

Abstract
The occupational chemical 4-vinylcyclohexene diepoxide (VCD) has been shown to cause selective destruction of ovarian small pre-antral (primordial and primary) follicles in rats and mice by accelerating the natural, apoptotic process of atresia. Chemicals that destroy primordial follicles are of concern to women because exposure can result in premature ovarian failure (early menopause). Initial studies using in vivo exposure of rats determined that VCD specifically targets primordial and primary (small pre-antral) follicles and that repeated dosing is required. Through a method of isolation of ovarian small follicles, biochemical and molecular studies determined that intracellular pro-apoptotic pathways are activated following VCD dosing in rats. Subsequently an in vitro system using cultured whole neonatal rat ovaries was developed to provide more mechanistic information. That approach was used to demonstrate that the cell survival c-kit/kit ligand signaling pathway is the direct target for VCD-induced ovotoxicity. Specifically, VCD directly interacts with the oocyte-associated c-kit receptor to inhibit its autophosphorylation, and thereby impair oocyte viability. The cellular and molecular approach developed to determine these findings is described in this article.
Keywords: follicle, ovary, toxicology, VCD, VCH
Introduction
In the ovary, the primary structure is the follicle, composed of a germ cell (oocyte) surrounded by somatic cells (granulosa and theca interna). In mammals during fetal (human) or early postnatal (rodents) development, oocytes are formed from dividing oogonia and become surrounded by a single layer of squamous shaped pre-granulosa cells. This structure forms the primordial follicle. Because oocytes in the primordial follicle pool are arrested in pro-phase of the first meiotic division, they are non-dividing and form the full cohort of germ cells the female will possess throughout her lifespan [Hoyer and Devine 2002]. Thus, environmental chemicals with the potential to destroy the primordial follicle pool in women are of concern because exposure to those chemicals could lead to early menopause. Early menopause would increase the risk of developing such menopause-associated disorders as cardiovascular disease, osteoporosis, metabolic syndrome, ovarian cancer, Alzheimer’s disease, and depression [Sowers et al. 1995; Carr 2003; Fernandez et al. 1998; Vanderhyden 2005; Waring 1999]. Conversely, environmental chemicals that target larger pre-antral (secondary) or antral follicles, but do not affect the primordial pool, might disrupt menstrual cyclicity causing reversible infertility. In that case, if exposure to those chemicals is removed, regular cyclicity and fertility might return to normal.
Several classes of chemicals have been shown to be associated with early ovarian failure (menopause) in women. One well known class of such chemicals is composed of chemotherapeutic agents in which in young women being treated for cancer experience irreversible infertility (sterility) [Sobrinho et al. 1971; Chapman 1983; Damewood and Grochow 1986]. Another observed cause of early menopause in women has been cigarette smoking, with a 1–4 year earlier onset compared with non-smokers [Mattison et al. 1989; Everson et al. 1986]. Cigarette smoke is a complex mixture of chemicals, however, evidence from animal studies supports that polycyclic aromatic hydrocarbons (PAHs) contained therein may contribute to early ovarian failure [Mattison and Thorgeirsson 1983; Borman et al. 2000]. Because chemotherapeutic agents and PAHs have been shown in animal studies to destroy follicles at all stages (small and large pre-antral, and antral) of development, including primordial, the early onset of menopause in exposed women is likely due, in part, to their effects on primordial follicles.
Certain occupational chemicals may also have the potential to cause early menopause in women due to a demonstrated potential in animal studies to destroy primordial follicles. One such chemical is 4-vinylcyclohexene (VCH) and its diepoxide metabolite (VCD). VCH is a dimer of 1,3-butadiene produced during the process of tire curing, whereas VCD is used commercially as a chemical intermediate and reactive diluent for diepoxides and epoxy resins [Huff 2001]. The National Toxicology Program (NTP) conducted two long term studies with VCH and VCD with the purpose of screening for carcinogenic potential in mice and rats [NTP 1986; 1989]. Animals were exposed to the chemicals over a two year period. Although the studies concluded that there was some carcinogenic potential with VCH and VCD following this lengthy exposure, an interesting observation was the appearance of ovarian and uterine atrophy in exposed mice within 13 weeks of exposure. Additionally, no visible ovarian follicles or corpora lutea were observed at the end of exposure. No similar effects were observed in rats. These findings suggested that these ovarian effects might result from direct damage to ovarian follicles. Because the apparent infertility was seen to be irreversible, this suggested further that one effect of VCH and VCD could be the result of destruction of the primordial follicle pool [Hoyer and Sipes 2007]. These reports prompted initiation of an ongoing investigation into possible ovarian effects of VCH and VCD in mice and rats.
In Vivo Dosing Studies
Initial studies were designed to investigate why mice developed ovarian damage from these chemicals, but rats appeared to be resistant. In a 30-day dosing experiment comparing VCH and VCD in mice and rats, VCH produced a dose-responsive loss of small ovarian follicles in mice, whereas, it was ineffective in rats [Smith et al. 1990]. However, both mice and rats were susceptible to the monoepoxide and diepoxide metabolites of VCH. Further, VCD caused follicle destruction at 2.5–3 times lower doses than did the monoepoxide in both species. This study suggested that VCD is the ovotoxic form and VCH represents the parent form of the compound (Fig. 1). A further structure–activity study supported that conclusion [Doerr et al. 1995]. Subsequently, studies addressing the metabolism of VCH and VCD led to the hypothesis that in mice VCH is more readily bioactivated to VCD, and VCD is less readily detoxified, as compared with rats [Hoyer and Sipes 2007]. Further studies have determined that the mouse and rat ovary possess the enzymatic capabilities to bioactivate and detoxify VCH and VCD, respectively [Cannady et al. 2002; 2003; Rajapaksa et al. 2007; Keating et al. 2008a; Keating et al. 2008b; Keating et al. 2010]. Therefore, the ovary itself may directly contribute to the degree of follicle damage produced by exposure to xenobiotic agents.
