

Abstract

The Pd- and Ni-promoted decarbonylation of amino acid thioesters proceeds smoothly to yield enamides. The synthesis of the (S)-(Z)-AviMeCys subunit of mersacidin, an MRSA-active lantibiotic, via this approach, is described.
The prevalence of antibiotic resistance poses a significant public health problem and contiunes to undermine the achievements that have led to the development of our current antibiotic arsenal. Among these agents, “last line” glycopeptide antibiotics such as vancomycin, have arguably received the most attention in light of the onset of methicillin-resistant S. aureus (MRSA), a particularly problematic nosocomial infection. However, the emergence of new resistance mechanisms, coupled with the paucity of novel antibiotics belonging to new structural classes, further underscores the need to revitalize the search for alternative therapeutics.
In an effort to address this need, we have initiated a broad research program based on the chemical synthesis and mechanistic study of several peptide antibiotics that inhibit bacterial peptidoglycan (PG) biosynthesis via mechanisms that are orthogonal to that utilized by vancomycin.1 One such candidate is mersacidin (1), a type B lantibiotic produced ribosomally by Bacillus sp. strain HIL Y-85,54728 and whose in vivo activity against MRSA is comparable to that of vancomycin.2,3 The discovery that mersacidin’s bioactivity is uninhibited by vancomycin despite also forming a 1:1 stoichiometric complex with lipid II suggests that its mode of action is distinct.
Its unique binding site as well as inhibition of the transglycosylation reaction make it an attractive target for development of novel therapeutics because; (i) transglycosylation occurs on the cell surface so cellular penetration of the antibiotic is not a requirement for activity, and (ii) the transglycosylation reaction utilized in PG biosynthesis is unique to bacterial cells. With the ultimate goal of delineating the exact mode of action of mersacidin and its variants, as well as developing novel analogues with increased efficacy, we have embarked on the total synthesis of 1.
The presence of the sensitive S-[(Z)-2-aminovinyl]-3-methyl-D-cysteine (AviMeCys) linkage in the CD ring system, as well as the unnatural amino acid (2S,3S)-β-methylcysteine (β-MeCys) and (2S,3S,6R)-β-methyllanthionine (β-MeLan) fragments in the A, B, and CD ring systems render mersacidin a challenging synthetic target. The AviMeCys subunit is present in other bioactive peptides including epidermin4, thioviridamide5, and the recently discovered microbisporicin, now considered the most potent lantibiotic to date.6 Still, at the time we initiated our studies, no synthesis of the AviMeCys functionality had been reported.7
Although little is known about the function of the enamide subunit, it appears to have a critical role for the biological activity of other agents.8,9 As a result, a wide range of methodologies has arisen to address the issues of chemo- and stereoselectivity associated with their chemical synthesis. Given that enamides possess intrinsically moderate reactivity, they may also serve as intermediates for accessing other functionality.10,11
Methods that have been developed for the synthesis of enamides include: the Curtius rearrangement,12 Peterson olefination,13 vinyl transfer,14 hydrometallation of alkynes,15 radical decarboxylation,16 and radical addition to ynamides.17 Most recently, several reports of transition metal-catalyzed cross-coupling reactions between amides and activated olefins have appeared.18,19,20,21 Despite its simplicity, however, this approach is hampered by its requirement of strongly basic conditions, which may not be suitable for all substrates. In addition, these methods may also require access to activated and/or stereodefined olefins.
Given our recent report of the concise synthesis of orthogonally protected β-MeCys and β-MeLan,22 we envisioned the direct synthesis of this AviMeCys unit via decarbonylation of an activated lanthionine precursor (Scheme 1). In continuation of our efforts towards the chemical synthesis of mersacidin, we now wish to report our advances in this area, specifically the expeditious synthesis of the AviMeCys unit directly from the β-MeLan unit.23
Scheme 1.

