

Abstract
Nuclear factor erythroid-derived factor 2-related factor 2 (Nrf2) is a cap-n-collar basic leucine zipper transcription factor that is involved in the cellular adaptive response to oxidative stress. Our previous study reported that targeted disruption of the Nrf2 gene in mice decreases adipose tissue mass and protects against obesity induced by a high-fat diet. Deficiency of Nrf2 in preadipocytes and mouse embryonic fibroblasts led to impaired adipogenesis. Consistent with these findings, the current study found that lack of Nrf2 in primary cultured mouse preadipocytes and 3T3-L1 cells hampered adipogenic differentiation induced by hormonal cocktails. Stable knockdown of Nrf2 in 3T3-L1 cells blocked the enhanced adipogenesis caused by deficiency of kelch-like ECH-associated protein 1 (Keap1), a Cul3-adapter protein that allows for Nrf2 to be ubiquinated and degraded by the 26S protesome complex. In addition, increased production of reactive oxygen species (ROS) and activation of Nrf2 occurred at the very early stage upon adipogenic hormonal challenge in 3T3-L1 cells, followed by an immediate induction of CCAAT/enhancer-binding protein β (C/EBPβ). Knockdown of Nrf2 led to reduced expression of C/EBPβ induced by adipogenic hormonal cocktails, chemical Nrf2 activators or Keap1 silencing. Cebpβ promoter-driven reporter assays and chromatin immunoprecipitation suggested that Nrf2 associates with a consensus antioxidant response element (ARE) binding site in the promoter of the Cebpβ gene during adipogenesis and upregulates its expression. These findings demonstrate a novel role of Nrf2 beyond xenobiotic detoxification and antioxidant response, and suggest that Nrf2 is one of the transcription factors that control the early events of adipogenesis by regulating expression of Cebpβ.
Keywords: Nrf2, C/EBPβ, Adipogenesis
Introduction
Adipogenesis (differentiation of adipogenic precursor cells, so called preadipocytes, into adipocytes) is regulated by a complex network of transcription factors that coordinate expression of hundreds of proteins responsible for establishing the mature fat-cell phenotype [1-4]. While it was firmly established that peroxisome proliferator-activated receptor γ (PPARγ) and CCAAT/enhancer-binding protein α (C/EBPα) are the terminal factors responsible for adipogenesis, the signaling cascade underlying the differentiation process, especially the early events, are only partially resolved.
C/EBPs, including C/EBPα, C/EBPβ, C/EBPγ, C/EBPδ, C/EBPε and C/EBPζ (also known as CHOP10), belong to basic region/leucine zipper (bZIP) transcription factors and are expressed in adipocytes [2]. C/EBPβ and C/EBPδ are transiently expressed and function at an early stage of differentiation by sensing adipogenic stimuli and initiating expression of PPARγ and C/EBPα [5, 6]. C/EBPα and PPARγ, forming a positive feedback loop by activating each other's expression, play roles at a later stage by inducing and maintaining expression of adipocyte-specific genes [7, 8]. Although the forced expression of C/EBPα in fibroblasts can trigger adipogenic differentiation, C/EBPα is incapable of inducing adipogenesis in the absence of PPARγ [9]. In contrast, PPARγ can induce adipogenic differentiation in C/EBPα-null cells indicating that PPARγ is sufficient in effecting adipogenesis [10]. C/EBPβ is thought to initiate mitotic clonal expansion of preadipocytes and to later coordinate the transcription network by turning on C/EBPα and PPARγ [11]. The induction of C/EBPβ occurs rapidly (within 2-4 h) on stimulation of differentiation [12], whereas the acquisition of DNA binding activity by C/EBPβ requires various posttranslational modifications, including phosphorylation, acetylation, disulfide bond formation and dimerization [11, 13-15]. Apart from cAMP response element-binding protein (CREB) [16], early growth response 2 (EGR2, also known as KROX20) [17] and Kruppel-like factor 4 (KLF4) [18], relatively few transcription factors have been identified that bind to the C/EBPβ promoter and positively regulate its transcription during adipogenesis. Other C/EBP isoforms, including C/EBPγ and CHOP10, seem to suppress adipogenesis [19].
Nuclear factor erythroid-derived factor 2-related factor 2 (Nrf2) belongs to the cap-n-collar subfamily of bZIP transcription factors [20], which also includes Nrf1 [21], Nrf3 [22] and the NF-E2 p45 subunit [23], as well as the more distantly related factor BTB and CNC homology 1 (BACH1) and BACH2 [24]. In response to oxidative/electrophilic stress Nrf2 heterodimerizes with small Maf proteins, as well as other bZIP proteins, bind to cis-acting element(s) termed antioxidant response elements (AREs) in the promoters of target genes [25], inducing transcriptional responses. ARE-dependent genes encode various enzymes, including detoxification enzymes such as glutathione S-transferases (GSTs), NAD(P)H quinone oxidoreductase 1 (NQO1), and heme oxygenase 1 (HO-1), as well as antioxidant enzymes such as catalase (CAT), sulfiredoxin (SRX), γ-glutamate cysteine ligase catalytic subunit (GCLC) and regulatory subunit (GCLM). Nrf2 therefore plays a central role in the cellular adaptive response to oxidative stress. Although Nrf2 is abundantly expressed in adipose tissue, its function in adipocyte biology has not been fully explored.
Our previous study [26] showed that mice deficient in Nrf2 display decreased fat mass in association with small adipocytes and are resistant to high-fat diet (HFD)-induced obesity. Mouse embryonic fibroblasts (MEFs) deficient in Nrf2 have impaired adipogenesis, and in 3T3-L1 and human subcutaneous preadipocytes, knockdown of Nrf2 expression inhibits adipogenic differentiation. Conversely, stable knockdown of kelch-like ECH-associated protein 1 (Keap1), which is a Cul3-adapter protein that allows for Nrf2 to be ubiquinated and degraded by the 26S protesome complex [27], leads to accelerated and enhanced adipogenesis in 3T3-L1 cells. In addition, we showed that the impaired adipogenesis induced by knockout or knockdown of Nrf2 is related at least in part to down-regulation of PPARγ expression. The current study explores the mechanisms further and reveals that Nrf2 is involved in transcriptional regulation of C/EBPβ expression during adipogenesis. These findings demonstrate a novel biologic role of Nrf2 beyond its participation in detoxification and antioxidant pathways, and suggest Nrf2 is one of the transcription factors that control the early events of adipogenesis by regulating expression of Cebpβ.
Materials and Methods
Reagents
Insulin solution (human, I9278), 3-isobutyl-1-methylxanthine (IBMX, I7018), dexamethasone (D1756), indomethacin (I7378), sulforaphane (SFN, S6317), tert-butylhydroquinone (tBHQ, #19986), sodium arsenite (iAs, #71287), Oil-red O (ORO, #75087), N-acetyl-L-cysteine (NAC, A7250), glutathione reduced ethyl ester (GSH-EE, G1404), diphenyleneiodonium (DPI, D2926), mesoxalonitrile 4-trifluoromethoxyphenylhydrazone (FCCP, C2920), diethyl maleate (DEM, W505005) and puromycin (P8833) were obtained from Sigma (St. Louis, MO). Rosiglitazone maleate was from SmithKline Beecham Pharmaceuticals (London, UK). Culture media, calf serum (CS), fetal bovine serum (FBS) and supplements were purchased from Invitrogen (Carlsbad, CA).
Animals
Nrf2-knockout (Nrf2-/-) mice was developed as described previously [28] and backcrossed onto C57BL/6J background for 10 generations using alternating male and female stock mice from The Jackson Laboratories (JAX Stock No. 000664). For experiments, age-matched (12-16 weeks old) mice were used for preadipocyte isolation. Animals were housed in virus-free facilities on a 12-h light/12-h dark cycle and were fed standard rodent food. Genotyping was performed by PCR using genomic DNA isolated from tail snips. All protocols for animal use were approved by the Institutional Animal Care and Use Committee of The Hamner Institutes and were in accordance with National Institutes of Health guidelines.
Cell culture and differentiation
Mouse primary preadipocytes were isolated from WAT as described previously [29] and cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 8.3 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. 3T3-L1 preadipocytes were obtained from ATCC (Manassas, VA) and maintained in high-glucose DMEM with 50 unit/ml penicillin, 50 μg/ml streptomycin, and 10% CS. Mouse primary preadipocytes and 3T3-L1 cells were differentiated 1 day after they became confluent (designated as day 0) using DMI or DMIRI protocols as detailed in Figure 1. DMI protocol, cells were differentiated by replacing CS growth medium with DMI differentiation medium containing 1 μM dexamethasone, 0.5 mM IBMX and 5 μg/ml insulin in DMEM with 10% FBS. After 48 h the medium was changed to DMEM with 10% FBS and 1 μg/ml insulin (Insulin medium). The cells were maintained for an additional 5 days and the medium was refreshed every two days; DMIRI protocol, cells were differentiated by replacing growth medium with DMIRI differentiation medium containing 1 μM dexamethasone, 0.5 mM IBMX, 5 μg/ml insulin, 1 μM rosiglitazone and 125 μM indomethacin in DMEM with 10% FBS. After 72 h, cells were maintained for an additional 2-4 days in the same medium without additives but 10% FBS, and fresh medium was replenished every 2 days. Differentiation of preadipocytes to mature adipocytes was confirmed by ORO staining of lipid vesicles as detailed previously [26]. All the cells were maintained at 37 °C in a 5% CO2 environment.
Figure 1.

Adipogenic differentiation protocols for mouse primary preadipocytes and 3T3-L1 cells. Cells were differentiated 1 day after confluence (designated as day 0) by replacing growth medium with differentiation medium as indicated (Black arrows). CS, DMEM with 10% calf serum; DMI: 1 μM dexamethasone, 0.5 mM IBMX and 5 μg/ml insulin in DMEM with 10% FBS; Insulin medium, 1 μg/ml insulin in DMEM with 10% FBS; DMIRI: DMI + 1 μM rosiglitazone and 125 μM indomethacin in DMEM with 10% FBS; FBS medium, DMEM with 10% FBS.
