

Abstract
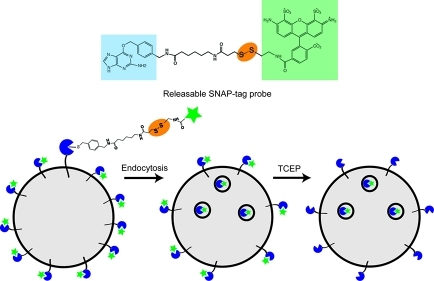
Site-specific labeling of cellular proteins with chemical probes is a powerful tool for live cell imaging of biological processes. One popular system, known as the SNAP-tag, is based on an engineered variant of the 20-kDa DNA repair protein O6-alkylguanine-DNA-alkyltransferase (AGT) that covalently reacts with O6-benzylguanine (BG) and can be derivatized with a number of reporter groups. For studying the endocytosis and recycling of cell surface proteins, the covalent nature of BG binding to the SNAP-tag is problematic, since removing excess noninternalized probe from the cell surface is not feasible. Here we describe a modification of the SNAP-tag technology that permits the rapid release of fluorescently labeled probes from the cell surface without affecting the population of labeled molecules sequestered within endosomes. This simple yet effective approach allows quantitative measurements of endocytosis and recycling in both imaging and biochemical assays and is especially useful when studying endosomal dynamics in live cells.
Methods to study endocytosis typically rely on the use of fluorescently labeled antibodies to follow uptake of membrane-bound cargo proteins into cells as well as to monitor recycling from internal compartments back to the cell surface.1−5 Alternatively, cargo proteins can be genetically tagged with fluorescent proteins (FP).6 Problems inherent with these approaches include the large size of antibodies, the noncovalent nature of the antibody–cargo interaction, the harsh treatments (e.g., low pH, high salt) necessary to remove excess cell surface antibodies, and the current inability to specifically follow cell surface populations of FP-tagged cargos. In addition, assays that quantify internalization and recycling are often indirect, based on measuring signal loss from or reappearance to the cell surface without actually measuring the intracellular population of labeled molecules.
Recently, a number of chemical labeling approaches have been characterized that alleviate some of these issues.7,8 One popular system, known as the SNAP-tag, is based on an engineered variant of the 20-kDa DNA repair protein O6-alkylguanine-DNA-alkyltransferase (AGT) that covalently reacts with O6-benzylguanine (BG), which can be derivatized with a number of reporter groups (e.g., fluorescent probes, biotin, etc.).9 Expression of genetically encoded fusions with the SNAP-tag, followed by reaction with BG probes allows temporal control of labeling, flexibility in the nature of the fluorophore, and given that binding is covalent, confidence that the fluorescence detected is associated with the fusion protein.
For studying the endocytosis and recycling of cell surface proteins, the covalent nature of BG binding to the SNAP-tag is problematic, since removing excess probe from the cell surface to reveal the intracellular endocytosed pool is not feasible. Here, we describe a modification of the SNAP-tag system that introduces a cleavable disulfide bond between the BG moiety and various fluorophores. After internalization of various BG-labeled SNAP-tag fusion proteins, the remaining cell surface associated fluorescence is effectively removed by application of a cell-impermeable reducing agent without affecting the population of labeled molecules sequestered within endosomes. This simple yet effective approach is especially useful when studying endosomal dynamics in live cells.
Results and Discussion
Characterization of Releasable SNAP-tag Probes
A disulfide linkage was introduced between O6-benzylguanine and various fluorescent dyes using standard synthesis and chromatography methods (see Methods and Supporting Information). The structure of BG-S-S-Alexa Fluor 488 (called BG-S-S-488) is depicted (Figure. 1, panel a). To examine the potential usefulness of this probe in labeling proteins for endocytosis, HeLa cells were transfected with the SNAP-tag fused to the N-terminus of the G protein-coupled β2 adrenergic receptor, β2ADR, which has been previously characterized.10 Cells were labeled with either BG-S-S-488 or a commercially available noncleavable probe “SNAP-Surface 488” at 4 °C for 30 min to label the cell surface. Internalization was allowed to proceed at 37 °C in the presence of the agonist isoproterenol. Cells were then treated with the cell-impermeable reducing agent tris(2-carboxyethyl)phosphine (TCEP) for 1 min at 37 °C before fixation. Cell surface staining was completely removed in cells labeled with BG-S-S-488, whereas no effect on cell surface fluorescence could be detected with SNAP-Surface 488, even at 10-fold higher TCEP concentrations (Figure 1, panel b). Cleavage by TCEP was also effective at 4 °C, although longer incubation times (15 min) and higher concentrations of TCEP (10 mM) were used.