Retrosynthetic analysis of the enamide unit.
The concept of a decarbonylative pathway to enamides is not novel, although its documentation is limited,24,25,26 probably because it has traditionally been viewed as an undesirable side reaction. As a result, we took a systematic approach for optimization of this strategy.27 undesirable side reaction. As a result, we took a systematic approach for optimization of this strategy.27
Our initial approach consisted of evaluating amino acid chlorides in light of Bubner’s observation that an enamide byproduct was isolated during the attempted Stille cross-coupling of Fmoc-Pro-Cl.24,28 Like Bubner, we also observed the lack of enamide formation in the absence of an organostannane. In search of additives that could enhance reactivity, we found that nBu4I was very efficient at promoting the catalytic decarbonylation of Fmoc-Pro-Cl, while only traces of the desired product were observed with NaI, presumably an artifact of its low solubility in organic solvents. The efficiency of this process is highlighted by the observation that Fmoc-Pro-OH could be activated in situ with diphenylphosphoryl chloride to give high yields of the pyrroline product. Unfortunately, when these conditions were applied to the synthesis of cysteine-derived enamides, only the enamine byproduct arising from β-elimination (i.e., desulfurization) was observed. CuI was added in an effort to sequester thiol byproducts and suppress the potential for poisoning of Pd catalyst. However, this only increased the amount of the elimination byproduct that was generated. Furthermore, only decomposition was observed in the attempted decarbonylation of Fmoc-Ser(OBn)-Cl and Fmoc-Val-Cl.
The isolation of dehydroalanine as a byproduct from decarbonylation reactions with cysteine-derived acid chlorides, coupled with the lack of generality of this process, prompted us to examine milder activating groups for the amino acid derived esters. Although a range of common activating groups, including −OSu, −OCOR, −OOBt, −OAt and −OPy gave only traces of product at best, the 2-pyridyl thioester showed promise. While trace amounts of the desired enamide were observed under Mukaiyama’s conditions,25 added DPEPhos as a ligand with Ni(COD)2 as the pre-catalyst yielded 15% of the vinyl sulfide along with 37% of the product resulting from sulfur abstraction.
Even more promising was thioester activation,29 which allowed for the efficient decarbonylation of Fmoc-Pro-SPh (4j). When these conditions were applied to Fmoc-Cys(Me)-SPh, high yields of the decarbonylation product were observed, albeit as the trapped thioaminal, along with reduced amounts of the undesired desulfurization by-product.
Inspired by Liebeskind’s work on the Cu(I) carboxylate activation of thioesters,23 we sought to limit the propensity for SPh deinsertion by taking advantage of the inherent thiophilicity of Cu(I). Although simple salts, such as Cu(I) and Cu(OAc) showed limited activity, high quantities of the desired enamide product 5 were isolated, along with a significant amount of the sulfur-abstraction product, when copper(I) 3-methylsalicylate (CuMeSal) was used. Similar yields were observed with an SiPr ester suggesting that steric hindrance may not play a significant role in the transition state leading to thioaminal formation. Under optimized conditions for decarbonylation of Fmoc-Cys-(Me)-SPh, thioaminal formation was kept to a minimum through use of PPh3 as ligand, providing the desired aminovinyl sulfide in 91% yield after 1 h at room temperature.
With these promising results in hand, we proceeded to examine the substrate scope as summarized in Table 1. We were pleased to find that Pd[P(OEt)3]4 efficiently catalyzed the formation of conjugated enamides (entries 1-9).
Table 1.
Scope and Limitations of the Decarbonylative Synthesis of Enamides from N-protected Amino Acids
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| entrya | amino acid thiolester | MLnb | CuX | t(°C) | Time (h) | product | yieldc | E:Z |
| 1 | Fmoc-Phe-SPh (4a) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |
|
90% | 7.1:1 |
| 2 | Cbz-Phe-SPh (4b) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
82% | 4.9:1 |
| 3 | Cbz-Phe-SPh (4c)d | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 | 77% | 7:1 | |
| 4 | Boc-Phe-SPh (4d) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |
|
92% | 5.6:1 |
| 5 | Phth-Phe-SPh (4e) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
33% | E only |
| 6 | Phth-Phe-SPh (4e) | 120% Ni(PPh3)4 | CuTC | rt | 22 | 94% | E only | |
| 7 | Fmoc-Trp(Boc)-SPh (4f) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
81% | 1:1.4 |
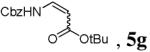
| 8 | Cbz-Asp(OtBu)-SPh (4g) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |
|
82% | 1:3.7 |
| 9 | Fmoc-Asp(SPh)-OMe(4h) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
trace | --- |
| 10 | Fmoc-Asp(SPh)-OMe(4h) | 120% Ni(PPh3)4 | CuTC | rt | 22 | 93% | --- | |
| 11 | Cbz-Pro-SPh (4i) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
33% | --- |
| 12 | Fmoc-Pro-SPh (4j) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
41% | --- |
| 13 | Fmoc-Pro-SPh (4j) | 120% Ni(PPh3)4 | CuTC | 50 | 5 | 89% | --- | |
| 14 | Tos-Pro-SPh (4k) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |
|
46%(68%)e | --- |
| 15 | Boc-Ser(Bn)-SPh (4l) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
42% | Z only |
| 16 | Fmoc-Cys(Me)-SPh (4m) | 120% Ni(PPh3)4 | CuMeSal | rt | 1 |
|
91% | Z only |
| 17 | Fmoc-Cys(tBu)-SPh (4n) | 120% Ni(PPh3)4 | CuMeSal | 50 | 3 |
|
53% | Z only |
| 18 | Fmoc-Cys(Trt)-SPh (4o) | 120% Ni(PPh3)4 | CuMeSal | 50 | 5 |
|
0% | --- |
| 19 | Fmoc-Val-SPh (4p) | 15% Pd[P(OEt)3]4 | CuMeSal | 50 | 3 |