Figure 1.
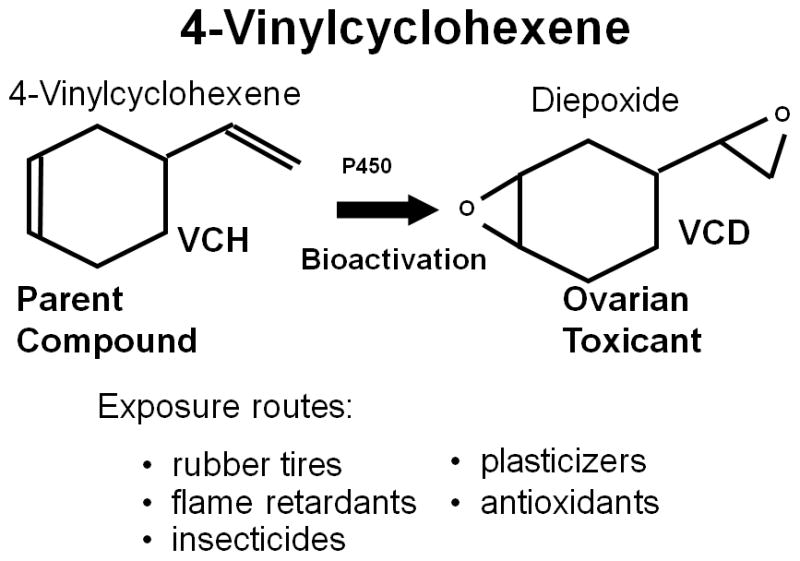
The occupational chemical, 4-vinylcyclohexene (VCH) is a source of exposure in the manufacture of rubber tires, flame retardants, insecticides, plasticizers, and antioxidants. VCH represents the parent form of the compound, and can be metabolized by cytochrome p450 enzymes to the diepoxide (VCD) which is the ovotoxic form of the chemical. Formation of the bioactive form (VCD) can occur in the liver, although the ovary itself expresses the enzymes capable of this conversion.
An early question in these studies was whether the observed ovotoxicity with VCH/VCD was due to direct ovarian targeting, or resulted from disruptions in hypothalamic-pituitary signaling. In the hypothalamic-pituitary-ovarian axis of regulation, gonadotropin releasing hormone (GnRH) released by the hypothalamus signals the pituitary to secrete luteinizing hormone (LH) and follicle stimulating hormone (FSH) [Hoyer and Devine 2002]. Consequently, ovarian hormones (17β-estradiol, progesterone, inhibin) provide a negative feedback on GnRH and LH/FSH. Thus, it was reasoned that if VCH/VCD targets the hypothalamus/pituitary, LH/FSH levels would drop and this would precede the observed loss of small pre-antral follicles. Conversely, if the chemicals target the ovary, follicle loss would precede an increase in LH/FSH (resulting from loss of negative feedback). In a long-term study following 30 days of dosing mice with VCH, substantial small pre-antral follicle loss was observed at the end of dosing, whereas, circulating FSH levels did not rise until 240 days after the onset of dosing [Hooser et al. 1994]. Therefore, it was concluded that VCH/VCD directly targets the ovary, and mechanistic investigations into VCD effects in the ovary were undertaken.
Dosing studies to that point had only investigated ovarian effects following 30 days of repeated dosing with VCH/VCD. Under those conditions, follicle populations in all stages of development (primordial, primary, secondary, antral) had been seen to be targeted. Thus, it was unknown whether VCH/VCD directly targets all sizes of ovarian follicles, or is selective for a specific population. Two time course studies identified that VCD directly targets primordial and primary follicles [Springer et al. 1996; Kao et al. 1999] (Fig. 2). Additionally, it was concluded that VCD-induced ovotoxicity requires repeated daily dosing [Springer et al. 1996; Borman et al. 1999].
Figure 2.