Lentiviral-based shRNA Transduction
MISSION shRNA lentiviral vectors were obtained from Sigma. The packaging of lentiviral particles was performed based on manufacturer's protocol. Lentiviral transduction of 3T3-L1 cells with particles for shRNA targeting Nrf2 (SHVRS-NM_010902), Keap1 (SHVRS_016679) or non-target negative control (SHC002V) was performed as described previously [26]. The non-target shRNA control vector activates RNA-induced silencing complex and RNA interference pathway, but does not silence any human or mouse genes. In the current study, we define the cells transduced with the non-target shRNA control vector as Scramble (Scr) cells.
Measurement of intracellular glutathione (GSH)
Cells were sonicated in cold PBS immediately after collection followed by centrifugation at 12,000 × g for 5 min. The resulting supernatants were used for measurement of GSH using BIOXYTECH GSH/GSSG-412 kit (OxisResearch, Portland, OR) [30].
Quantitative real-time RT–PCR
Total RNA was isolated with TRIzol (Invitrogen) according to manufacturer's instructions and then subjected to cleanup using RNase-Free DNase Set and RNeasy Mini kit (Qiagen, Valencia, CA). The resultant DNA-free RNA was diluted in RNase-free H2O and quantified by Nanodrop (Thermo, Wilmington, DE) at 260 nm. RNA samples were stored at −80 °C until use. Total RNA was reverse transcribed with MuLV reverse transcriptase and Oligo d(T) primers (Applied Biosystems, Foster City, CA). The SYBR Green PCR Kit (Qiagen) was used for quantitative real-time RT-PCR analysis. The primers were designed using Primer Express (Applied Biosystems) and synthesized by MWG-BIOTECH Inc. (High Point, NC). The primer sequences are listed in Supplementary Table S1. Relative differences in gene expression between groups were determined from cycle time (Ct) values. These values were first normalized to 18S in the same sample (ΔCt) and expressed as fold-change over control (2−ΔΔCt). Realtime fluorescence detection was carried out using an ABI PRISM 7900 HT Sequence Detector (Applied Biosystems).
Western blot analysis
Isolation of cell fractions and Western blotting were performed as detailed previously [31, 32]. Antibodies for Nrf2 (sc-13032; 1:500), Keap1 (sc-15246; 1:500), C/EBPβ (sc-7962; 1:500) and MafF/G/K (sc-22831; 1:500) were from Santa Cruz, Inc. (Santa Cruz, CA). Antibodies for C/EBPδ (#2318; 1:1000), PPARγ (#2435; 1:1000), CREB (#9197; 1:1000), p-CREB (#9198; 1:1000), c-Jun (#9165; 1:1000), c-Fos (#2250; 1:1000) and CHOP10 (#2895; 1:1000), p-AKT (#9271; 1:1000) and AKT (#4685; 1:1000) were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Antibodies for HO1 (H4535; 1:500), LAMIN A (L1293; 1:2500) and β-ACTIN (A1978; 1:2000) were purchased from Sigma. Antibody for GCLC (RB-1697; 1:800) was from Thermo Fisher Scientific Inc. (Pittsburg, PA). Antibody for KLF5 (ab24331; 1:1000) was from Abcam (Cambridge, UK). Antibody for KROX20 (PRB-236p; 1:1000) was from Covance, Inc. (Princeton, NJ). The molecular weight (MW) of each protein shown on immunoblot was estimated based on the MagicMark™ XP Western Protein Standard (Invitrogen) on 4-12% or 12% Tris-Glycine Gel (Invitrogen).
Constructs and mutations
Serial 5′-deleted Cebpβ promoter-driven luciferase reporters were designed as described previously [16]. -1036 bp, -556 bp and -218 bp inserts were amplified by PCR using mouse (C57BL/6J) genomic DNA as template and the EcoRV- and HindIII-ended primers (-1036 bp forward: 5′-tgtaacactGATATCcacctaggagtggca gaa-3′; -556 bp forward: 5′-tgtaacactGATATCgttctgcttcccaggagttg-3′; -218 bp forward: 5′-tgtaacact GATATCtcagcctcccgcgcgagga-3′; Common reverse: 5′-aaacggtaaAAGCTTggctccgctgcgtcccgg-3′; see Online Materials Figure S1). -116 bp and -m116 constructs ended with EcoRV/HindIII sites were synthesized by MWG-BIOTECH Inc. In the -m116 insert, the core “GC” site of the ARE was replaced with “AA”. These promoter sequences were digested with EcoRV/HindIII and ligated into EcoRV/HindIII-digested pGL4.10[luc2] vector (Promega, Madison, WI).
ARE-luciferase reporter assay
Cignal Lenti ARE reporter, which expresses a luciferase gene driven by multiple ARE (TCACAGTGACTCAGCAAAATT) repeats, was obtained from SABiosciences (Frederick, MD). Lentiviral transduction of 3T3-L1 cells was performed as described previously [31]. Cells were grown to ∼90% confluency and sub-cultured in medium containing 1.0 μg/ml of puromycin. The luciferase activity was measured by Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. The luciferase activity was normalized to cell viability which was determined using a Non-Radioactive Cell-Proliferation Assay Kit (Promega).
Intracellular ROS determination
Intracellular ROS levels in 3T3-L1 cells were measured by flow cytometry (Becton Dickinson FACSCanto II, Becton Dickinson, San Jose, CA) using the fluorescent probe 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Invitrogen) as described previously [33]. CM-H2DCFDA reacts with a variety of cellular oxidants including nitric oxide, peroxynitrite, and hypochloride in addition to H2O2. The final concentration of CM-H2DCFDA used was 4 μM and loading time was 30 min.
Chromatin immunoprecipitation (ChIP) assay
ChIP analyses were performed using the EZ ChIP kit (Upstate Biotechnology, Temecula, CA) according to the manufacture's protocol. In brief, 3T3-L1 cells were treated with DMI for 2 h, after which cells were cross-linked with 1% formaldehyde for 10 min at 37 °C. After washing twice with ice-cold PBS containing protease inhibitors, scraping and centrifugation, cell pellets were resuspended in SDS lysis buffer. After incubation for 5 min on ice, the cell lysates were sonicated 6 times with each time being 10 s using the XL-2000 Ultrasonic liquid Processor (Misonix Inc., Farmingdale, NY). Cell debris was removed by centrifugation for 10 min at 12,000 rpm. The supernatant was diluted 10 times in ChIP dilution buffer followed by preclearing with 60 μl of protein G-agarose beads for 1 h at 4 °C with rotation. A portion of the precleared lysate (25μl) was preserved for input, and the left was immunoprecipitated using antibodies for non-specific IgG and anti-Nrf2 (sc-13032, Santa Cruz Inc.) overnight at 4 °C with rotation. Immune complexes were recovered by the addition of 60 μl of protein G-agarose beads for 1 h at 4 °C with rotation. Agarose beads were pelleted by gentle centrifugation (1,000 rpm at 4 °C). The beads were sequentially washed with low and high salt buffer, LiCl buffer, and finally twice with TE buffer. To reverse the cross-linking of DNA, 8 μl of 5 M NaCl were added and incubated overnight at 65 °C. After treatment with proteinase K for 1 h at 45 °C, DNA was recovered using spin columns. The DNA pellets were resuspended in 50 μl of elution buffer. PCR amplification was carried out for 40 cycles with 5 μl of sample DNA solution, and PCR products were separated on 3% agarose gels in 1× TAE buffer. Two primers were used to amplify the segment flanking the ARE site (-115 bp ∼ -107 bp) on Cebpβ promoter with forward primer 5′-GTGGCCGGGCAATGA-3′ and reverse primer 5′-GGCTCCGCTGCGTCCCGGTC-3′, which generate a 155 bp product.
Statistical analyses
All statistical analyses were performed using Graphpad Prism 4 (GraphPad Software, San Diego, CA), with p < 0.05 considered as significant. More specific indices of statistical significance are indicated in individual figure legends. Data are expressed as mean ± SEM. For comparisons between two groups, a Student's t-test was performed. For comparisons among groups, one-way or two-way ANOVA with Bonferroni post hoc testing was performed.
Results
Deficiency of Nrf2 impairs adipogenic differentiation in mouse preadipocytes
To determine the regulatory role of Nrf2 in adipogenesis, preadipocytes derived from white adipose tissues (WAT) of Nrf2-knockout (Nrf2-/-) and wildtype (Nrf2+/+) mice were isolated, cultured and differentiated in vitro. As shown in Figure 2A, on the day 5 of hormonal cocktail DMIRI-induced adipogenesis Nrf2-/- preadipocytes showed substantially reduced levels of lipid accumulation, revealing lack of Nrf2 expression impairs adipogenic differentiation in preadipocytes. Consistent with the reduction in lipid accumulation, Nrf2-/- preadipocytes exhibited significantly decreased expression of adipogenic genes, including Pparγ1, Pparγ2, adiposin and adipocyte fatty acid-binding protein 4 (Fabp4) (Figure 2B).
Figure 2.