|
trace | --- |
| 20 | Fmoc-Val-SPh (4p) | 120% Ni(PPh3)4 | CuTC | 50 | 5 | 73% | --- | |
| 21 | Fmoc-Ile-SPh (4q) | 120% Ni(PPh3)4 | CuTC | 50 | 5 |
|
67% | 1.2:1 |
| 22 | Boc-a-Me-Val-SPh (4r) | 120% Ni(PPh3)4 | CuTC | 50 | 5 |

|
0% | --- |
| 23 | Fmoc-β-Ala-SPh (4s) | 120% Ni(PPh3)4 | CuTC | rt | 22 |
|
82% | --- |
| 24 | FmocNHPh- CH2CH2CH2COSPh(4t) |
120% Ni(PPh3)4 | CuMeSal | rt | 20 |
|
94%f | --- |
| 25 | BocNH(CH2)10COSPh (4u) | 120% Ni(PPh3)4 | CuTC | rt | 22 |
|
95% | --- |
| 26 | Boc-Ala-Phe-SPh (4v) | 120% Ni(PPh3)4 | CuMeSal | rt | 12 |

|
89% | 17:1 |
| 27 | Fmoc-Phe-Cys(Me)-SPh (4w) |
120% Ni(PPh3)4 | CuTC | 35 | 18 |