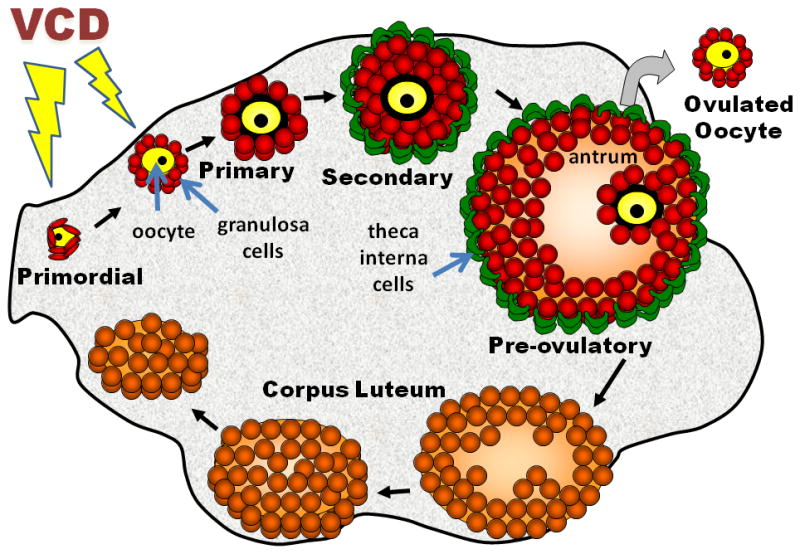
The mammalian ovary is a heterogeneous mixture of dynamic structures. The basic unit is the ovarian follicle which contains an oocyte surrounded by somatic cells. The most immature form of the follicle is termed primordial (surrounded by a single layer of squamous granulosa cells). Upon activation, the follicle goes through several stages of development: primordial to primary (layer of cuboidal granulosa cells); primary to secondary (granulosa cell layer proliferates, theca interna cells form); and secondary to pre-ovulatory (antrum develops). The pre-ovulatory follicle is responsible for production of 17β-estradiol. Following ovulation, remaining cells differentiate to become a corpus luteum which produces progesterone required for implantation and maintenance of pregnancy. 4-vinylcyclohexene diepoxide (VCD) specifically targets primordial and primary follicles in producing its ovotoxic effects.
An investigation into the nature of VCD’s effects on ovarian follicles was made. Two types of death, necrosis and apoptosis, can be a mechanism by which cells are destroyed. Cell death by necrosis usually occurs in response to injury and elicits an inflammatory response in surrounding tissue. Conversely, apoptosis is a physiological process of programmed cell death [Wyllie et al. 1980]. Thus, it was of interest to determine whether VCD causes necrosis (toxic response) or apoptosis (programmed cell death) in ovarian follicles. A morphological investigation determined that VCD causes accelerated atresia (apoptotic cell death) rather than necrosis [Springer et al. 1996]. In reaching that conclusion, it was postulated that due to the specificity for primordial and primary follicles, and because cell death is via a natural process (atresia), women who might be exposed to similar chemicals would only experience early menopause without first experiencing disruptions in menstrual cyclicity. Therefore, VCD appeared to be an ideal model chemical to study selective effects of xenobiotics on primordial and primary follicles.
To confirm that VCD-induced ovotoxicity is via accelerated atresia (apoptosis), more mechanistic investigations were required. Because VCD selectively targets primordial and primary follicles, a method to isolate those small follicles from the greater untargeted amount of ovarian tissue was needed. Thus, a method for isolation of small pre-antral follicles from the ovaries of animals that had been dosed with VCD was developed [Flaws et al. 1994]. By that method, ovaries are collected from rats that have been dosed daily with VCD. Following gentle dissociation of the ovaries with collagenase, intact follicles of all sizes are recovered in suspension. The dissociate is passed through a 250μm pore filter to exclude larger antral follicles (non-targets). Pre-calibrated Pasteur pipettes are then used to hand sort follicles in the filtrate into two populations, fraction 1 (primordial and primary, targeted by VCD) and fraction 2 (secondary, non-targets). The result is a fraction highly enriched in the target population of follicles, as well as a fraction containing non-targeted follicles (used to distinguish effects that are specifically due to VCD).
Having the ability to prepare a cellular fraction containing primordial and primary follicles, biochemical analyses of the effects of VCD on apoptotic signaling pathways were conducted. In some cell types, an apoptotic signal for apoptosis occurs at an intracellular checkpoint involving the Bcl-2 family of proto-oncogenes [Reed 1997]. Several members of this family can heterodimerize to modulate cellular apoptosis versus survival. Pro-apoptotic Bax and Bad can direct a cell death signal, whereas, Bcl-2 and its homolog Bcl-xl function in an anti-apoptotic manner. Using fraction 1 follicles isolated from rats dosed (15 days) with vehicle control or VCD (80 mg/kg), the following experiments determined that, compared with controls, the Bad/Bax response was increased by VCD [Hu et al, 2001a], and this culminated in activation of caspase-3 activity (pro-apoptotic executioner protease) [Hu et al. 2001b]. Another intracellular pathway associated with apoptosis is the mitogen activated protein kinase family (MAPK) [Marshall 1995]. The pro-apoptotic branch of this family included c-Jun-N-terminal kinase (JNK). Using the isolated follicle approach this pathway was also investigated [Hu et al. 2002]. As with Bcl-2, the pro-apoptotic branch of MAPK involving JNK was also seen to be activated by VCD. In both pathways, the VCD response was selective for fraction 1 follicles (target population) as compared with fraction 2 follicles which were insensitive. This helped confirm that the observed effects were those specifically elicited by VCD.
At this point, it was determined that an improved method of investigating mechanistic information using an in vitro approach would be necessary. In 2007, a detailed review of studies investigating VCD-induced ovotoxicity concluded with a series of still unanswered questions [Hoyer and Sipes 2007]. Those questions were: “Can we confirm whether the oocyte is directly targeted by VCD? Why are primordial and primary follicles selectively targeted? Can we identify a cellular component that directly interacts with VCD to initiate the apoptotic cascade?” Subsequent mechanistic studies utilizing an in vitro whole ovary culture system have been used to answer those questions.