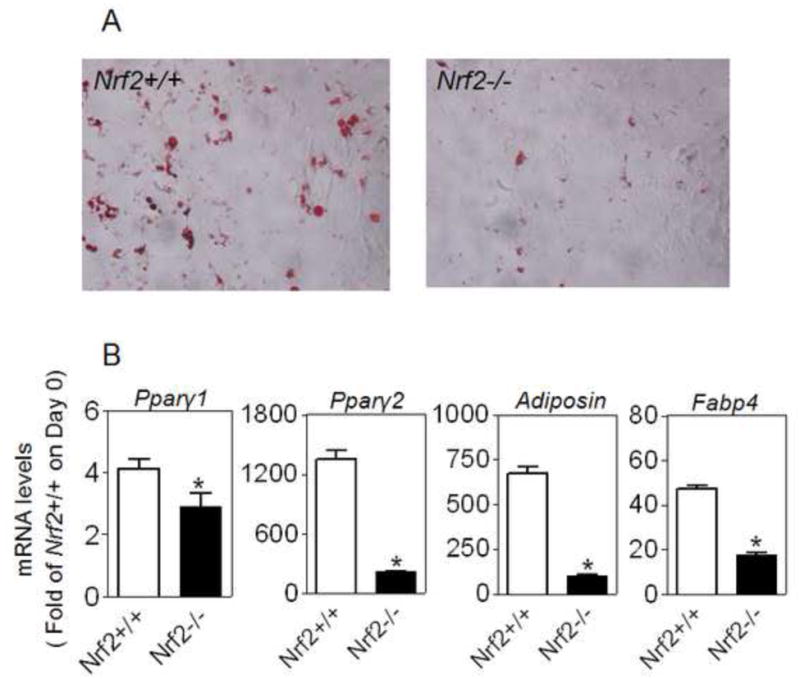
Ablation of Nrf2 gene results in reduced adipogenesis in mouse WAT-derived preadipocytes. Preadipocytes isolated from WAT of Nrf2-/- and Nrf2+/+ mice were cultured to confluency and differentiated for 5 days using DMIRI protocol. Cells were then stained with ORO staining to visualize lipid accumulation (A) or used to measure the expression of adipogenic genes (B). n = 3; *p < 0.05 vs Nrf2+/+.
Our previous study [26] indicated that knockdown of Keap1 in 3T3-L1 cells results in activation of Nrf2 and an enhanced DMIRI-induced adipogenesis. Although Keap1 is a well known negative regulator for activation of Nrf2, it is still possible that the enhanced adipogenesis caused by knockdown of Keap1 may be a result independent of Nrf2. To rule out this possibility, we examined the effect of Nrf2- and Keap1-double knockdown on adipogenesis. As shown in Figure 3A-D, 3T3-L1 cells transduced with Nrf2-shRNA showed efficient silencing effect on Nrf2 expression under basal or iAs-challenged condition, whereas Keap1-KD cells exhibited reduced Keap1 expression and enhanced Nrf2 protein level in comparison to Scramble cells. In contrast, the cells with Nrf2- and Keap1-double knockdown showed substantially reduced Nrf2 and Keap1 expression. Of note, reduction of Keap1 expression in Nrf2-KD cells suggests a feedback regulatory loop between Nrf2 and Keap1 may exist. Consistent with our previous report [26], ORO staining demonstrated reduced lipid accumulation in Nrf2-KD cells, whereas Keap1-KD cells showed increased adipogenesis (Figure 3E). As expected, the enhanced DMIRI-induced adipogenesis of Keap1-KD cells was totally blocked by knockdown of Nrf2 (Figure 3E), confirming that the enhanced adipogenesis in Keap1-KD cells resulted from the activation of Nrf2. It should be noted that no spontaneous adipogenesis was observed in Keap1-KD cells. In addition, PPARγ ligand rosiglitazone alone cannot significantly increase the adipogenesis in Keap1-KD cells (not shown), suggesting that the potentially enhanced expression of PPARγ by Keap1 knockdown, as reported previously [26], is not sufficient for adipogenic differentiation.
Figure 3.
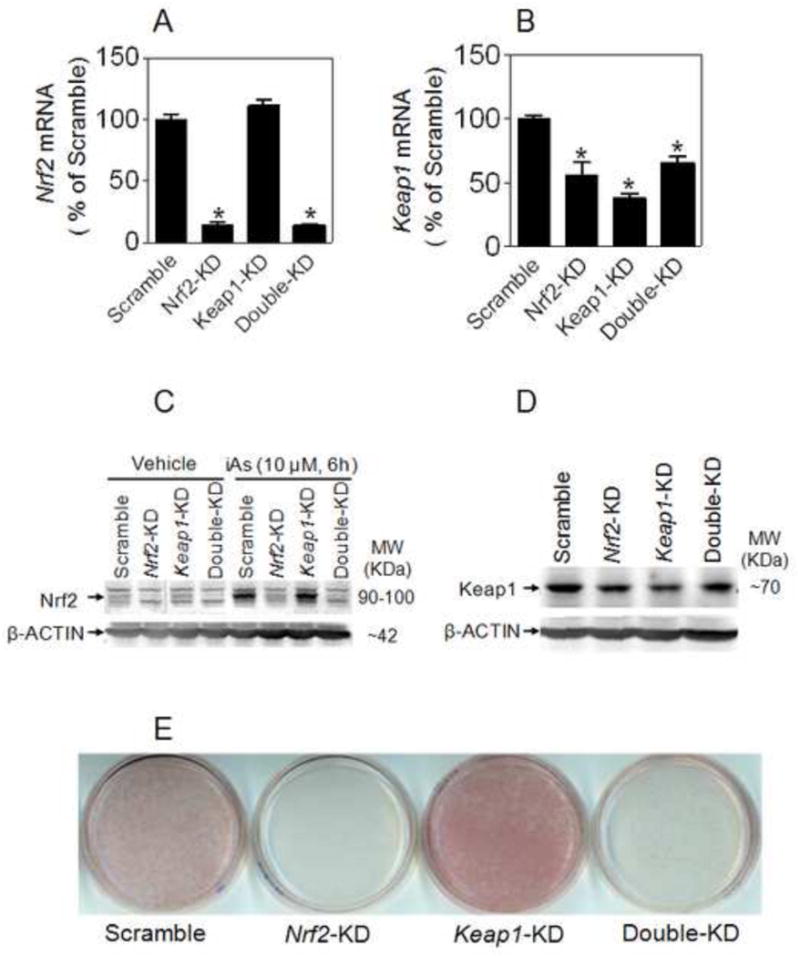
Effect of knockdown of Nrf2 and/or Keap1 on adipogenesis in 3T3-L1 preadipocytes. mRNA expression of Nrf2 (A) and Keap1 (B) in 3T3-L1 cells transduced with shRNA lentivirus targeted against mouse Nrf2 and/or Keap1. Nrf2-KD, Nrf2-knockdown; Keap1-KD, Keap1- knockdown; Double-KD, Keap1- and Nrf2-double knockdown. n = 3; *p < 0.05 vs Scramble. (C) Protein expression of Nrf2 in 3T3-L1 cells under basal and iAs-treated conditions. (D) Protein expression of Keap1 in 3T3-L1 cells with Nrf2-KD, Keap1-KD or Double-KD. (E) Knockdown of Nrf2 blocks the enhanced adipogenesis caused by deficiency of Keap1 in 3T3-L1 cells. Cells were differentiated for 7 days by DMIRI protocol.
ARE–driven transcription during hormone-induced adipogenesis in 3T3-L1 cells
To determine the involvement of Nrf2 in adipogenesis, we measured the nuclear accumulation of Nrf2 and its transcriptional activity in 3T3-L1 cells in response to challenge of hormonal cocktails. In Scramble cells increase of Nrf2 in nuclear fractions occurred as early as 1 h and peaked at 2-4 h post DMIRI treatment. In contrast, Nrf2-KD cells exhibited marginal Nrf2 accumulation during the process (Figure 4A-B). Of note, single, double or multi-band Nrf2 is observed when using the Nrf2 antibody for this study. The resolution of the number of Nrf2 bands appears to be dependent on protein separation condition, cell type and treatment. While we have reported that Nrf2 exhibited multi bands in several human cell lines, including HaCaT, HepG2, H69, G361 and primary cultured human umbilical vein endothelial cells [34], in mouse protein samples used here we cannot exclude the possibility that the antibody may detect some unknown proteins which exhibit similar MW on Tris-Glycine gel as Nrf2 and are also activated by iAs and other prototypic Nrf2 activators.
Figure 4.
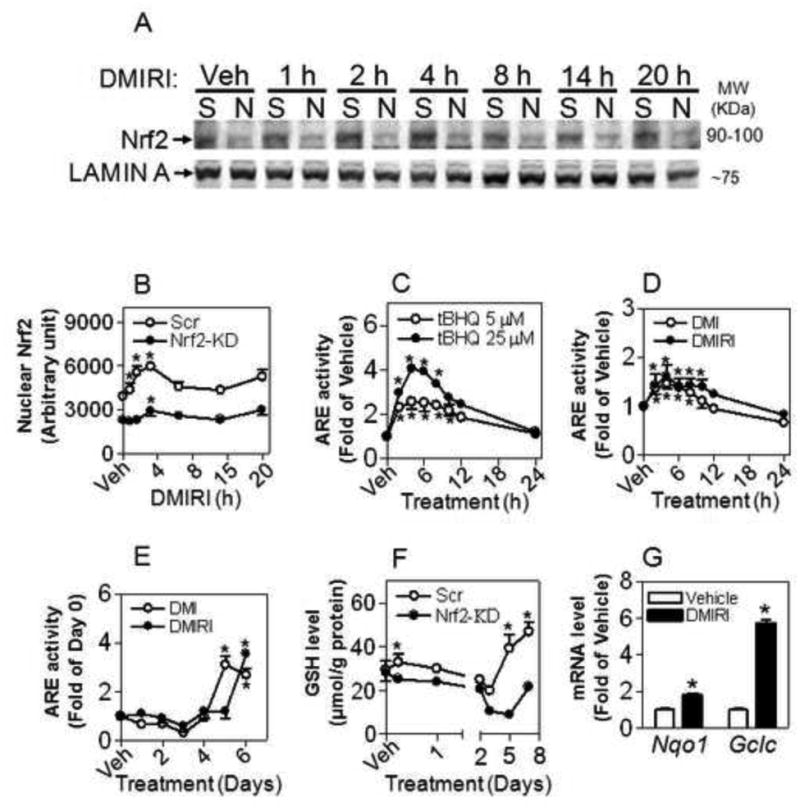
Activation of Nrf2 occurs during adipogenesis in 3T3-L1 cells. (A) Nrf2 protein levels in nuclear fractions. Cells were treated with DMIRI for indicated time. S, Scramble; N, Nrf2- knockdown; Veh, Vehicle (medium). (B) Quantification of Nrf2 level in (A). *, p < 0.05 vs respective cells with Vehicle. (C) ARE-luciferase reporter stably expressed in 3T3-L1 cells is responsive to tBHQ challenge. *, p < 0.05 vs Vehicle. (D) and (E) ARE-luciferase activity during adipogenesis induced by DMI or DMIRI protocol; *, p < 0.05 vs Vehicle. (F) Intracellular GSH levels in Nrf2-KD and Scramble cells during adipogenesis induced by DMIRI. *, p < 0.05 vs respective cells with Vehicle. (G) DMIRI-induced expression of Nrf2-target genes. The cells were treated with DMIRI for 6 h. n = 3-4; * p < 0.05 vs Vehicle.