Figure 1.

Characterization of a releasable SNAP-tag probe. (a) Schematic of BG-S-S-488, with O6-benzylguanine (blue), Alexa Fluor 488 (green), and disulfide linkage (red) highlighted. (b) Comparison of sensitivity of BG-S-S-488 versus SNAP-Surface 488 (New England Biolabs) to TCEP in HeLa cells transfected with SNAP-β2ADR. Scale bar, 10 μm. (c) Comparison of the ability of unesterified TCEP versus tmTCEP to cleave intracellular BG-S-S-488. Free label is extracted by TX-100 from endosomes in cells incubated with tmTCEP. Scale bar, 10 μm. (d) Fluorescence time course showing sensitivity of BG-S-S-488 to different TCEP concentrations in live COS cells transfected with SNAP-β2ADR. Measurements were taken in the absence of endocytosis, so that only cell surface fluorescence was quantified.
Although TCEP is highly charged and would not be expected to cross cell membranes,11 we wished to formally test whether TCEP treatment would cleave the intracellular population of BG-S-S-488 labeled SNAP-β2ADR, releasing free dye within the endosomal lumen. As a positive control, we synthesized the trimethyl ester form of TCEP (tmTCEP), previously shown to penetrate artificial lipid bilayers.11 Cells expressing SNAP-β2ADR labeled with BG-S-S-488 were allowed to endocytose receptors (as in Figure 1, panel b) and then were treated with TCEP or tmTCEP prior to fixation. Both reducing agents effectively removed cell surface associated fluorescence, as expected (Figure 1, panel c). Subsequent membrane permeabilization with 0.1% Triton X-100 (v/v) had little effect on the fluorescence signal in cells treated with TCEP, whereas free dye was almost fully extracted in cells pretreated with tmTCEP, demonstrating its access to endosomal lumens (Figure 1, panel c). Thus, under the conditions shown here, TCEP did not gain access to SNAP-tagged proteins in endosomes. These results are also consistent with evidence that the endosomal system is generally nonreducing.12,13 Using this permeabilization protocol, we did observe some reduction of BG-S-S-488 during extended incubations (>10 min) with TCEP at 37 °C, demonstrating that endocytosed fluid can access labeled SNAP-β2ADR. Thus, for longer term imaging studies, TCEP should be washed out or neutralized (see below).
Treatment of cells with TCEP might be expected to reduce disulfide bonds within endogenous cell surface proteins. However, we observed no effect of TCEP on antibody binding or uptake of disulfide bonded cell surface MHC class I molecules that constitutively internalize via clathrin-independent endocytosis (data not shown).14 This is likely due to the decreased accessibility of TCEP to protein disulfides when compared to DTT at neutral pH.11 Longer-term treatment of cells with higher concentrations of TCEP (>20 min at 10 mM) resulted in changes to cell shape, which was prevented by adding an excess of a disulfide-linked substrate such as oxidized glutathione or by washing out the TCEP.
The most obvious benefit in removing cell surface fluorescence would be in live cell imaging. As only a fraction of receptors are actually internalized during uptake experiments, removal of surface fluorescence would dramatically reduce the background when studying endosomal dynamics. This can be illustrated in Supplemental Videos 1 and 2, which show time lapse movies of COS cells expressing SNAP-β2ADR, labeled with BG-S-S-488 or “SNAP-Surface 488” and allowed to internalize labeled receptor for 30 min at 37 °C. TCEP treatment led to the rapid (within seconds) and almost quantitative removal of cell surface fluorescence such that only receptors within endosomal compartments remained labeled. TCEP concentration curves demonstrate the effectiveness of fluorescence removal from the plasma membrane (Figure 1, panel d).