In Vitro Culture Studies
An in vitro culture system has been established to assess the precise mechanisms underlying the accelerated follicle loss induced by VCD [Devine et al. 2002]. The culture system consists of ovaries from postnatal day (PND) 4 rats placed on a membrane floating in 0.5 ml culture medium. This allows the ovary sufficient access to both oxygen and nutrients from the medium. Environmental toxins, like VCD, can then be added to the medium and the cellular mechanism of follicular loss can be evaluated. Since the PND4 rat ovary is highly enriched in primordial and primary follicles and VCD selectively targets these early follicular stages, the neonatal rat ovarian culture system is especially useful for evaluating ovotoxicity by VCD. In vivo studies must also contend with metabolic contributions from other tissues, such as the liver, to clearance of VCD, which adds complexity to those studies and is avoided in the culture system. The in vitro studies have demonstrated that the ovarian organ cultures mimic the physiological response in vivo, making it a valuable tool for mechanistic studies [Devine et al. 2002]. A time course of VCD exposure has also shown that a depletion of both primordial and primary follicles occurs following 6 days in culture [Keating et al. 2009].
A diverse group of growth factors has been recognized as important for follicular survival and development. Investigations into the ability of some of these growth factors to override VCD-induced ovotoxicity have been made. Those growth factors include: granulosa cell-associated factors, kit ligand (KITLG), leukemia inhibitory factor (LIF), growth and differentiation factor 9 (GDF9), and bone morphogenic factor 4(BMP4); oocyte-associated factors, glial cell line-derived neurotrophic factor (GDNF), platelet-derived growth factor isoform B (PDGFB), fibroblast growth factor 2 (FGF2), and an ovarian thecal cell factor, fibroblast growth factor 7 (FGF7) [Mark-Kappeler et al. 2011; Fernandez et al. 2008]. Amongst all of the growth factors tested, only KITLG demonstrated an ability to attenuate VCD-induced ovotoxicity (Fig. 3). The growth factor KITLG binds to its oocyte-associated receptor, KIT, which plays an important role in follicular survival, and is able to act as an anti-apoptotic factor in oocytes of primordial follicles [Parrott and Skinner 1999; Jin et al. 2005]. Because only endogenous KITLG was shown to protect against VCD-induced ovotoxicity, this suggested that the KIT/KITLG signaling pathway is involved in the ability of VCD to target primordial and primary follicles.
Figure 3.

The effect of exogenous growth factors on VCD-induced ovotoxicity. A variety of growth factors have been shown to be important for development and survival in small pre-antral follicles. Postnatal day 4 rat ovaries were exposed to growth factors ± VCD (30 μM) for 8 days. Growth factors tested were: kit ligand (KITLG), leukemia inhibitory factor (LIF), glial cell line-derived neurotrophic factor (GDNF), platelet-derived growth factor isoform B (PDGFB), growth factor 9 (GDF9), and bone morphogenic factor 4 (BMP4). Black bars=oocyte targeted. Grey bars=granulosa cell targeted. Primordial follicles were counted and represented as a % of control (*P < 0.05, different from control).
KITLG and KIT interaction plays an important role in the communication between the oocyte and surrounding granulosa cells by activating downstream pathway members. This plays a vital role in oocyte survival [Liu et al. 2006] (Fig. 4). Therefore, investigations into the effect of VCD on KIT/KITLG and members of its cellular signaling cascade were undertaken. Relative to controls, on day 4 of VCD exposure there was a decrease in mRNA encoding Kit and on day 6 an increase in mRNA encoding Kitlg [Fernandez et al. 2008]. VCD also decreased levels of KIT protein on the oocyte pericytoplasmic membrane following 4 days of exposure [Keating et al. 2011]. The binding of KITLG to KIT has been shown to activate the PI3K signaling pathway [Reddy et al. 2005]. AKT functions as an important downstream molecule in the PI3K signaling pathway. Once phosphorylated, activated ovarian AKT translocates to the nucleus, and plays a role in primordial to primary follicle activation and recruitment [Reddy et al. 2005; Liu et al. 2006]. In assessing VCD exposure, a decrease in oocyte nuclear pAKT protein was observed on day 2 of culture supporting a decrease in its activity. This demonstrated an early downstream response to VCD interaction with KIT receptor signaling [Keating et al. 2011].
Figure 4.
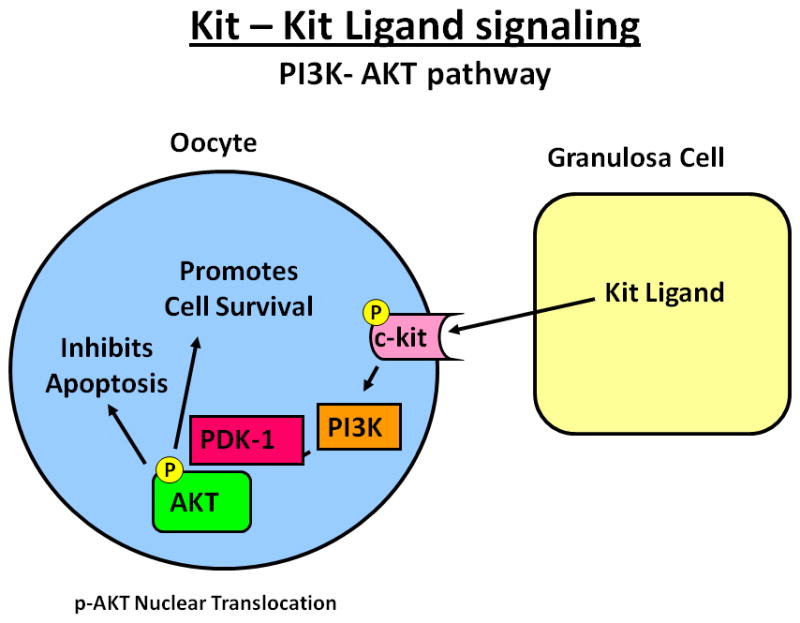
The KIT/KITLG pathway in primordial and small primary follicles participates in oocyte viability and survival. Kit ligand (KITLG) produced by the granulosa cells binds to KIT (tyrosine kinase receptor) on the oocyte. Binding of KITLG to KIT leads to activation of protein kinase activity, KIT autophosphorylation, followed by a series of cellular responses via the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Active PI3K signals to 3-phosphoinositide-dependent protein kinase-1 (PDK-1), which phosphorylates and activates the protein kinase AKT. pAKT then translocates to the nucleus and activates a variety of pathways important for inhibition of apoptosis and promotion of cell survival.