To determine the transcriptional activity of Nrf2 during adipogenesis, the Cignal Lenti ARE reporter, which is designed to monitor the activity of the antioxidant response signal transduction pathway in cultured cells, was employed. As shown in Figure 4C, 3T3-L1 preadipocytes stably transduced with the ARE reporter showed a dose- and time-dependent induction of luciferase activity upon Nrf2 activator tBHQ treatment, confirming that the cells are responsive to activation of Nrf2. We next examined the ARE activity during DMI- and DMIRI-induced adipogenic differentiation. After the cells were acutely challenged with the hormonal cocktails, significant increases in ARE activity appeared after 2 hr and peaked at 4 hr post treatment (Figure 4D), which is consistent with the result of Nrf2 nuclear accumulation (Figure 4A-B). Interestingly, when the cells were differentiated further, a slight reduction of ARE reporter activity on day 2 and 3 was followed by a late stage induction of much larger magnitude (Figure 4E). Consistent with the notion that Nrf2 is a key regulator for GSH synthesis, intracellular GSH levels in Scramble cells followed a similar pattern as Nrf2-ARE activity, whereas knockdown of Nrf2 blocked the stimulatory effect of adipogenic hormones on the induction of intracellular GSH (Figure 4F). This finding provides additional support that activation of Nrf2 occurs in the early stage (< 12 hr) of adipogenesis. Additionally, significant induction of Nrf2 target genes Nqo1 and Gclc was observed at 6 hr post DMIRI treatment (Figure 4G).
ROS are involved in adipogenic hormones-induced adipogenesis
Consistent with previous report [35] that ROS facilitate adipocyte differentiation, transient treatment of 3T3-L1 cells with DMI or DMIRI resulted in a significant increased ROS production, which was suppressed by antioxidant NAC (Figure 5A). In contrast to the reduced expression of antioxidant enzyme GCLC (Figure 6B and C) and decreased GSH levels (Figure 4F), Nrf2-KD cells exhibited diminished levels of intracellular ROS in response to acute DMI treatment (Figure 5B), suggesting a compensatory mechanism may exist in response to the deficiency of Nrf2. To further verify the involvement of ROS in adipogenesis, the effects of antioxidant agents NAC and GSH-EE, inhibitor of NADPH oxidase (NOX) DPI, uncoupler of mitochondrial oxidative phosphorylation FCCP and sulfhydryl depletor DEM on adipogenic differentiation were investigated. As shown in Figure 5C, NAC and GSH-EE dose-dependently attenuated DMI-induced adipogenesis in 3T3-L1 cells, confirming the inhibitory effect of antioxidants on adipogenesis [35-37]. The suppression of DPI and FCCP on adipogenesis (Figure 5C) suggests NOX and mitochondria may be the sources of intracellular ROS that are related to adipogenesis. Since DPI also inhibits nitric oxide synthetase, the detailed mechanisms for the inhibitory effect of DPI on adipogenesis need further clarification. In addition, sulfhydryl depletor DEM exhibited a strong inhibitory effect on adipogenesis (Figure 5C), suggesting GSH is involved in adipogenic process.
Figure 5.
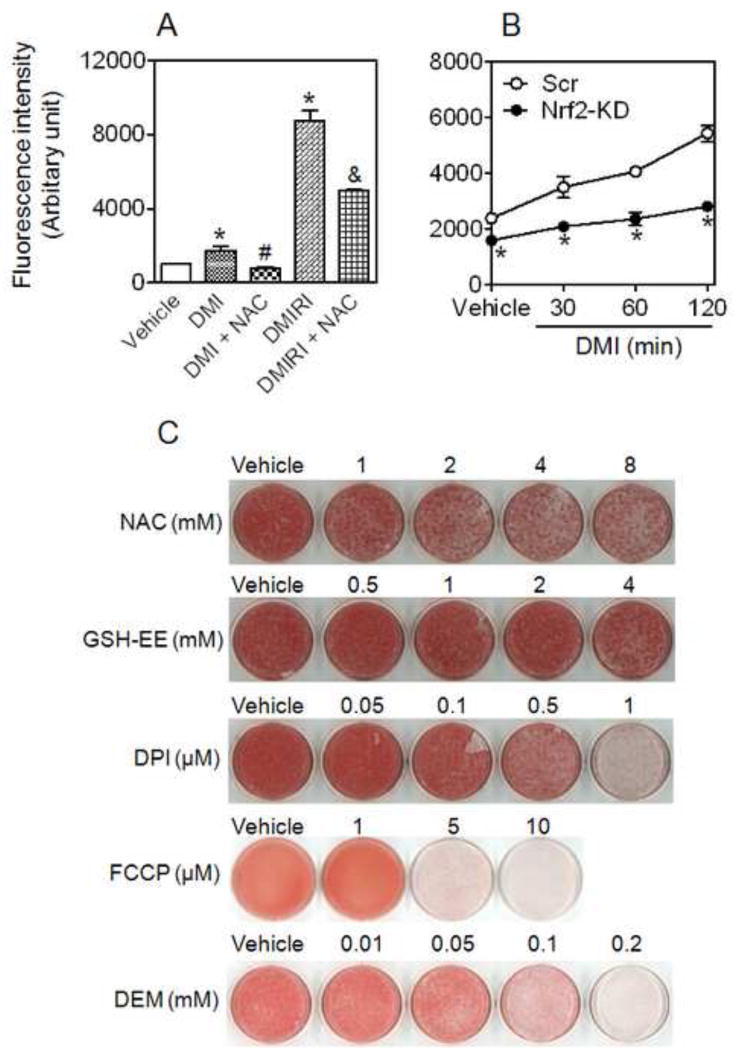
ROS are involved in hormonal cocktail-induced adipogenesis in 3T3-L1 preadipocytes. (A) Adipogenic hormonal cocktails stimulate ROS production. Cells were treated with DMI and DMIRI with or without NAC for 2 hrs followed by intracellular peroxide measurement using CM-H2DCFDA. * p < 0.05 vs Vehicle; # p < 0.05 vs DMI; & p < 0.05 vs DMIRI. (B) Nrf2-KD cells exhibit reduced ROS levels in response to adipogenic hormonal treatments. * p < 0.05 vs Scr with the same treatment. (C) Inhibitory effects of exogenous antioxidants, DPI, FCCP and DEM on adipogenesis in 3T3-L1 cells. Cells were differentiated 1 day after confluence by DMI protocol. Lipid vesicles were stained by ORO on Day 8. NAC, GSH-EE, DPI, FCCP and DEM were added by indicated concentrations during Day 1 and 2 of differentiation.
Figure 6.
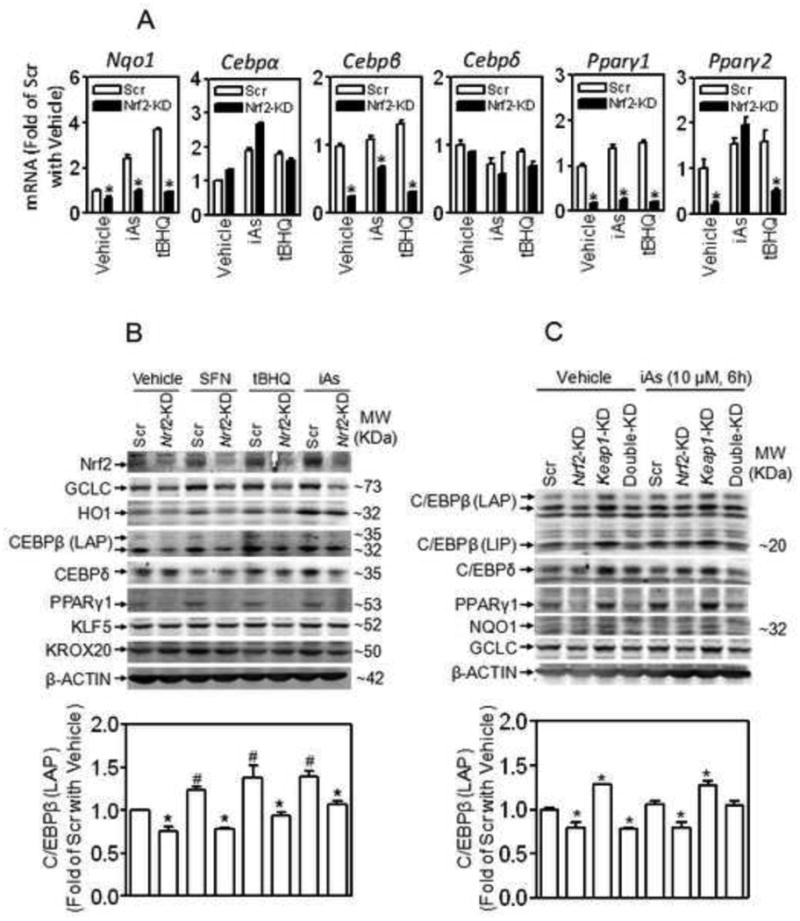
Expression of C/EBPβ is dependent on Nrf2 in 3T3-L1 cells. (A) Knockdown of Nrf2 suppresses expression of Cebpβ under basal and oxidative stressed conditions. Confluent cells were treated with iAs (10 μM) or tBHQ (25 μM) for 6 h. Scr, Scramble; Nrf2-KD, Nrf2-knockdown; n = 3; *p < 0.05 vs Scr with the same treatment. (B) Knockdown of Nrf2 suppresses protein expression of C/EBPβ under basal and oxidative stressed conditions. Upper panel: Confluent cells were treated with SFN (10 μM), tBHQ (25 μM) or iAs (10 μM) for 6 h. LAP, liver-enriched activating protein. Lower panel: quantification of C/EBPβ (LAP). n = 3-4; *p < 0.05 vs Scr with the same treatment; #p < 0.05 vs Scr with Vehicle (medium). (C) Knockdown of Keap1 activates Nrf2 and increases C/EBPβ expression. Keap1-KD, Keap1-knockdown; LIP, liver-enriched inhibitory protein. Upper panel: representative images of immunoblots; Lower panel: quantification of C/EBPβ (LAP). n = 3-4; *p < 0.05 vs Scr with the same treatment.