|
74% | Z only |
For reaction conditions see Supporting Information.
Prepared from either Ni(COD)2 and PPh3 or Pd(OAc)2 and P(OEt)3.
Isolated yields.
Prepared in situ with DCC/PhSH.
Based on recovered starting material.
Mixture of isomers (External:Internal (E):Internal (Z) = 10:5.8:0.1).
However, moderate yields were observed with amino acids such as Pro (entries 11-14) and Ser (entry 15). In spite of near quantitative recovery of starting material, enamide yields plateaued to a similar level regardless of whether the reaction was run at or above room temperature. Moreover, added CuMeSal did not have any positive effect on the yield, while only slightly increased yields were observed when more Pd catalyst was added to the reaction mixture.
In most instances, the Ni-based approach proved to be superior and, in general, yielded enamides in good to excellent yields; some reactions proceeded very efficiently at room temperature (entries 23-27). This is further highlighted by the contrast between the good yields obtained for the Asp and Val-derived enamides with Ni that were unattainable with the use of Pd catalyst (entries 9, 10, 19, 20). To the best of our knowledge, these conditions provide a route to enamides under non-oxidative and non-basic conditions not reported previously. We did observe, however, that steric hindrance in the substrate may be a limiting factor for successful application of this methodology (c.f., entries 18 and 22).
A series of thioesters (4s-u) were prepared in order to assess the relationship between the proximity of the protected nitrogen to the thioester and the overall efficiency of the decarbonylation reaction. The data reveal the possibility of a modest trend toward improved yields with increasing distance between the two functional groups. In general, excellent yields of alkene products were observed with simple alkyl thioesters (entries 23 and 24), an indication that this approach may be general for the synthesis of alkenes from thioester precursors.
The decarbonylation of dipeptide substrate 4w returned an excellent yield of the desired (Z)-enamide. This result was critically dependent on the choice of Cu(I) salt and ligand (see Table S2 in the Supporting Information). While reactions utilizing copper(I) thiophene-2-carboxylate (CuTC) reproducibly returned good yields of the desired product, reactions employing CuMeSal resulted in isolation of an alkene byproduct arising from a desulfurization process.
Product selectively (i.e., enamide vs. desulfurization) was also strongly influenced by choice of ligand. Reactions employing PPh3 provided the desired enamide in good yields (CuTC additive) while reactions using (o-Tol)3P provide the desulfurization product exclusively. Bulky, as well as sigma-donating, ligands also returned higher yields of the desulfurization byproduct.
Under the optimized conditions, we were pleased to find that lanthionine 6 could be decarbonylated smoothly to yield aminovinyl cysteine 7 in 75% yield (Scheme 2). More importantly, the synthetically more relevant subunit 2 could be prepared in satisfactory yields directly from lanthionine 8 and can be used directly for the synthesis of mersacidin.
Scheme 2.

Synthesis of the AviMeCys unit of mersacidin.
In conclusion, we have developed a mild alternative to the synthesis of enamides that makes use of readily available amino acid starting materials. This has allowed efficient access to the AviMeCys fragment of mersacidin. Future work directed toward the total synthesis of mersacidin as well as attempts to determine the function of the AviMeCys subunit will be presented in due course.
Supplementary Material
Figure 1.