Autophosphorylation of KIT activates its signaling cascade. Therefore, the effect of VCD on phospho-KIT (pKIT) was also investigated. A decrease in pKIT protein was observed with VCD exposure after 2 days of culture. This observation supported that VCD-induced ovotoxicity is initiated by direct interaction with KIT. Additional experiments were conducted to further analyze a possible interaction between VCD and KIT using an anti-mouse KIT2 (ACK2) antibody. ACK2 has been shown to bind to KIT and block its signaling activity. This results in inhibited oocyte growth, and increased follicular atresia [Packer et al. 1994; Carlsson et al. 2006]. There was no effect of VCD or ACK2 on total KIT protein following 2 days in culture, however, ACK2±VCD caused a decrease in pKIT protein [Mark-Kappeler et al. 2011]. This suggests a similar mechanism of interaction with KIT between ACK2 and VCD. Additionally, the effects of an anti-mouse KIT4 (ACK4) antibody on the ovary were evaluated and compared with those of ACK2. ACK4 recognizes the KIT receptor, but it is directed against a different epitope than ACK2. While ACK2 is an antagonist of KIT, ACK4 binds to the receptor but does not block its function in hemopoietic progenitor cells [Ogawa et al. 1991]. On day 2 of in vitro incubation of ovaries with ACK4 and VCD there was a partial attenuation of the decreased phosphorylation of KIT protein caused by VCD [Mark-Kappeler et al., 2011]. This suggests that ACK4, by binding to KIT, can protect it from interacting with VCD. Alternatively, ACK4 binding to KIT may change the conformation of KIT and interfere with its ability to be targeted by VCD. Overall, these results provided further evidence for a direct interaction between VCD and KIT.
In summary, the collective findings reveal that VCD interacts directly with membrane-bound KIT and its downstream signaling pathway in the oocyte to cause follicular destruction. Initially, there is a decrease in phosphorylation of KIT and AKT on day 2 of VCD exposure, relative to controls (post-translational signaling effects). Subsequently, on day 4 of VCD exposure there is a decrease in expression of KIT (mRNA and protein), as well as, a decrease in mRNA encoding AKT (transcriptional effects). Lastly, there is an increase in KITLG mRNA on day 6 of VCD exposure, relative to controls. All of these effects on the KIT/KITLG signaling pathway show that post-translational signaling effects of VCD precede transcriptional effects and the result is small follicle loss on day 6 of VCD exposure (Fig. 5).
Figure 5.
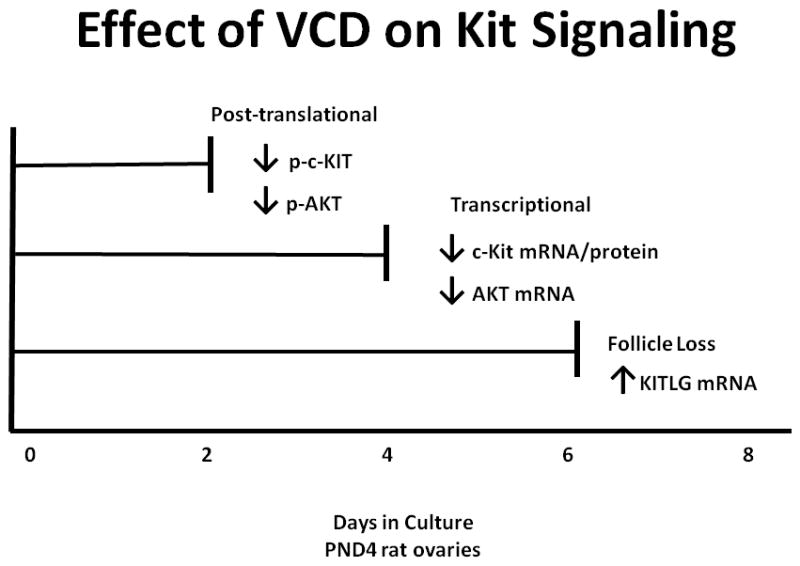
Time course of the effect of VCD on the KIT signaling pathway. mRNA and protein samples were examined from neonatal rat ovaries ± VCD (30 μM) on days 2, 4, 6, and 8 of culture. Relative to controls, two days of VCD exposure decreased post-translational events (phosphorylation of KIT and AKT). Relative to controls, four days of VCD exposure decreased in mRNA encoding KIT, along with a decrease in AKT mRNA (post-transcriptional effects). Relative to controls, six days of VCD exposure increased in mRNA encoding KITLG and caused small follicle loss.