Expression of C/EBPβ is largely dependent on Nrf2 in 3T3-L1 cells under normal and oxidative stressed conditions
To investigate the regulatory mechanism of Nrf2 in adipogenesis, the expression of major adipogenic factors was determined in Nrf2-KD and Scramble 3T3-L1 cells under various Nrf2-activated conditions. As with Nrf2 downstream targets, such as NQO1, GCLC and HO1, the expression of C/EBPβ and PPARγ, but not C/EBPα, C/EBPδ, KLF5 and KROX20, was highly dependent on Nrf2 (Figure 6). In Nrf2-KD cells, mRNA expression of Cebpβ, Pparγ1 and Pparγ2 under normal and tBHQ- or iAs-challenged conditions was significantly lower than those in Scramble cells (Figure 6A). Acute exposure to Nrf2 activators, including sulforaphane (SFN), tBHQ and iAs, led to increased Nrf2 accumulation and induction of GCLC, HO1 and C/EBPβ protein in Scramble cells. In contrast, no enhanced Nrf2 expression and much less induction of C/EBPβ was detected in Nrf2-KD cells (Figure 6B). Consistent with this finding, stable knockdown of Keap1 resulted in a significant induction of C/EBPβ, NQO1, GCLC and PPARγ1 (Figure 6C). These results suggest that Nrf2 is a key regulator of Cebpβ expression. Of note, the protein expression of PPARγ2 (57 KDa) is too low to measure by immunoblotting in 3T3-L1 preadipocytes.
Knockdown of Nrf2 in 3T3-L1 cells results in reduced C/EBPβ induction during adipogenesis
Upon initiation of differentiation by DMI or DMIRI in Scramble cells, a significant increase in Nrf2 accumulation occurred in 30 min (Figure 7A), and was followed by an immediate induction of C/EBPβ (Figure 7A-C). In Nrf2-KD cells, DMI- or DMIRI-induced C/EBPβ expression was significantly lower than those in Scramble cells at each time point (Figure 7A-C), suggesting Nrf2 is critical in regulating the expression of C/EBPβ in the early stage of adipogenesis. Only until 3 days post hormonal challenge was significant induction of Pparγ in Scramble cells observed (Figure 7C).
Figure 7.
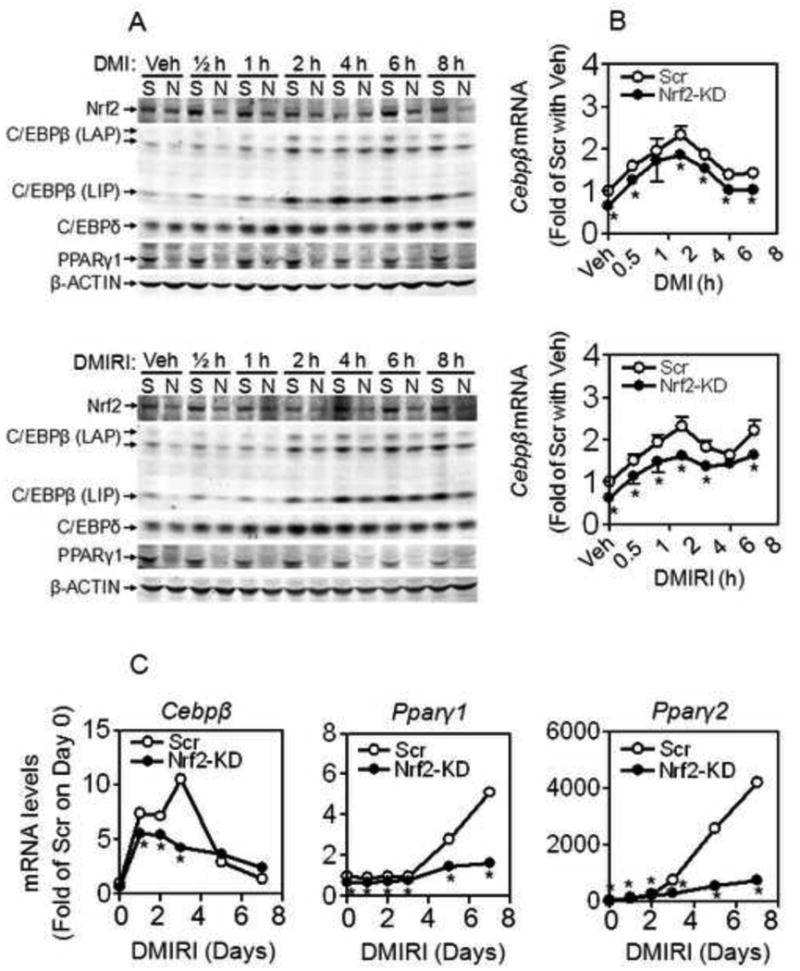
Knockdown of Nrf2 suppresses C/EBPβ induction in 3T3-L1 cells. (A) Protein expression of Nrf2, C/EBPβ, C/EBPδ and PPARγ in the early stage of adipogenesis induced by DMI (upper panel) or DMIRI (lower panel). S, Scramble; N, Nrf2- knockdown. (B) Transcription of C/EBPβ in the early stage of adipogenesis induced by DMI (upper panel) or DMIRI (lower panel). Scr, Scramble; Nrf2-KD, Nrf2- knockdown. n = 3; *, p < 0.05 vs Scr with the same treatment. (C) Expression of Cebpβ and Pparγ during adipogenesis induced by DMIRI. n = 3; *, p < 0.05 vs Scr with the same treatment.
Nrf2 regulates transcription from the Cebpβ gene promoter
To investigate whether Nrf2 is involved in the transcriptional regulation of Cebpβ, we searched for Nrf2-binding motif in the promoter region of Cebpβ and identified a consensus ARE site between -115 bp and -107 bp (Online Materials Figure S1). To determine the activity of the ARE site identified, a series of 5′-truncated Cebpβ promoter-driven luciferase reporters were generated and the reporter activities were analyzed in 3T3-L1 cells under basal and DMI-treated conditions. As shown in Figure 8A, deletions between -1036 bp and -116 bp, which do not disturb the identified ARE site (Online Materials Figure. S1), showed no inhibitory effect on reporter activity. The GC base pairs of the ARE, which contact small maf proteins, are critical for induction of genes in response to oxidative stimuli [38, 39], and are what distinguish the ARE from AP-1 or CRE binding sites (Online Materials Figure S1). When the “GC” box in the ARE is replaced with “AA” the activity of -116 bp reporter showed ∼97% reduction (Figure 8A), indicating the ARE site (-115∼-107 bp) in the Cebpβ promoter is crucial for its transcriptional activity. To further demonstrate that Nrf2 may bind the ARE and upregulate Cebpβ expression, the effect of NAC, which inhibits intracellular ROS accumulation and Nrf2 activation [33, 40], on the activity of -116 bp Cebpβ-promoter induced by DMI was investigated. As shown in Figure 8B, a substantial inhibitory effect of NAC on DMI-induced activation of Cebpβ-promoter was observed. In addition, a ChIP assay using primers surrounding the ARE site revealed that Nrf2 binds to the region in response to adipogenic stimulation (Figure 8C).
Figure 8.
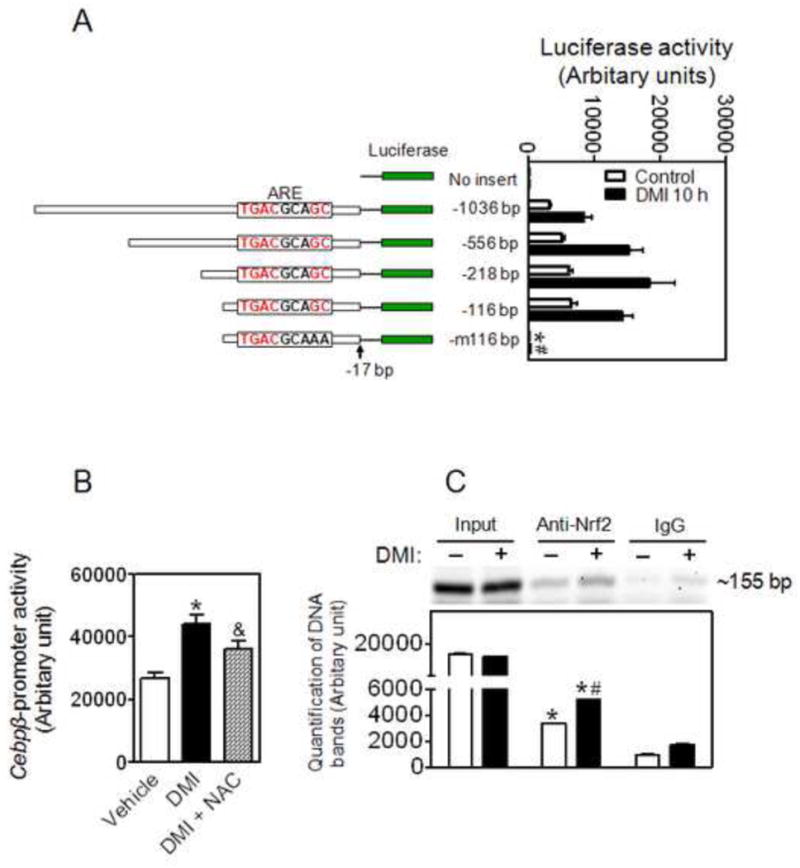
An ARE site on Cebpβ promoter is critical for its expression during adipogenesis. (A) Cebpβ promoter-reporter assay indicates an ARE site (-114∼-105 bp) in the Cebpβ promoter is critical for its expression. Left: Structures of Cebpβ promoter-luciferase reporters; Right: Activity of luciferase reporters. 3T3-L1 cells were transfected with the Cebpβ promoter-luciferase constructs when the cells reach 60-70% confluence. After 2 days confluent cells were treated with DMI for 10 h. -m116 bp, the “GC” in the ARE site were replaced with “AA”. n = 3-6; *p < 0.05 vs -116 bp Control; #p < 0.05 vs -116 bp DMI 10 h. (B) Inhibitory effect of NAC on the Cebpβ promoter activity induced by DMI. Cells expressed -116 bp Cebpβ promoter-luciferase reporter were treated with DMI with or without 5 mM NAC for 10 hrs. * p < 0.05 vs Vehicle; & p = 0.08 vs DMI alone. (C) ChIP assay indicates a physical association between Nrf2 and the Cebpβ promoter region with the ARE site. DMI, the cells were treated with DMI for 2 h. ChIP assays were performed on 3T3-L1 chromatin using either Nrf2 antibody or pre-immune antisera as negative control. Non-immunoprecipitated chromatin (1%) was used as an input control. The lower panel is the quantification of the image. n = 3; *p < 0.05 vs negative control (IgG); #p < 0.05 vs Anti-Nrf2 without DMI.