Structures of bioactive peptides incorporating the aminovinyl cysteine subunit.
Acknowledgment
Financial support provided by the National Institutes of Health (AI 059327) and Indiana University is gratefully acknowledged. P.G.-R thanks Eli Lilly and Company for fellowship support. We also thank Professor Lanny Liebeskind (Emory University) and Professor Kenneth Caulton (Indiana University) for helpful discussions.
Footnotes
Supporting Information Available: Experimental details and spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1).Narayan RS, VanNieuwenhze MS. Eur. J. Org. Chem. 2007;2007:1399. doi: 10.1002/ejoc.200600750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chatterjee S, Chatterjee DK, Jani RH, Blumbach J, Ganguli BN, Klesel N, Limbert M, Seibert GJ. Antibiot. 1992;45:832. doi: 10.7164/antibiotics.45.839. [DOI] [PubMed] [Google Scholar]
- (3).For recent reviews on lantibiotics, see: Willey JM, van der Donk WA. Annu. Rev. Microbiol. 2007;61:477–501. doi: 10.1146/annurev.micro.61.080706.093501. Chatterjee C, Paul M, Xie L, van der Donk WA. Chem. Rev. 2005;105:633–683. doi: 10.1021/cr030105v.
- (4).Allgaier H, Jung G, Werner RG, Schneider U, Zähner H. Angew Chem. Int. Ed. 1985;24:1051. [Google Scholar]
- (5).Hayakawa Y, Sasaki K, Adachi H, Furihata K, Nagai K, Shin-ya K. J Antibiot. 2006;59:1. doi: 10.1038/ja.2006.1. [DOI] [PubMed] [Google Scholar]
- (6).Castiglione F, Lazzarini A, Carrano L, Corti E, Ciciliato I, Gastaldo L, Candiani P, Losi D, Marinelli F, Selva E, Parenti F. Chemistry & Biology. 2008;15:22. doi: 10.1016/j.chembiol.2007.11.009. [DOI] [PubMed] [Google Scholar]
- (7).For a recent review of AviCys-containing lantibiotics, see: Sit CS, Yoganathan S, Vederas JC. Acc. Chem. Res. 2011;44:261–268. doi: 10.1021/ar1001395.
- (8).Yet L. Chem. Rev. 2003;103:4283. doi: 10.1021/cr030035s. [DOI] [PubMed] [Google Scholar]
- (9).Wu Y, Liao X, Wang R, Xie X-S, De Brabander JK. J. Am. Chem. Soc. 2002;124:3245. doi: 10.1021/ja0177713. [DOI] [PubMed] [Google Scholar]
- (10).Carbery DR. Organic & Biomolecular Chemistry. 2008;6:3455. doi: 10.1039/b809319a. [DOI] [PubMed] [Google Scholar]
- (11).Matsubara R, Kobayashi S. Acc. Chem. Res. 2008;41:292. doi: 10.1021/ar700098d. [DOI] [PubMed] [Google Scholar]
- (12).Kuramochi K, Osada Y, Kitahara T. Tetrahedron. 2003;59:9447. [Google Scholar]
- (13).Furstner A, Brehm C, Cancho-Grande Y. Org. Lett. 2001;3:3955. doi: 10.1021/ol016848p. [DOI] [PubMed] [Google Scholar]
- (14).Brice JL, Meerdink JE, Stahl SS. Org. Lett. 2004;6:1845. doi: 10.1021/ol0494360. [DOI] [PubMed] [Google Scholar]
- (15).Buissonneaud D, Cintrat J-C. Tetrahedron Lett. 2006;47:3139. [Google Scholar]
- (16).Wang X, Porco JA. J. Org. Chem. 2001;66:8215. doi: 10.1021/jo0158027. [DOI] [PubMed] [Google Scholar]
- (17).Banerjee B, Litvinov DN, Kang J, Bettale JD, Castle SL. Org. Lett. 2010;12:2650. doi: 10.1021/ol1008679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dehli JR, Legros J, Bolm C. Chem. Commun. 2005;973 doi: 10.1039/b415954c. [DOI] [PubMed] [Google Scholar]
- (19).Wallace DJ, Klauber DJ, Chen C.-y., Volante RP. Org. Lett. 2003;5:4749. doi: 10.1021/ol035959g. [DOI] [PubMed] [Google Scholar]
- (20).Cesati RR, Dwyer G, Jones RC, Hayes MP, Yalamanchili P, Casebier DS. Org. Lett. 2007;9:5617. doi: 10.1021/ol7025729. [DOI] [PubMed] [Google Scholar]
- (21).Yuri B, Robert AB. Angewandte Chemie. 2008;120:2139. [Google Scholar]
- (22).Narayan RS, VanNieuwenhze MS. Org. Lett. 2005;7:2655. doi: 10.1021/ol0507930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).For a review on recent developments in lantibiotic synthesis, see: Tabor AB. Org. Biomol. Chem. 2011;9:7606–7628. doi: 10.1039/c1ob05946g.
- (24).Crisp GT, Bubner TP. Synth. Commun. 1990;20:1665. [Google Scholar]
- (25).Goto T, Onaka M, Mukaiyama T. Chem. Lett. 1980;9:709. [Google Scholar]
- (26).Yang H, Li H, Wittenberg R, Egi M, Huang W, Liebeskind LS. J. Am. Chem. Soc. 2007;129:1132. doi: 10.1021/ja0658719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Min GK, Hernández D, Lindhardt AT, Skrysdstrup T. Org. Lett. 2010;12:4716–4719. doi: 10.1021/ol101620r. [DOI] [PubMed] [Google Scholar]
- (28).We note that reference 27, highlighting a qualitatively analogous approach using palladium catalysis, appeared during the course of our study. Our method requires lower reaction temperatures (rt with most nickel catalyst systems) and utilized simple phosphine ligands. In addition, we also observed the desired (Z)-selectivity for cysteine- and serine-based substrates in contrast to the results reported in reference 27.
- (29).Wenkert E, Chianelli DJ. Chem. Soc., Chem. Commun. 1991;627 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.