Conclusions
Thus, with the combined in vivo and in vitro approach using morphological, cellular, biochemical, and molecular methods, strong evidence to answer the remaining questions posed in 2007 and stated above [Hoyer and Sipes 2007] has been provided. “Can we confirm whether the oocyte is directly targeted by VCD?” The plasma membrane of the oocyte appears to be the direct target for VCD. “Why are primordial and primary follicles selectively targeted?” Kit is expressed and in the ovary functions as a survival factor largely in primordial and primary follicles. “Can we identify a cellular component that directly interacts with VCD to initiate the apoptotic cascade?” The initiating event appears to be direct inhibition of autophosphorylation of Kit (impairing downstream survival signaling events). Proteomic studies are currently underway to identify the molecular site of Kit targeting by VCD. In the future, VCD will be used as a tool to identify intracellular decisions which influence primordial follicle survival versus activation. Findings using this approach will improve our understanding of the complex events that dictate the fate of the primordial follicle cohort within the mammalian ovary, and will help elucidate factors that regulate the dynamic balance that is achieved in determination of the normal reproductive lifespan in women.
Acknowledgments
The authors wish to thank Dr. Minetaro Ogawa at Kumamoto University (Kumamoto, Japan) for the generous gift of anti-mouse KIT4 (ACK4).
Abbreviations
- VCD
4-vinylcyclohexene diepoxide
- VCH
4-vinylcyclohexene
- PAHs
polycyclic aromatic hydrocarbons
- NTP
National Toxicology Program
- GnRH
gonadotropin releasing hormone
- LH
luteinizing hormone
- FSH
follicle stimulating hormone
- MAPK
mitogen activated protein kinase family
- PND
postnatal day
- JNK
c-Jun-N-terminal kinase
- KITLG
kit ligand
- LIF
leukemia inhibitory factor
- GDF9
growth and differentiation factor 9
- BMP4
bone morphogenic factor 4(BMP4)
- GDNF
glial cell line-derived neurotrophic factor
- PDGFB
platelet-derived growth factor isoform B
- FGF2
fibroblast growth factor 2
- FGF7
fibroblast growth factor 7
- ACK
anti-mouse KIT
- pKIT
phospho-KIT
Footnotes
Declaration of Interest: This research was supported by training grant ES007091, grant R01 ES09246, and Center grant ES06694. The authors report no declarations of interest.
References
- Borman SM, VanDePol BJ, Kao S, Thompson KE, Sipes IG, Hoyer PB. A single dose of the ovotoxicant 4-vinylcyclohexene diepoxide is protective in rat primary ovarian follicles. Toxicol Appl Pharmacol. 1999;158:244–252. doi: 10.1006/taap.1999.8702. [DOI] [PubMed] [Google Scholar]
- Borman SM, Christian PJ, Sipes IG, Hoyer PB. Ovotoxicity in female Fischer rats and B6 mice induced by low-dose exposure to three polycyclic aromatic hydrocarons: Comparison through calculation of an ovotoxic index. Toxicol Appl Pharmacol. 2000;167:191–198. doi: 10.1006/taap.2000.9006. [DOI] [PubMed] [Google Scholar]
- Cannady EA, Dyer CA, Christian PJ, Sipes IG, Hoyer PB. Expression and activity of microsomal epoxide hydrolase in follicles isolated from mouse ovaries. Toxicol Sci. 2002;68:24–31. doi: 10.1093/toxsci/68.1.24. [DOI] [PubMed] [Google Scholar]
- Cannady EA, Dyer CA, Christian PJ, Sipes IG, Hoyer PB. Expression and activity of cytochromes P450 2E1, 2A, and 2B in the mouse ovary: the effect of 4-vinylcyclohexene and its diepoxide metabolite. Toxicol Sci. 2003;73:423–430. doi: 10.1093/toxsci/kfg077. [DOI] [PubMed] [Google Scholar]
- Carlsson I, Laitinen MP, Scott J, Louhio H, Velentzis L, Tuuri T, et al. Kit ligand and c-kit are expressed during early human ovarian follicular development and their interaction is required for the survival of follicles in long-term culture. Reprod. 2006;131:641–649. doi: 10.1530/rep.1.00868. [DOI] [PubMed] [Google Scholar]
- Carr MC. The emergence of the metabolic syndrome with menopause. J Clin Endocrinol Metabol. 2003;88:2404–2411. doi: 10.1210/jc.2003-030242. [DOI] [PubMed] [Google Scholar]
- Chapman RM. Gonadal injury resulting from chemotherapy. Am J Ind Med. 1983;4:149–161. [PubMed] [Google Scholar]
- Damewood MD, Grochow LB. Prospects for fertility after chemotherapy or radiation for neoplastic disease. Fertil Steril. 1986;45:443–459. doi: 10.1016/s0015-0282(16)49268-x. [DOI] [PubMed] [Google Scholar]
- Devine PJ, Sipes IG, Skinner MK, Hoyer PB. Characterization of a rat in vitro ovarian culture system to study the ovarian toxicant 4-vinylcyclohexene diepoxide. Toxicol Appl Pharmacol. 2002;184:107–115. [PubMed] [Google Scholar]