To further ascertain the regulatory mechanism of Nrf2 in Cebpβ transcription, the expression of several Nrf2 binding partners and purported Cebpβ regulatory protein, CREB [16], was determined in 3T3-L1 cell models. As shown in Figure 9, no significant difference in expression of MafG/F/K, c-Jun and c-Fos was observed between Nrf2-KD and Scramble cells in 8 h post hormonal challenge, though a dramatic induction of c-Fos occurred at 1-2 h in response to DMI treatment in both Nrf2-KD and Scramble cells. Consistent with previous work [16], DMI or DMIRI treatment in Scramble cells led to a substantial increase in phosphorylated CREB (p-CREB) levels. Interestingly, the levels of p-CREB and total CREB in response to acute DMI or DMIRI challenge were much lower in Nrf2-KD cells than that in Scramble cells, suggesting Nrf2 may be involved in regulating CREB activation during adipogenesis. In addition, there was no significant difference in phosphorylation of AKT at S473 (p-AKT) and total AKT between Scramble and Nrf2-KD cells in the process, although DMI induced a dramatic increase in AKT phosphorylation in the cells.
Figure 9.
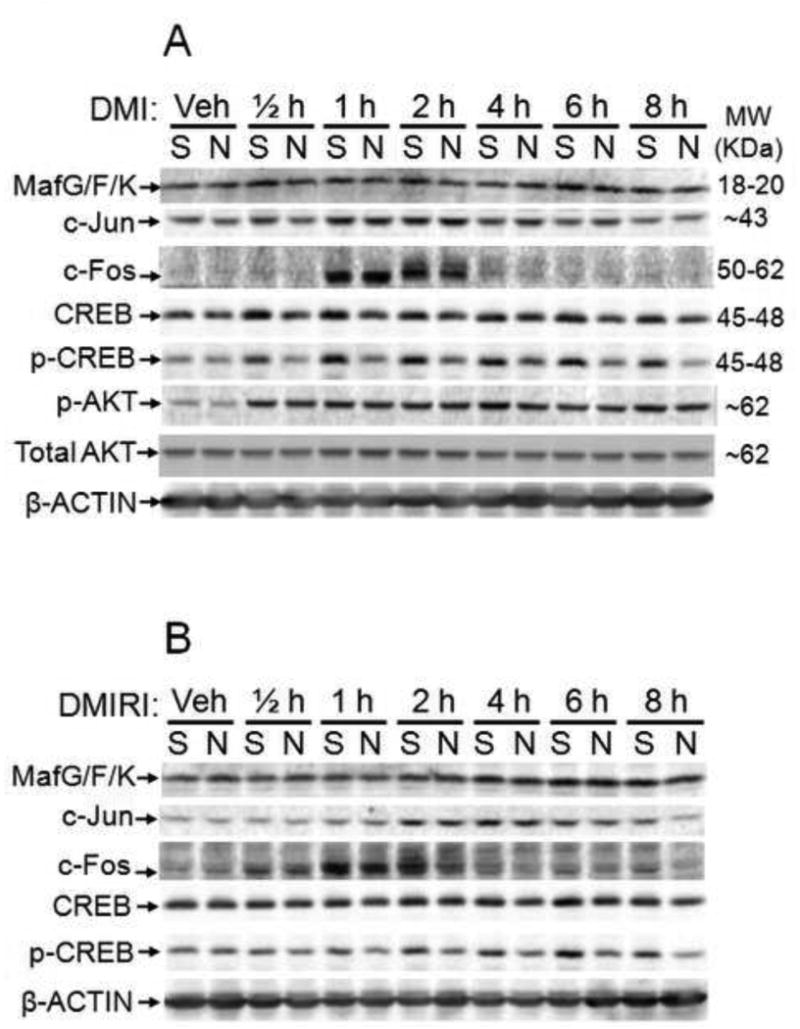
Expression of Nrf2-related transcription factors in the early stage of adipogenesis in 3T3-L1 cells induced by DMI (upper panel) or DMIRI (lower panel). Veh, vehicle (medium); S, Scramble; N, Nrf2- knockdown.
Discussion
The current model for adipogenesis begins with increased expression of C/EBPβ presumably through transcriptional activation by CREB, KLF4 and KROX20 [16-18, 41]. C/EBPβ then induces expression of PPARγ and C/EBPα, which leads to the expression of factors associated with a mature adipocyte phenotype. Therefore, identifying the factors that regulate C/EBPβ expression as well as other factors that cooperate with other C/EBPs should provide additional insight into the mechanisms regulating adipogenesis. Apart from CREB, KLF4 and KROX20, relatively few transcription factors have been described to bind to the Cebpβ promoter and positively regulate its transcription during adipogenesis. In the present investigation, we identified and characterized the regulatory role of Nrf2 in the induction of Cebpβ early in the adipocyte differentiation program. We found that the expression of Cebpβ at the early stage of adipogenesis is highly dependent on Nrf2, which possibly binds to an ARE site in the Cebpβ promoter and increases its transcription. The data presented here provide additional insight into adipogenesis in which Nrf2 may be one of the earliest transcription factors involved in adipogenic differentiation.
Adipogenic differentiation of preadipocytes is commonly enhanced by treating postconfluent cells with a differentiation cocktail DMI, a combination of dexamethasone, IBMX, insulin and FBS [42, 43]. Addition of thiazolidinedione drugs, such as rosiglitazone and indomethacin, to the DMI cocktail promotes and accelerates adipogenesis. The use of insulin is classically viewed as a promoter of adipogenesis as it increases expression of CREB and PPARγ [2, 3, 44]. Dexamethasone is a potent enhancer of adipogenic differentiation by binding to glucocorticoid receptor (GR) and upregulating C/EBPδ [45]. IBMX and indomethacin increase cAMP, promote CREB phosphorylation, induce KLF4 and thus upregulate expression of C/EBPβ [16, 18, 46]. Serum stimulates expression of KROX20, a zinc finger-containing transcription factor, and promotes C/EBPβ expression [17]. In addition, FBS contains fatty acids, insulin, insulin-like growth factor (IGF) and other cytokines, all of which are involved in regulation of adipocyte differentiation. Interestingly, dexamethasone has been shown to induce Nrf2 downstream genes, including CAT, glutathione peroxidase and superoxide dismutase [47], suggesting dexamethasone is one of the key components to activate Nrf2 during adipogenesis. Insulin/IGF1 are active stimulators of NADPH-dependent ROS generation [48, 49], and can directly regulate SKN-1, a worm homolog of Nrf2 [50]. In addition, insulin has been shown to induce HO1 through PI3K/AKT pathway and Nrf2 [51]. Indomethacin has been revealed to activate NOX, which enhances intracellular ROS production and thus triggers activation of Nrf2 [40, 52]. Furthermore, significant inductions of H-ferritin [53] and metallothionein-II [36], both of which are Nrf2 target genes [54, 55], have been observed during differentiation of 3T3-L1 cells. These studies together with our previous [26] and current findings clearly demonstrate that adipogenic hormones, individually or in combination, may activate Nrf2-ARE pathway during adipogenesis.
Obesity has been recognized as a state of chronic oxidative stress and inflammation [56-59]. However, the literature remains controversial regarding the effect of ROS and antioxidants on adipogenesis. There is increasing evidence for ROS, a primary trigger of activation of Nrf2 [60], to promote the conversion from preadipocytes to mature adipocytes [35]. It has been reported that ROS are involved in insulin action [61, 62] and facilitate adipocyte maturation by enhancing the activation of C/EBPβ [35]. In contrast, N-acetylcysteine and α-lipoic acid, potent antioxidants and inhibitors of activation of Nrf2 [32, 33], suppress adipogenesis [35-37]. Our current studies also demonstrated that ROS may serve as an important mediator for adipogenic hormonal cocktails-induced Nrf2 activation, Cebpβ induction and adipogenesis. Although the exact sources for adipogenic hormones-triggered ROS production and the mechanisms for Nrf2 activation in response to DMI or DMIRI still need further investigation, the present study suggests that NOX- and mitochondria-derived ROS may be critical for adipogenesis. Considering the oxidative stress occurred in T2D and obesity is of a low-level chronic type, whether Nrf2-mediated cellular adaptive antioxidant response is involved in adipogenesis and lipogenesis, as well as the effect of chronic oxidative stress on Nrf2-mediated expression of Cebpβ and Pparγ still requires intensive investigations.