Quantitation of Endocytosis and Recycling
The rapid removal of surface label with TCEP led us to use this assay to quantify uptake of labeled receptor over time (Figure 2, panel a). HEK293A cells stably expressing SNAP-β2ADR were labeled with BG-S-S-488 and induced to endocytose the receptor in the presence of isoproterenol. The ratio of total integrated fluorescence intensity of an entire field of cells before and after TCEP treatment was measured and quantified to assess the percentage of receptor uptake over time (Figure 2, panel b, purple bars) (see Methods). Initial endocytosis of receptors followed first-order kinetics (ke 0.013 ± 0.001 min–1; R2 = 0.98) that increased from <4% (background value) to ∼25% within 30 min. Similar values were obtained when individual cells rather than fields of cells were measured (data not shown), or when receptor endocytosis was monitored indirectly by measuring loss of cell surface receptor (Supplemental Figure 1), one standard method for measuring GPCR endocytosis.4 An advantage to this method of detection is we are not relying on simply one measured parameter (e.g., loss of signal from the surface), but rather that both total (before TCEP) and internal (after TCEP) cell fluorescence are directly determined from the same cells.
Figure 2.

Endocytosis and recycling of BG-S-S-488 labeled SNAP-β2ADR. (a) Internalization in the presence of isoproterenol for indicated times. Images show the same cells before and 30 s after addition of 10 mM TCEP. Scale bar, 10 μm. (b) Quantitation of uptake based on total fluorescence intensity before and after TCEP. Error bars, SEM (n = 14–16 independent time courses for untreated and n = 5 for monensin treated cells). (c) Representative images showing recycling before and after 10 mM TCEP treatment. Scale bar, 10 μm. (d) Quantitation of recycling based on total fluorescence intensity after continuous exposure to TCEP. Error bars, SEM (n = 16 independent time courses for untreated and n = 7 for monensin treated cells).
In the above experiments, cells were treated with TCEP for 30 s to remove cell surface associated fluorescence. Interestingly, longer treatments resulted in the continual loss of cell fluorescence due to recycling of labeled receptor back to the surface and cleavage of newly exposed disulfide linked fluorophores. Loss of fluorescence was not due to photobleaching since a similar time course in cells not treated with TCEP showed no loss of fluorescence signal (data not shown). A time series of continued exposure to TCEP indicated that recycling was very rapid, contributing to ∼40% loss of signal over 10 min (Figure 2, panels c and d). Receptor recycling followed first order kinetics, giving a rate constant (kr) of 0.062 ± 0/005 min–1 (R2 =0.98). One caveat to this approach is that the incubation with TCEP for extended periods of time could result in some cleavage of the labeled probe in endosomes (see above). This could lead to an underestimation of the SNAP-β2ADR recycling rate, assuming the membrane-impermeant 488 dye would remain in the fluid phase of endosomes without recycling to the cell surface. However, the recycling rates shown here are comparable to those using indirect cell surface labeling approaches.15 Pretreatment of cells with the carboxylic ionophore monensin prevented loss of the fluorescence signal (Figure 2, panel d), consistent with its effects as an inhibitor of β2ADR recycling,15 leading to an apparent net increase in rate of uptake (ke 0.025 ± 0.002 min–1; R2 = 0.99) that may reflect the true endocytosis rate of SNAP-β2ADR in the absence of recycling (Figure 2, panel b, pink bars). Thus using a single assay, both uptake and recycling can be directly quantified in live cells.
In addition to live cell imaging, releasable SNAP-tag probes were tested for use in biochemical pulse-chase uptake experiments using fluorescence in-gel detection. In this case, a BG-S-S-800 probe was synthesized (see Methods) and tested in SNAP-β2ADR stable HEK293A cells. Cells were labeled at 4 °C and then chased for various periods of time at 37 °C in the presence or absence of isoproterenol. One set of samples was left untreated, and the other was treated with TCEP to release cell surface dye. After TCEP neutralization, cell lysates were separated by SDS-PAGE and directly imaged. As shown in Supplemental Figure 2, the proportion of labeled TCEP-resistant SNAP-β2ADR increased over time in the presence but not absence of agonist, reflecting agonist-induced endocytosis. Quantitation yielded results similar to those determined through live cell imaging (see Figure 2, panel b), thus validating the use of releasable SNAP-tag probes in both live cell and biochemical assays.
