Hindricks G, Prendergast B, Heidbuchel H, Alfieri O, Angelini A, Atar D, Colonna P, De Caterina R, De Sutter J, Goette A, Goronek B, Heldal M, Hohloser SH, Kolh P, Le Heuzey JY, Ponikowski P, Rutten FH. Guidelines for the management of atrial fibrillation. The task force for the management of atrial fibrillation of the European Society of Cardiology (ESC) Eur Heart J. 2010;31:2369–2429. doi: 10.1093/eurheartj/ehq278. [DOI] [PubMed] [Google Scholar]
- 4.Cao F, Huang CX, Wang T, Li X, Jiang H, Bao MW. Effects of carvedilol on rabbit atrial cell electrophysiology. Heart Rhythm. 2006;3(Suppl 1):S178. Abstract. [Google Scholar]
- 5.Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- 6.Courtemanche M, Ramirez RJ, Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol. 1998;275:H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301. [DOI] [PubMed] [Google Scholar]
- 7.Courtemanche M, Ramirez RJ, Nattel S. Ionic targets for drug therapy and atrial fibrillation-induced electrical remodeling: insights from a mathematical model. Cardiovasc Res. 1999;42:477–489. doi: 10.1016/s0008-6363(99)00034-6. [DOI] [PubMed] [Google Scholar]
- 8.Dawodu AA, Monti F, Iwashiro K, Schiariti M, Chiavarelli R, Puddu PE. The shape of human atrial action potential accounts for different frequency-related changes in vitro. Int J Cardiol. 1996;54:237–249. doi: 10.1016/0167-5273(96)02605-8. [DOI] [PubMed] [Google Scholar]
- 9.Dobrev D, Ravens U. Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res Cardiol. 2003;98:137–148. doi: 10.1007/s00395-003-0409-8. [DOI] [PubMed] [Google Scholar]
- 10.Escande D, Coulombe A, Faivre JF, Coraboeuf E. Two types of transient outward currents in adult human atrial cells. Am J Physiol. 1987;252:H142–H148. doi: 10.1152/ajpheart.1987.252.1.H142. [DOI] [PubMed] [Google Scholar]
- 11.Fermini B, Wang Z, Duan D, Nattel S. Differences in rate dependence of transient outward current in rabbit and human atrium. Am J Physiol. 1992;263:H1747–H1754. doi: 10.1152/ajpheart.1992.263.6.H1747. [DOI] [PubMed] [Google Scholar]
- 12.Fink M, Giles WR, Noble D. Contributions of inwardly rectifying K+ currents to repolarization assessed using mathematical models of human ventricular myocytes. Philos Trans R Soc A -Math Phys Eng Sci. 2006;364:1207–1222. doi: 10.1098/rsta.2006.1765. [DOI] [PubMed] [Google Scholar]
- 13.Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, Leger J, Charpentier F, Christ T, Dobrev D, Escande D, Nattel S, Demolombe S. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation. 2005;112:471–481. doi: 10.1161/CIRCULATIONAHA.104.506857. [DOI] [PubMed] [Google Scholar]
- 14.Greenstein JL, Wu R, Po S, Tomaselli GF, Winslow RL. Role of the calcium-independent transient outward current Ito1 in shaping action potential morphology and duration. Circ Res. 2000;87:1026–1033. doi: 10.1161/01.res.87.11.1026. [DOI] [PubMed] [Google Scholar]
- 15.Greiser M, Halaszovich CR, Frechen D, Boknik P, Ravens U, Dobrev D, Luckhoff A, Schotten U. Pharmacological evidence for altered src kinase regulation of ICa,L in patients with chronic atrial fibrillation. Naunyn-Schmiedeberg’s Arch Pharmacol. 2007;375:383–392. doi: 10.1007/s00210-007-0174-6. [DOI] [PubMed] [Google Scholar]
- 16.Karle CA, Zitron E, Zhang W, Wendt-Nordahl G, Kathofer S, Thomas D, Gut B, Scholz E, Vahl CF, Katus HA, Kiehn J. Human cardiac inwardly-rectifying K+ channel Kir2.1b is inhibited by direct protein kinase C-dependent regulation in human isolated cardiomyocytes and in an expression system. Circulation. 2002;106:1493–1499. doi: 10.1161/01.cir.0000029747.53262.5c. [DOI] [PubMed] [Google Scholar]
- 17.Kong W, Po S, Yamagishi T, Ashen MD, Stetten G, Tomaselli GF. Isolation and characterization of the human gene encoding Ito: further diversity by alternative mRNA splicing. Am J Physiol. 1998;275:H1963–H1970. doi: 10.1152/ajpheart.1998.275.6.H1963. [DOI] [PubMed] [Google Scholar]