Nrf2 is known to heterodimerize with small Maf proteins and bind to AREs in the promoters of target genes [25]. In addition, Nrf2 may interact, structurally or functionally, with other bZIP proteins, such as AP-1 family, and regulate ARE-driven gene expression [25]. In the current study, we found that adipogenic hormones increase Nrf2 protein level, but cannot affect the expression of MafG/F/K. Interestingly, the expression of c-Fos and c-Jun, in particular c-Fos, is transiently upregulated in response to DMIRI challenge, although deficiency of Nrf2 does not affect their expression. Importantly, the induction of c-Fos and c-Jun by DMIRI is coincident with the ARE activation occurring in the early stage of adipogenesis. This finding suggests that these two transcription factors may interact with Nrf2 to regulate Cebpβ expression. This new aspect of adipogenesis still needs further study.
In 2004, Zhang et al. [16] identified dual cAMP response element (CRE)-like elements (termed TGA sites) within the promoter of the Cebpβ to which CREB family members, i.e. CREB and activating transcription factor 1 (ATF1), may bind and activate transcription. However, individual depletion of CREB or ATF1, cAMP responsive element modulator, c-Jun or ATF2 cannot diminish hormone-induced Cebpβ expression, but combined knockdown of those factors resulted in loss of Cebpβ expression [41], suggesting multiple transcription factors may be involved in Cebpβ expression. In the current study, we identified a consensus ARE site in the region of Cebpβ promoter with transcriptional activity and provided evidence that Nrf2 may bind with the ARE and upregulate Cebpβ expression in response to oxidative stress and hormonal stimulation. The evidence that the ARE site we identified in the Cebpβ promoter (Online Materials Figure S1) overlaps with the TGA2 site discovered before [16] suggests a potential interaction between Nrf2 and CREB or ATF1 in the regulation of Cebpβ expression. To test this hypothesis we attempted to investigate the association between Nrf2 and CREB or ATF1 using immunoprecipitation. However, no clear CREB and Nrf2 binding was detectable in 12 hrs post DMI treatment (not shown), possibly due to the low expression of Nrf2 in the cells. In contrast, when we enriched the expression of Nrf2, CREB, p-CREB or p-ATF1 using arsenite or forskolin treatment, an interaction between these two proteins was identified (not shown). The substantial reduction of CREB and p-CREB in Nrf2-KD cells in the early stage of adipogenesis (Figure 9) suggests another possibility that Nrf2 regulates Cebpβ expression through CREB/p-CREB. However, whether Creb is a direct downstream target of Nrf2 and how the expression of Creb is regulated in the early stage of adipogenesis still need further clarification.
Nrf2 and C/EBPs belong to bZIP family. Although direct interactions between Nrf2 and C/EBPs are still under investigation, NRF1, another member of CNC-bZIP transcription factors, has been reported to interact with C/EBPβ to regulate odontoblast differentiation [63]. In hepatocytes, the expression of phase II detoxification enzymes, such as GSTA2, is transcriptionally activated partly through activation of both Nrf2 and C/EBPβ via their binding to the respective response elements in the promoter regions of the target genes [64, 65]. In addition, several nuclear factors, including CREB, ATF and silencing mediator for retinoid and thyroid hormone receptors, have been shown to regulate and/or interact with C/EBPβ or δ [16, 41, 66, 67]. Importantly, these nuclear factors are also implicated in Nrf2-mediated antioxidant response [68-73], suggesting that there may be a regulatory relationship between Nrf2 and C/EBPβ and/or δ through these factors. In our previous study [26], we have proposed that Nrf2 is a key transcription factor in regulating PPARγ expression during adipogenesis. Although the delayed induction of PPARγ in adipogenesis implies that Nrf2 may not directly regulate its transcription in the early stage of adipogenesis, the late stage induction of ARE-reporter activity in response to hormonal challenge (Figure 4E) suggests that Nrf2 may coordinate with other adipogeneic factors and regulate the late stage induction of PPARγ.
Understanding the regulatory mechanisms of adipogenesis is the basis for research on metabolic diseases and the respective pharmaceutical intervention. Based upon our findings that disruption of the Nrf2 gene leads to impaired adipogenesis, it raises a question as to whether inhibiting expression or activity of Nrf2 is an appropriate therapeutic approach for obesity to reduce body weight. Considering the unexpected discovery that the antidiabetic thiazolidinediones are ligands for PPARγ, downregulation of C/EBPβ and PPARγ in preadipocytes and adipocytes by inhibition of Nrf2 may result in impaired adipose function and induce glucose intolerance. On the other hand, suppression of Nrf2 in cardiovascular tissues may reduce fat accumulation in the tissues and prevent the deleterious cardiovascular effects of thiazolidinedione therapies. Given the paradoxical roles for Nrf2 in obesity and diabetes that now exist, a better understanding of the role of Nrf2 in the function of adipocyte and pathogenesis of obesity and diabetes is needed.
Supplementary Material
Acknowledgments
The content is solely the responsibility of the authors. All authors have agreed to its content and there are no financial or other conflicts of interest. Y.H., P.X., C.G.W, K.Y., J.F., Q.Z., M.E.A. and J.P. are employees of The Hamner Institutes for Health Sciences. The Hamner is a 501(c)3 not-for-profit organization that has a diverse research portfolio that includes funding from the American Chemistry Council, a trade association that represents chemical manufacturers. This work was supported in part by the National Institutes of Health Grants DK76788 (to J.P.) and ES016005 (to J.P.), the American Chemistry Council-Long Range Research Initiative (to M.E.A.) and the DOW Chemical Company (to M.E.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 2.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 3.Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefterova MI, Lazar MA. New developments in adipogenesis. Trends Endocrinol Metab. 2009;20:107–114. doi: 10.1016/j.tem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 6.Yeh WC, Cao Z, Classon M, McKnight SL. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:168–181. doi: 10.1101/gad.9.2.168. [DOI] [PubMed] [Google Scholar]
- 7.Shao D, Lazar MA. Peroxisome proliferator activated receptor gamma, CCAAT/enhancer-binding protein alpha, and cell cycle status regulate the commitment to adipocyte differentiation. J Biol Chem. 1997;272:21473–21478. doi: 10.1074/jbc.272.34.21473. [DOI] [PubMed] [Google Scholar]
- 8.Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE, McKeon C, Darlington GJ, Spiegelman BM. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell. 1999;3:151–158. doi: 10.1016/s1097-2765(00)80306-8. [DOI] [PubMed] [Google Scholar]
- 9.Freytag SO, Paielli DL, Gilbert JD. Ectopic expression of the CCAAT/enhancer-binding protein alpha promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes Dev. 1994;8:1654–1663. doi: 10.1101/gad.8.14.1654. [DOI] [PubMed] [Google Scholar]
- 10.Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ, Spiegelman BM. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16:22–26. doi: 10.1101/gad.948702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang QQ, Otto TC, Lane MD. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci U S A. 2003;100:850–855. doi: 10.1073/pnas.0337434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang QQ, Lane MD. Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev. 1999;13:2231–2241. doi: 10.1101/gad.13.17.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Kim JW, Gronborg M, Urlaub H, Lane MD, Tang QQ. Role of cdk2 in the sequential phosphorylation/activation of C/EBPbeta during adipocyte differentiation. Proc Natl Acad Sci U S A. 2007;104:11597–11602. doi: 10.1073/pnas.0703771104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Molina H, Huang H, Zhang YY, Liu M, Qian SW, Slawson C, Dias WB, Pandey A, Hart GW, Lane MD, Tang QQ. O-linked N-acetylglucosamine modification on CCAAT enhancer-binding protein {beta&rcub: role during adipocyte differentiation. J Biol Chem. 2009;284:19248–19254. doi: 10.1074/jbc.M109.005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JW, Tang QQ, Li X, Lane MD. Effect of phosphorylation and S-S bond-induced dimerization on DNA binding and transcriptional activation by C/EBPbeta. Proc Natl Acad Sci U S A. 2007;104:1800–1804. doi: 10.1073/pnas.0611137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang JW, Klemm DJ, Vinson C, Lane MD. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J Biol Chem. 2004;279:4471–4478. doi: 10.1074/jbc.M311327200. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. 2005;1:93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Birsoy K, Chen Z, Friedman J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008;7:339–347. doi: 10.1016/j.cmet.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darlington GJ, Ross SE, MacDougald OA. The role of C/EBP genes in adipocyte differentiation. J Biol Chem. 1998;273:30057–30060. doi: 10.1074/jbc.273.46.30057. [DOI] [PubMed] [Google Scholar]
- 20.Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15:4184–4193. doi: 10.1128/mcb.15.8.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biswas M, Chan JY. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol Appl Pharmacol. 2010;244:16–20. doi: 10.1016/j.taap.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi A, Ito E, Toki T, Kogame K, Takahashi S, Igarashi K, Hayashi N, Yamamoto M. Molecular cloning and functional characterization of a new Cap‘n’ collar family transcription factor Nrf3. J Biol Chem. 1999;274:6443–6452. doi: 10.1074/jbc.274.10.6443. [DOI] [PubMed] [Google Scholar]