Dual Color Imaging
As an example of the utility of releasable SNAP-tag probes for dual color imaging, we examined the relationship of different classes of GPCRs with the adaptor β-arrestin. Upon agonist treatment, GPCRs rapidly associate with members of the arrestin family of adaptors16,17 but exhibit two distinct patterns of β-arrestin interaction.18,19 The β2 adrenergic receptor rapidly dissociates from β-arrestin upon internalization, whereas the neurokinin-1 receptor (NK-1R) forms stable receptor-β-arrestin complexes in endocytic vesicles.20,21
HEK293A stably expressing SNAP-β2ADR or SNAP-NK1R were transiently transfected with β-arrestin2 fused at its C-terminus with monomeric TagRFP (see Methods). Upon addition of agonist, β-arrestin2-TagRFP rapidly (<40 s) translocated from the cytoplasm to the cell surface in cells expressing both SNAP-tagged GPCRs (Figure 3, panel a; Supplemental Videos 3 and 4) and accumulated in punctate structures that colocalized with AP2 (Supplemental Figure 3). Internalization of both receptors increased over time, with endosomal populations of SNAP-NK1R showing persistent colocalization with β-arrestin2-TagRFP. In contrast, SNAP-β2ADR was found alone in endosomes, while β-arrestin2-TagRFP remained on the plasma membrane.22 Visualizing this distinction was greatly enhanced after TCEP treatment (Figure 3, panel b), allowing us to follow the trafficking of the different populations of receptor/β-arrestin complexes.
Figure 3.

Live cell imaging of HEK293A cells stably expressing SNAP-β2ADR or SNAP-NK1R transfected with β-arrestin2-TagRFP. From Supplementary Videos 3 and 4. (a) Images show the redistribution of β-arrestin2-TagRFP from the cytosol to the plasma membrane in the presence of isoproterenol in cells expressing SNAP-β2ADR. Boxes highlight the redistribution to the cell surface in adjacent cells. (b) Comparison of the colocalization of SNAP-β2ADR or SNAP-NK1R with β-arrestin2-TagRFP after agonist-induced uptake (isoproterenol for SNAP-β2ADR or Substance P for SNAP-NK1R) and 5 mM TCEP treatment. β-Arrestin2-TagRFP colocalizes with SNAP-NK1R labeled endosomes but not with SNAP-β2ADR. Scale bar, 10 μm.
Although we show here the use of releasable SNAP-tag probes in GPCR trafficking, the method can be used to study any cell surface protein. The capacity to rapidly and effectively distinguish intracellular from cell surface pools in both imaging and biochemical assays greatly simplifies current approaches in studying endocytosis and may be useful, for example, in analyzing synaptic vesicle uptake and recycling in neurons.23 These probes enhance the versatility of the SNAP-tag system and are of general use for tracking the intracellular fate of internalized molecules.
Methods
Plasmids and Cell Lines
Expression plasmids encoding SNAP-tagged beta-2 adrenergic receptor (SNAP-β2ADR) and CLIP-tagged neurokinin-1 receptor (SNAP-NK1R) were from Covalys Biosciences AG (now available from New England Biolabs). The β2ADR coding region was replaced with NK1R by digesting both plasmids with SbfI and BamHI. SNAP-β2ADR and SNAP-NK1R were both cloned into pcDNA3.1(−). The human cDNA clone for β-arrestin2 (NM_004313) was from Origene and cloned into HindIII and BamHI sites of pTagRFP-N (from Evrogen) using gene specific primers.
All cell lines used in this study were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Lonza) with 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/mL streptomycin and grown in a 5% CO2 atmosphere at 37 °C. Cells were transiently transfected using Fugene 6 (Roche Molecular Biochemicals). Stable HEK293A cell lines were selected in G418 (500 μM) but were routinely cultured following selection in its absence without loss of expression.
Synthesis of Releasable SNAP-tag Probes
tmTCEP Synthesis
Unesterified TCEP (in methanol) was stirred overnight with HCl-treated and washed Dowex 50W cation exchange resin (Bio-Rad), in methanol, according to Cline et al.11 Trimethyl ester formation was characterized by ESI-MS, with over 90% yield.