- Doerr JK, Hooser SB, Smith BJ, Sipes IG. Ovarian toxicity of 4- vinylcyclohexene and related olefins in B6C3F1 mice: Role of diepoxides. Chem Res Toxicol. 1995;8:963–969. doi: 10.1021/tx00049a010. [DOI] [PubMed] [Google Scholar]
- Everson BR, Sandler DP, Wilcox AJ, Schreinemachhers D, Shore DL, Weinberg CR. Effect of passive exposure to smoking in age at natural menopause. Br Med J. 1986;293:792. doi: 10.1136/bmj.293.6550.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E, LaVecchia C, Braga C, Talamini R, Negri E, Prazzini F, Franceschi S. Hormone replacement therapy and risk of colon and rectal cancer. Cancer Epidemiol Viomarkers Prev. 1998;7:329–333. [PubMed] [Google Scholar]
- Fernandez SM, Keating AF, Christian PJ, Sen N, Hoying JB, Brooks HL, et al. Involvement of the KIT/KITLG signaling pathway in 4-vinylcyclohexene diepoxide-induced ovarian follicle loss in rats. Biol Reprod. 2008;79:318–327. doi: 10.1095/biolreprod.108.067744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaws JA, Salyers KL, Sipes IG, Hoyer PB. Reduced ability of rat pre-antral follicles to metabolize 4-vinylcyclohexene diepoxide. Toxicol Appl Phrmacol. 1994;126:286–294. doi: 10.1006/taap.1994.1118. [DOI] [PubMed] [Google Scholar]
- Hooser SB, Douds DA, Hoyer PB, Sipes IG. Long-term ovarian and hormonal alterations due to the ovotoxin 4-vinylcyclohexene. Reprod Toxciol. 1994;8:315–323. doi: 10.1016/0890-6238(94)90047-7. [DOI] [PubMed] [Google Scholar]
- Hoyer PB, Devine PJ. Endocrine toxicology: The female reproductive system. In: Derelanko MJ, editor. Handbook of Toxicology. 2. CRC Press; FL USA: 2002. pp. 573–595. [Google Scholar]
- Hoyer PB, Sipes IG. Development of an animal model for ovotoxicity using 4- vinylcyclohexene: a case study. Birth Defects Res B Dev Reprod Toxicol. 2007;80:113–125. doi: 10.1002/bdrb.20103. [DOI] [PubMed] [Google Scholar]
- Hu X, Christian PJ, Sipes IG, Hoyer PB. Expression and redistribution of cellular bad, bax and bcl-xl protein is associated with VCD-induced ovotoxicity in rats. Biol Reprod. 2001a;65:1489–1495. doi: 10.1095/biolreprod65.5.1489. [DOI] [PubMed] [Google Scholar]
- Hu X, Christian PJ, Thompson KE, Sipes IG, Hoyer PB. Apoptosis induced in rats by 4-vinylcyclohexene diepoxide is associated with activation of the caspase cascades. Biol Reprod. 2001b;65:87–93. doi: 10.1095/biolreprod65.1.87. [DOI] [PubMed] [Google Scholar]
- Hu X, Flaws JA, Sipes IG, Hoyer PB. Activation of mitogen-activated protein kinases and AP-1 transcription factor in ovotoxicity induced by 4-vinylcyclohexene diepoxide in rats. Biol Reprod. 2002;67:718–724. doi: 10.1095/biolreprod.102.004259. [DOI] [PubMed] [Google Scholar]
- Huff J. Carcinogenicity bioassays of bisphenol A, 4-vinylcyclohexene diepoxide, and 4-vinylcyclohexene. Toxicol Sci. 2001;64:282–283. doi: 10.1093/toxsci/64.2.282. [DOI] [PubMed] [Google Scholar]
- Jin X, Han CS, Yu FQ, Wei P, Hu ZY, Liu YX. Anti-apoptotic action of stem cell factor on oocytes in primordial follicles and its signal transduction. Mol Reprod Dev. 2005;70:82–90. doi: 10.1002/mrd.20142. [DOI] [PubMed] [Google Scholar]
- Kao S, Sipes IG, Hoyer PB. Early effects of ovotoxicity induced by 4-vinylcyclohexene diepoxide in rats and mice. Reprod Toxicol. 1999;13:67–75. doi: 10.1016/s0890-6238(98)00061-6. [DOI] [PubMed] [Google Scholar]
- Keating AF, Sipes IG, Hoyer PB. Expression of ovarian microsomal epoxide hydrolase and glutathione-S-transferase during onset of VCD-induced ovotoxicity in B6C3F1 mice. Toxicol Appl Pharmacol. 2008a;230:109–116. doi: 10.1016/j.taap.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating AF, Rajapaksa K, Sipes IG, Hoyer PB. Effect of CYP2E1 gene deletion in mice on expression of microsomal epoxide hydrolase in response to VCD exposure. Toxicol Sci. 2008b;105:351–359. doi: 10.1093/toxsci/kfn136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating AF, Mark CJ, Sen N, Sipes IG, Hoyer PB. Effect of phosphatidylinositol-3 kinase inhibition on ovotoxicity caused by 4-vinylcyclohexene diepoxide and 7, 12-dimethylbenz[a]anthracene in neonatal rat ovaries. Toxicol Appl Pharmacol. 2009;241:127–134. doi: 10.1016/j.taap.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating AF, Sen N, Sipes IG, Hoyer PB. Dual protective role for glutathione-S-transferase class pi against VCD-induced ovotoxicity in the rat ovary. Toxicol Appl Pharmacol. 2010;247:71–75. doi: 10.1016/j.taap.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating AF, Fernandez SM, Mark-Kappeler CJ, Sen N, Sipes IG, Hoyer PB. Inhibition of PI3K signaling pathway members by the ovotoxicant 4-vinylcyclohexene diepoxide in rats. Biol Reprod. 2011;84:743–751. doi: 10.1095/biolreprod.110.087650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Rajareddy S, Liu L, Jagarlamundi K, Boman K, Selstam G, et al. Control of mammalian oocyte growth and early follicular development by the oocyte PI3K pathway: new roles for an old timer. Dev Biol. 2006;299:1–11. doi: 10.1016/j.ydbio.2006.07.038. [DOI] [PubMed] [Google Scholar]