- 23.Andrews NC, Erdjument-Bromage H, Davidson MB, Tempst P, Orkin SH. Erythroid transcription factor NF-E2 is a haematopoietic-specific basic-leucine zipper protein. Nature. 1993;362:722–728. doi: 10.1038/362722a0. [DOI] [PubMed] [Google Scholar]
- 24.Oyake T, Itoh K, Motohashi H, Hayashi N, Hoshino H, Nishizawa M, Yamamoto M, Igarashi K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol Cell Biol. 1996;16:6083–6095. doi: 10.1128/mcb.16.11.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motohashi H, O'Connor T, Katsuoka F, Engel J, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294:1. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- 26.Pi J, Leung L, Xue P, Wang W, Hou Y, Liu D, Yehuda-Shnaidman E, Lee C, Lau J, Kurtz TW, Chan JY. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J Biol Chem. 285:9292–9300. doi: 10.1074/jbc.M109.093955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekhar KR, Rachakonda G, Freeman ML. Cysteine-based regulation of the CUL3 adaptor protein Keap1. Toxicol Appl Pharmacol. 2010;244:21–26. doi: 10.1016/j.taap.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 29.Cannon BaNJ. Adipose tissues protocols. Totowa, NJ: Humana Press; 2001. [Google Scholar]
- 30.Fu J, Woods CG, Yehuda-Shnaidman E, Zhang Q, Wong V, Collins S, Sun G, Andersen ME, Pi J. Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: involvement of cellular adaptive response to oxidative stress. Environ Health Perspect. 118:864–870. doi: 10.1289/ehp.0901608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woods CG, Fu J, Xue P, Hou Y, Pluta LJ, Yang L, Zhang Q, Thomas RS, Andersen ME, Pi J. Dose-dependent transitions in Nrf2-mediated adaptive response and related stress responses to hypochlorous acid in mouse macrophages. Toxicol Appl Pharmacol. 2009;238:27–36. doi: 10.1016/j.taap.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–245. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- 33.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 34.Pi J, Bai Y, Reece JM, Williams J, Liu D, Freeman ML, Fahl WE, Shugar D, Liu J, Qu W, Collins S, Waalkes MP. Molecular mechanism of human Nrf2 activation and degradation: role of sequential phosphorylation by protein kinase CK2. Free Radic Biol Med. 2007;42:1797–1806. doi: 10.1016/j.freeradbiomed.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee H, Lee YJ, Choi H, Ko EH, Kim JW. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J Biol Chem. 2009;284:10601–10609. doi: 10.1074/jbc.M808742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JR, Ryu HH, Chung HJ, Lee JH, Kim SW, Kwun WH, Baek SH, Kim JH. Association of anti-obesity activity of N-acetylcysteine with metallothionein-II down-regulation. Exp Mol Med. 2006;38:162–172. doi: 10.1038/emm.2006.20. [DOI] [PubMed] [Google Scholar]
- 37.Cho KJ, Moon HE, Moini H, Packer L, Yoon DY, Chung AS. Alpha-lipoic acid inhibits adipocyte differentiation by regulating pro-adipogenic transcription factors via mitogen-activated protein kinase pathways. J Biol Chem. 2003;278:34823–34833. doi: 10.1074/jbc.M210747200. [DOI] [PubMed] [Google Scholar]
- 38.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 39.Kusunoki H, Motohashi H, Katsuoka F, Morohashi A, Yamamoto M, Tanaka T. Solution structure of the DNA-binding domain of MafG. Nat Struct Biol. 2002;9:252–256. doi: 10.1038/nsb771. [DOI] [PubMed] [Google Scholar]
- 40.Sekhar KR, Spitz DR, Harris S, Nguyen TT, Meredith MJ, Holt JT, Gius D, Marnett LJ, Summar ML, Freeman ML, Guis D. Redox-sensitive interaction between KIAA0132 and Nrf2 mediates indomethacin-induced expression of gamma-glutamylcysteine synthetase. Free Radic Biol Med. 2002;32:650–662. doi: 10.1016/s0891-5849(02)00755-4. [DOI] [PubMed] [Google Scholar]
- 41.Fox KE, Fankell DM, Erickson PF, Majka SM, Crossno JT, Jr, Klemm DJ. Depletion of cAMP-response element-binding protein/ATF1 inhibits adipogenic conversion of 3T3-L1 cells ectopically expressing CCAAT/enhancer-binding protein (C/EBP) alpha, C/EBP beta, or PPAR gamma 2. J Biol Chem. 2006;281:40341–40353. doi: 10.1074/jbc.M605077200. [DOI] [PubMed] [Google Scholar]
- 42.Pantoja C, Huff JT, Yamamoto KR. Glucocorticoid signaling defines a novel commitment state during adipogenesis in vitro. Mol Biol Cell. 2008;19:4032–4041. doi: 10.1091/mbc.E08-04-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novakofski J. Adipogenesis: usefulness of in vitro and in vivo experimental models. J Anim Sci. 2004;82:905–915. doi: 10.2527/2004.823905x. [DOI] [PubMed] [Google Scholar]
- 44.Janderova L, McNeil M, Murrell AN, Mynatt RL, Smith SR. Human mesenchymal stem cells as an in vitro model for human adipogenesis. Obes Res. 2003;11:65–74. doi: 10.1038/oby.2003.11. [DOI] [PubMed] [Google Scholar]
- 45.Wu Z, Bucher NL, Farmer SR. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol Cell Biol. 1996;16:4128–4136. doi: 10.1128/mcb.16.8.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev. 1998;78:783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 47.Jose HJ, Berenice SG, Cecilia VR. Induction of antioxidant enzymes by dexamethasone in the adult rat lung. Life Sci. 1997;60:2059–2067. doi: 10.1016/s0024-3205(97)00193-8. [DOI] [PubMed] [Google Scholar]
- 48.Krieger-Brauer HI, Kather H. The stimulus-sensitive H2O2-generating system present in human fat-cell plasma membranes is multireceptor-linked and under antagonistic control by hormones and cytokines. Biochem J. 1995;307(Pt 2):543–548. doi: 10.1042/bj3070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem. 2001;276:48662–48669. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- 50.Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–1038. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harrison EM, McNally SJ, Devey L, Garden OJ, Ross JA, Wigmore SJ. Insulin induces heme oxygenase-1 through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in renal cells. Febs J. 2006;273:2345–2356. doi: 10.1111/j.1742-4658.2006.05224.x. [DOI] [PubMed] [Google Scholar]
- 52.Sekhar KR, Crooks PA, Sonar VN, Friedman DB, Chan JY, Meredith MJ, Starnes JH, Kelton KR, Summar SR, Sasi S, Freeman ML. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–5645. [PubMed] [Google Scholar]
- 53.Festa M, Ricciardelli G, Mele G, Pietropaolo C, Ruffo A, Colonna A. Overexpression of H ferritin and up-regulation of iron regulatory protein genes during differentiation of 3T3-L1 pre-adipocytes. J Biol Chem. 2000;275:36708–36712. doi: 10.1074/jbc.M004988200. [DOI] [PubMed] [Google Scholar]
- 54.Pietsch EC, Chan JY, Torti FM, Torti SV. Nrf2 mediates the induction of ferritin h in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem. 2003;278:2361–2369. doi: 10.1074/jbc.M210664200. [DOI] [PubMed] [Google Scholar]
- 55.Yeh CT, Yen GC. Effect of sulforaphane on metallothionein expression and induction of apoptosis in human hepatoma HepG2 cells. Carcinogenesis. 2005;26:2138–2148. doi: 10.1093/carcin/bgi185. [DOI] [PubMed] [Google Scholar]
- 56.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 58.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nathan C. Epidemic inflammation: pondering obesity. Mol Med. 2008;14:485–492. doi: 10.2119/2008-00038.Nathan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Q, Pi J, Woods CG, Andersen ME. A systems biology perspective on Nrf2- mediated antioxidant response. Toxicol Appl Pharmacol. 2010;244:84–97. doi: 10.1016/j.taap.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7:1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Narayanan K, Ramachandran A, Peterson MC, Hao J, Kolsto AB, Friedman AD, George A. The CCAAT enhancer-binding protein (C/EBP)beta and Nrf1 interact to regulate dentin sialophosphoprotein (DSPP) gene expression during odontoblast differentiation. J Biol Chem. 2004;279:45423–45432. doi: 10.1074/jbc.M405031200. [DOI] [PubMed] [Google Scholar]
- 64.Kang KW, Cho IJ, Lee CH, Kim SG. Essential role of phosphatidylinositol 3- kinase-dependent CCAAT/enhancer binding protein beta activation in the induction of glutathione S-transferase by oltipraz. J Natl Cancer Inst. 2003;95:53–66. doi: 10.1093/jnci/95.1.53. [DOI] [PubMed] [Google Scholar]
- 65.Ko MS, Lee SJ, Kim JW, Lim JW, Kim SG. Differential effects of the oxidized metabolites of oltipraz on the activation of CCAAT/enhancer binding protein-beta and NF-E2-related factor-2 for GSTA2 gene induction. Drug Metab Dispos. 2006;34:1353–1360. doi: 10.1124/dmd.106.009514. [DOI] [PubMed] [Google Scholar]
- 66.Belmonte N, Phillips BW, Massiera F, Villageois P, Wdziekonski B, Saint-Marc P, Nichols J, Aubert J, Saeki K, Yuo A, Narumiya S, Ailhaud G, Dani C. Activation of extracellular signal-regulated kinases and CREB/ATF-1 mediate the expression of CCAAT/enhancer binding proteins beta and -delta in preadipocytes. Mol Endocrinol. 2001;15:2037–2049. doi: 10.1210/mend.15.11.0721. [DOI] [PubMed] [Google Scholar]
- 67.Choy L, Derynck R. Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J Biol Chem. 2003;278:9609–9619. doi: 10.1074/jbc.M212259200. [DOI] [PubMed] [Google Scholar]
- 68.He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276:20858–20865. doi: 10.1074/jbc.M101198200. [DOI] [PubMed] [Google Scholar]
- 69.Bakin AV, Stourman NV, Sekhar KR, Rinehart C, Yan X, Meredith MJ, Arteaga CL, Freeman ML. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding Phase II detoxifying proteins. Free Radic Biol Med. 2005;38:375–387. doi: 10.1016/j.freeradbiomed.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 70.Kang KW, Lee SJ, Kim SG. Molecular mechanism of nrf2 activation by oxidative stress. Antioxid Redox Signal. 2005;7:1664–1673. doi: 10.1089/ars.2005.7.1664. [DOI] [PubMed] [Google Scholar]
- 71.Brown SL, Sekhar KR, Rachakonda G, Sasi S, Freeman ML. Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)-regulated stress pathway. Cancer Res. 2008;68:364–368. doi: 10.1158/0008-5472.CAN-07-2170. [DOI] [PubMed] [Google Scholar]
- 72.Ki SH, Cho IJ, Choi DW, Kim SG. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol. 2005;25:4150–4165. doi: 10.1128/MCB.25.10.4150-4165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 2001;6:857–868. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.