Cell Labeling
The procedure is similar to labeling with commercially available SNAP ligands from New England Biolabs. The fluorescent ligand was diluted to 1–5 μM into medium (DMEM containing 10% FBS) and incubated at 4 °C for 30–45 min with cells transiently or stably expressing SNAP-tag fusion proteins. Performing the labeling reaction at 4 °C prevents uptake of unliganded probe via fluid phase endocytosis. The probe reacts with the SNAP-tag forming a stable covalent thioether bond. Cells were rinsed with Hanks’ balanced salt solution (HBSS) (Gibco) twice then incubated at 4 °C for 15 min with HBSS/10% FBS/20 μM BG-NH2 to block unlabeled SNAP-tag fusions. Labeled cells were incubated in HBSS/10% FBS or media at 37 °C for various periods of time to allow internalization of labeled proteins into the cell. Cells were rinsed once with HBSS, then treated with 1–10 mM TCEP (tris(2-carboxyethyl)phosphine) in HBSS for 1–3 min at 37 °C. Cleavage of the disulfide bond within the probe released unbound fluorophore into the medium. Since TCEP is highly charged and will not cross the plasma membrane, only ligands remaining on the cell surface will be cleaved. Release of cell surface ligand was generally complete within 2 min at 37 °C with 1 mM TCEP. For TCEP treatment at 4 °C, we used 10 mM TCEP for 15 min. At this point, cells can be returned to complete medium for imaging or further processing or fixed for fluorescence analysis. TCEP treatment also removed cell surface fluorescence after cells have been fixed in formaldehyde, albeit with reduced efficiency.
Immunofluorescence Staining
Cells were treated, as indicated, then fixed in 2% formaldehyde/PBS for 20 min at RT. Cells were rinsed twice in 10% FBS/PBS/0.02% sodium azide. Primary and species-specific secondary antibodies were diluted in 10% FBS/PBS containing 0.2% saponin, and incubated for 1 h at RT. Cells were rinsed three times after antibody incubations, rinsed once with PBS, and then mounted in Fluoromount G (Southern Biotechnologies) for imaging. Mouse antialpha adaptin (AP6) (Thermo Scientific) was used for staining the adaptor AP2.
Live Cell Imaging
Uptake and recycling experiments using HEK293A cells stably expressing SNAP-β2ADR and SNAP-tag-NK1R were performed on a 510 LSM confocal microscope (Axiovert 200M; Carl Zeiss). Images were acquired using a 40x Plan-Neofluar 1.3 numerical aperture (NA) objective with the pinhole wide open (<12.4 μm). Optical sectioning demonstrated that these cells had a thickness of 8–10 μm, so that the signal obtained represented most if not all of the total cellular fluorescence. An environmental chamber (Zeiss, XL-3) enclosing the microscope stand kept the temperature at 37 °C. For live cell imaging, cells were plated onto poly l-lysine treated Lab-Tek coverglass 8-well chambers (1.0, Nalge Nunc International) and, when necessary, transfected with the indicated constructs using Fugene 6 (Roche). At 18–40 h after transfection, cells were imaged in HBSS/10% FBS or DMEM without phenol red/10% FBS. After the cells were warmed to 37 °C for 5 min, agonist (isoproterenol for SNAP-β2ADR or Substance P SNAP-tag-NK1R) for was added to the media and allowed to diffuse through the solution. The final concentration was 200 μM isoproterenol or 5 μM Substance P. After the indicated period of time, a single image was taken. TCEP was then added to a final concentration of 10 mM for exactly 30 s. This period was determined to result in almost complete loss of surface fluorescence at the zero time point. At this point a second image was collected. For recycling experiments, after 30 min uptake and addition of TCEP, a series of sequential images was taken at 1 min intervals for 10 min. All time course measurements were with identical microscope settings and below saturation levels. For two-color videos, 488 Ar (BP 505-530) and He/Ne 543 laser (LP 560) lasers were used as light sources for BG-S-S-488 and β-arrestin2-TagRFP signals, respectively. Images were acquired every 20 s. For rapid imaging, (Figure 1D and Supplemental Videos 1 and 2), 5 s images were acquired on a LSM 5 Live confocal microscope (Carl Zeiss) equipped with a diode 488–100 laser, and a Plan-Apochromat 63 × 1.4 N.A. objective. A heated stage maintained the temperature of the solution bathing the cells at ∼28 °C. All videos were generated using MetaMorph (Molecular Devices)
Fluorescence Quantitation
For quantitation of uptake and recycling, images were imported into MetaMorph, and total fluorescence intensity measurements were collected. Zero time point images after TCEP treatment were used for determining inclusive threshold values. To remove bias from thresholding, the lower threshold limit was positioned at the right base of the histogram bar, and gray values above this value were collected. This threshold range was use for all measurements within a particular time course. Images typically contained 15–30 cells. The internalization and recycling curves were modeled using nonlinear regression to obtain first-order rate constants for endocytosis (ke) and recycling (kr) as described.24 Kinetic values were determined using Prism 5 (GraphPad Software). For quantifying uptake via loss of receptor from the cell surface (Supplemental Figure 1), cells were treated for various times at 37 °C in the presence of 200 μM isoproterenol and then labeled with BG-S-S-488 for 30 min at 4 °C. Cells were then rinsed and fixed. Total integrated intensity/area measurements were determined for individual cells using MetaMorph. Thresholding was set from the zero uptake time point, and was positioned similarly to uptake experiments (see above).