- Mark-Kappeler CJ, Sen N, Lukefahr A, McKee L, Sipes IG, Konhilas J, et al. Inhibition of ovarian KIT phosphorylation by the ovotoxicant 4-vinylcyclohexene diepoxide in rats. Biol Reprod. 2011 Jun 15; doi: 10.1095/biolreprod.111.092742. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaliing: transient versus sustained extracellular signal-related kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Mattison DR, Thorgeirsson SS. Ovarian arly hydrocarbon hydroxylase activity and primordial oocyte toxicity of polycyclic aromatic hydrocarbons in mice. Cancer Res. 1979;39:3471–3475. [PubMed] [Google Scholar]
- Mattison DR, Plowchalk BS, Meadows MJ, Miller MM, Malek A, London S. The effect of smoking on oogenesis, fertilization, and implantation. Semin Reprod Endocrinol. 1989;7:291–304. [Google Scholar]
- NTP. Tech Report No TR-303. Research Triangle Park, NC USA: National Toxicology Program; 1986. Toxicology and carcinogenesis studies of 4-vinylcyclohexene (CAS No. 100-40-3) in F344/N rats and B6C23F1 mice (gavage studies) [PubMed] [Google Scholar]
- NTP. Tech Report No TR-362. Research Triangle Park, NC USA: National Toxicology Program; 1989. Toxicology and carcinogenesis studies of 4-vinylcyclohexne diepoxide (CAS No. 106-87-6) in F344/N rats and B6C3F1 mice (dermal studies) [PubMed] [Google Scholar]
- Ogawa M, Matsuzaki Y, Nishikawa S, Hayahi SI, Kunisada T, Sudo T, et al. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med. 1991;174:63–71. doi: 10.1084/jem.174.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer A, Hsu Y, Besmer P, Bachvarova R. The ligand of the c-kit receptor promotes oocyte growth. Dev Biol. 1994;161:194–205. doi: 10.1006/dbio.1994.1020. [DOI] [PubMed] [Google Scholar]
- Parrott JA, Skinner MK. Kit-ligand/stem cell factor induces primordial follicle development and initiates folliculogenesis. Endocrinol. 1999;140:4262–4271. doi: 10.1210/endo.140.9.6994. [DOI] [PubMed] [Google Scholar]
- Rajapaksa KS, Cannady EA, Sipes IG, Hoyer PB. Involvement of CYP2E1 enzyme in ovotoxicity caused by 4-vinylcyclohexene and its metabolites. Toxicol Appl Pharmacol. 2007;221:215–221. doi: 10.1016/j.taap.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Shen L, Ren C, Boman K, Lundin E, Ottander U, et al. Activation of Akt (PKB) and suppression of FKHRL1 in mouse and rat oocytes by stem cell factor during follicular activation and development. Dev Biol. 2005;281:160–170. doi: 10.1016/j.ydbio.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Reed J. Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin Hematol. 1997;34:9–19. [PubMed] [Google Scholar]
- Smith BJ, Mattison DR, Sipes IG. The role of epoxidation in 4-vinylcyclohexene-induced ovarian toxicity. Toxicol Appl Pharmacol. 1990;105:372–381. doi: 10.1016/0041-008x(90)90141-g. [DOI] [PubMed] [Google Scholar]
- Sobrinho LG, Levine RA, DeConti RC. Amenorrhea in patients with Hodgkin’s disease treated with antineoplastic agents. Amer J Obstet Gynecol. 1971;109:135–139. doi: 10.1016/0002-9378(71)90848-9. [DOI] [PubMed] [Google Scholar]
- Springer LN, McAsey ME, Flaws JA, Tilly JL, Sipes IG, Hoyer PB. Involvement of apopotosis in 4-vinylcyclohexene diepoxide-induced ovotoxicity in rats. Toxicol Appl Phrmacol. 1996;139:394–401. doi: 10.1006/taap.1996.0180. [DOI] [PubMed] [Google Scholar]
- Sowers MFR, LaPietra MT. Menopause: Its epidemiology and potential association with chronic diseases. Epidemiol Rev. 1995;17:287–302. doi: 10.1093/oxfordjournals.epirev.a036194. [DOI] [PubMed] [Google Scholar]
- Vanderhyden BC. Loss of ovarian function and the risk of ovarian cancer. Cell Tiss Res. 2005;322:117–124. doi: 10.1007/s00441-005-1100-1. [DOI] [PubMed] [Google Scholar]
- Waring SC, Rocca WA, Petersen RC, O’Brien PC, Tangalos EG, Kokmen E. Postmenopausal estrogen replacement therapy and risk of AD: A population-based study. Neruol. 1999;52:965–970. doi: 10.1212/wnl.52.5.965. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–305. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]