- 18.Koumi S, Backer CL, Arentzen CE, Sato R. β-adrenergic modulation of the inwardly rectifying potassium channel in isolated human ventricular myocytes. Alteration in channel response to β-adrenergic stimulation in failing human hearts. J Clin Invest. 1995;96:2870–2881. doi: 10.1172/JCI118358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J. 1996;15:1004–1011. [PMC free article] [PubMed] [Google Scholar]
- 20.Li GR, Feng J, Wang Z, Fermini B, Nattel S. Adrenergic modulation of ultrarapid delayed rectifier K+ current in human atrial myocytes. Circ Res. 1996;78:903–915. doi: 10.1161/01.res.78.5.903. [DOI] [PubMed] [Google Scholar]
- 21.Liu QY, Rosen MR, McKinnon D, Robinson RB. Sympathetic innervation modulates repolarizing K+ currents in rat epicardial myocytes. Am J Physiol. 1998;274:H915–H922. doi: 10.1152/ajpheart.1998.274.3.H915. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Sendon J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, Tendera M, Waagstein F, Kjekshus J, Lechat P, Torp-Pedersen C. Expert consensus document on β-adrenergic receptor blockers. The task force on beta-blockers of the European Society of Cardiology. Eur Heart J. 2004;25:1341–1362. doi: 10.1016/j.ehj.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 23.Maguy A, Hebert TE, Nattel S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc Res. 2006;69:798–807. doi: 10.1016/j.cardiores.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 24.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 25.Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol. 2000;525:285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 27.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandit SV, Berenfeld O, Anumonwo JMB, Zaritski RM, Kneller J, Nattel S, Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raine AEG, Vaughan Williams EM. Adaptation to prolonged β-blockade of rabbit atrial, Purkinje, and ventricular potentials, and of papillary muscle contraction. Time-course of development of and recovery from adaptation. Circ Res. 1981;48:804–812. doi: 10.1161/01.res.48.6.804. [DOI] [PubMed] [Google Scholar]
- 30.Redpath CJ, Rankin AC, Kane KA, Workman AJ. Anti-adrenergic effects of endothelin on human atrial action potentials are potentially anti-arrhythmic. J Mol Cell Cardiol. 2006;40:717–724. doi: 10.1016/j.yjmcc.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Schotten U, Haase H, Frechen D, Greiser M, Stellbrink C, Vazquez-Jimenez JF, Morano I, Allessie MA, Hanrath P. The L-type Ca2+-channel subunits α1C and β2 are not downregulated in atrial myocardium of patients with chronic atrial fibrillation. J Mol Cell Cardiol. 2003;35:437–443. doi: 10.1016/s0022-2828(03)00012-9. [DOI] [PubMed] [Google Scholar]
- 32.Workman AJ. Cardiac adrenergic control and atrial fibrillation. Naunyn-Schmied Arch Pharmacol. 2010;381:235–249. doi: 10.1007/s00210-009-0474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res. 2001;52:226–235. doi: 10.1016/s0008-6363(01)00380-7. [DOI] [PubMed] [Google Scholar]
- 34.Workman AJ, Kane KA, Rankin AC. Cellular bases for human atrial fibrillation. Heart Rhythm. 2008;5:S1–S6. doi: 10.1016/j.hrthm.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Workman AJ, Kane KA, Russell JA, Norrie J, Rankin AC. Chronic beta-adrenoceptor blockade and human atrial cell electrophysiology: evidence of pharmacological remodelling. Cardiovasc Res. 2003;58:518–525. doi: 10.1016/s0008-6363(03)00263-3. [DOI] [PubMed] [Google Scholar]
- 36.Workman AJ, Smith GL, Rankin AC. Mechanisms of termination and prevention of atrial fibrillation by drug therapy. Pharmacol Ther. 2011;131:221–241. doi: 10.1016/j.pharmthera.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao RP, Zhu W, Zheng M, Cao C, Zhang Y, Lakatta EG, Han Q. Subtype-specific α1- and β-adrenoceptor signaling in the heart. Trends Pharmacol Sci. 2006;27:330–337. doi: 10.1016/j.tips.2006.04.009. [DOI] [PubMed] [Google Scholar]