Biochemistry
For uptake experiments, BG-amide-NH2 (compound 4, Supplemental Figures 2 and 4) was conjugated to IRDye 800CW NHS Ester, similar to synthesis of the BG-S-S-488 probe. HEK293A cells stably expressing SNAP-β2ADR were plated onto poly-l-lysine coated 6-well dishes (Biocoat, Becton Dickinson) and labeled with BG-S-S-800 (0.5–2 μM) on ice for 30 min. Cells were rinsed with HBSS twice and then incubated on ice for 15 min with HBSS/10% FBS/20 μM BG-NH2 to block unlabeled SNAP-β2ADR. Cells were then warmed to 37 °C in the absence or presence of isoproterenol for the indicated periods of time. Cells were placed on ice, and 20 mM TCEP was added to one well at each time point. Cells were incubated on ice for 30 min, rinsed twice with HBSS, and then incubated in the presence of 2 mM oxidized glutathione to neutralize the remaining TCEP. Finally, cells were rinsed twice in HBSS and solubilized in lysis buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% CHAPS) containing protease inhibitors (Roche, Complete tablets), and 20 mM iodoacetamide. Lysates were spun at 13,000 × g for 5 min to remove insoluble material. Protein determinations were by BCA assay (Thermo Scientific). SDS sample buffer without reducing agent was added and lysates incubated for 30 min at RT. Equal amounts of protein were separated by SDS-PAGE and directly imaged on an Odyssey infrared scanner (Li-Cor).
Acknowledgments
We thank G. Griffiths and A. Dulcey at the Imaging Probe Development Center, NHLBI/NIH for help during the initial phases of this work and thank C. Combs of the Light Microscopy Core Facility/NHLBI for help with image quantitation. We acknowledge the Biochemistry Core Facility (NHLBI) and especially D.-Y. Lee and R. Levine for help with compound synthesis and mass spectrometry analysis. We also thank C. Waterman and L. Greene (both NHLBI) and G. Patterson (NIBIB) for comments on the manuscript. The Intramural Research Program in the National Heart, Lung, and Blood Institute at the NIH supported this work.
Supporting Information Available
Additional data, probe synthesis scheme, and four videos. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Arancibia-Carcamo I. L., Fairfax B. P., Moss S. J., Kittler J. T. (2006) Studying the Localization, Surface Stability and Endocytosis of Neurotransmitter Receptors by Antibody Labeling and Biotinylation Approaches, Iin The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology (Kittler J. T., Moss S. J., Eds.), CRC Press, Boca Raton, FL. [PubMed] [Google Scholar]
- Hinkle P. M.; Puskas J. A. (2004) Detection of G protein-coupled receptors by immunofluorescence microscopy. Methods Mol. Biol. 237, 127–134. [DOI] [PubMed] [Google Scholar]
- Temkin P.; Lauffer B.; Jager S.; Cimermancic P.; Krogan N. J.; von Zastrow M. (2011) SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 13, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seachrist J. L.; Anborgh P. H.; Ferguson S. S. (2000) beta 2-adrenergic receptor internalization, endosomal sorting, and plasma membrane recycling are regulated by rab GTPases. J. Biol. Chem. 275, 27221–27228. [DOI] [PubMed] [Google Scholar]
- Oakley R. H.; Laporte S. A.; Holt J. A.; Barak L. S.; Caron M. G. (1999) Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem. 274, 32248–32257. [DOI] [PubMed] [Google Scholar]
- Bohme I.; Beck-Sickinger A. G. (2009) Illuminating the life of GPCRs. Cell Commun. Signaling 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wombacher R.; Cornish V. W. (2011) Chemical tags: applications in live cell fluorescence imaging. J. Biophotonics 4, 391–402. [DOI] [PubMed] [Google Scholar]
- Hinner M. J.; Johnsson K. (2010) How to obtain labeled proteins and what to do with them. Curr. Opin. Biotechnol. 21, 766–776. [DOI] [PubMed] [Google Scholar]
- Keppler A.; Gendreizig S.; Gronemeyer T.; Pick H.; Vogel H.; Johnsson K. (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89. [DOI] [PubMed] [Google Scholar]
- Maurel D.; Comps-Agrar L.; Brock C.; Rives M. L.; Bourrier E.; Ayoub M. A.; Bazin H.; Tinel N.; Durroux T.; Prezeau L.; Trinquet E.; Pin J. P. (2008) Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat. Methods 5, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline D. J.; Redding S. E.; Brohawn S. G.; Psathas J. N.; Schneider J. P.; Thorpe C. (2004) New water-soluble phosphines as reductants of peptide and protein disulfide bonds: reactivity and membrane permeability. Biochemistry 43, 15195–15203. [DOI] [PubMed] [Google Scholar]
- Austin C. D.; Wen X.; Gazzard L.; Nelson C.; Scheller R. H.; Scales S. J. (2005) Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proc. Natl. Acad. Sci. U.S.A. 102, 17987–17992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Chen H.; Vlahov I. R.; Cheng J. X.; Low P. S. (2006) Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl. Acad. Sci. U.S.A. 103, 13872–13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson J. G.; Williams D. B. (2009) Intracellular assembly and trafficking of MHC class I molecules. Traffic 10, 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippig S.; Andexinger S.; Lohse M. J. (1995) Sequestration and recycling of beta 2-adrenergic receptors permit receptor resensitization. Mol. Pharmacol. 47, 666–676. [PubMed] [Google Scholar]
- Hanyaloglu A. C.; von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568. [DOI] [PubMed] [Google Scholar]
- Marchese A.; Paing M. M.; Temple B. R.; Trejo J. (2008) G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmacol. Toxicol. 48, 601–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Barak L. S.; Anborgh P. H.; Laporte S. A.; Caron M. G.; Ferguson S. S. (1999) Cellular trafficking of G protein-coupled receptor/beta-arrestin endocytic complexes. J. Biol. Chem. 274, 10999–11006. [DOI] [PubMed] [Google Scholar]
- Oakley R. H.; Laporte S. A.; Holt J. A.; Caron M. G.; Barak L. S. (2000) Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210. [DOI] [PubMed] [Google Scholar]
- Luttrell L. M.; Lefkowitz R. J. (2002) The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465. [DOI] [PubMed] [Google Scholar]
- Drake M. T.; Shenoy S. K.; Lefkowitz R. J. (2006) Trafficking of G protein-coupled receptors. Circ. Res. 99, 570–582. [DOI] [PubMed] [Google Scholar]
- Puthenveedu M. A.; von Zastrow M. (2006) Cargo regulates clathrin-coated pit dynamics. Cell 127, 113–124. [DOI] [PubMed] [Google Scholar]
- Denker A.; Bethani I.; Krohnert K.; Korber C.; Horstmann H.; Wilhelm B. G.; Barysch S. V.; Kuner T.; Neher E.; Rizzoli S. O. (2011) A small pool of vesicles maintains synaptic activity in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 17177–17182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison K. J.; Moore R. H.; Carsrud N. D.; Trial J.; Millman E. E.; Tuvim M.; Clark R. B.; Barber R.; Dickey B. F.; Knoll B. J. (1996) Repetitive endocytosis and recycling of the beta 2-adrenergic receptor during agonist-induced steady state redistribution. Mol. Pharmacol. 50, 692–699. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.