

Abstract
Huntington's disease (HD) is a neurodegenerative disorder caused by polyglutamine (polyQ) expansion in Huntingtin protein (Htt). PolyQ expansion in Httexp causes selective degeneration of striatal medium spiny neurons (MSNs) in HD patients. Striatal MSN neurons receive glutamatergic input from the cortex and dopaminergic input from the substantia nigra. In previous studies with the yeast artificial chromosome (YAC128) transgenic HD mouse model, we established a connection between glutamate receptor activation, disturbed calcium (Ca2+) signaling, and apoptosis of HD MSNs (Tang et al., 2005). Here, we used the same YAC128 mouse model to investigate the role of dopaminergic signaling in HD. We discovered that glutamate and dopamine signaling pathways act synergistically to induce elevated Ca2+ signals and to cause apoptosis of YAC128 MSNs in vitro. We demonstrated that potentiating effects of dopamine are mediated by D1-class dopamine receptors (DARs) and not by D2-class DARs. Consistent with in vitro findings, in whole-animal experiments we found that persistent elevation of striatal dopamine levels exacerbated the behavioral motor deficits and MSN neurodegeneration in YAC128 mice. We further discovered that the clinically relevant dopamine pathway inhibitor tetrabenazine alleviated the motor deficits and reduced striatal cell loss in YAC128 mice. Our results suggest that dopamine signaling pathway plays an important role in HD pathogenesis and that antagonists of dopamine pathway such as tetrabenazine or dopamine receptor blockers may have a therapeutic potential for treatment of HD beyond well established “symptomatic” benefit.
Keywords: dopamine, calcium signaling, Huntington's disease, transgenic mouse, stereology, rotarod, tetrabenazine
Introduction
Huntington's disease (HD) is a dominantly inherited neurodegenerative disorder that has onset usually between 35 and 50 years of age with chorea, psychiatric disturbances, and gradual but inexorable intellectual decline to death 15–20 years after onset (Vonsattel and DiFiglia, 1998). Neuropathological analysis of HD brains reveals selective and progressive neuronal loss of the GABAergic medium spiny neurons (MSNs) in the striatum (Vonsattel and DiFiglia, 1998). The cause of HD is an expanded polyglutamine (polyQ) in the N terminus of Huntingtin (Htt), a 350 kDa ubiquitously expressed cytoplasmic protein of unknown function (The Huntington's Disease Collaborative Research Group, 1993). The cellular mechanisms underlying the cause of MSN neurodegeneration in HD are not clear, although most experimental evidence indicates that polyQ expansion in Httexp leads to a “toxic gain of function” (Tobin and Signer, 2000). Recent results suggested that abnormal Ca2+ signaling may play an important role in HD pathogenesis (Bezprozvanny and Hayden, 2004). It has been demonstrated that Httexp facilitates activity of the NR2B subtype of NMDA receptors (Chen et al., 1999; Sun et al., 2001; Zeron et al., 2002; Song et al., 2003) and the type 1 inositol 1,4,5-trisphosphate receptors (InsP3R1s) (Tang et al., 2003b). Recent studies with the MSN neurons from the yeast artificial chromosome (YAC) transgenic HD mouse models supported the connection between activation of glutamate receptors, Ca2+ overload, and apoptosis of MSNs in HD (Zeron et al., 2004; Tang et al., 2005; Shehadeh et al., 2006).
In addition to glutamatergic stimulation from the cortex, the striatum is the predominant target of dopaminergic neurons that originate from the substantia nigra (Gerfen, 1992). The connection between dopaminergic signaling and HD was uncovered when over one-third of asymptomatic relatives of individuals with HD developed dyskinesias in response to l-DOPA (Klawans et al., 1970). Since then, an increasing body of experimental evidence has pointed to a potential role for the dopaminergic system in HD (Jakel and Maragos, 2000). Moreover, a recent short-term clinical trial demonstrated that dopamine antagonist tetrabenazine (TBZ) efficiently reduced chorea in HD patients compared with placebo group (Huntington Study Group, 2006). Despite multiple experimental and clinical evidence suggesting a potential importance of dopaminergic signaling in HD, the mechanistic basis for these observations is poorly understood. Here, we employ the YAC128 HD mouse model (Slow et al., 2003) and use Ca2+ imaging techniques, in vitro apoptosis assays, behavioral assessments, and quantitative neuropathological analysis to evaluate the contribution of dopamine signaling to degeneration of HD MSNs. Based on the obtained results, we conclude that dopamine and glutamate signaling pathways act synergistically to potentiate Ca2+ signals and to induce apoptosis of HD MSNs. Furthermore, we demonstrate that dopamine inhibitors protect HD MSNs from cell death both in vitro and in vivo.
Materials and Methods
Primary neuronal cultures.
Generation and breeding of YAC128 HD transgenic mice (FVBN/NJ background strain) has been described previously (Slow et al., 2003). Heterozygous male YAC128 mice were crossed with wild-type (WT) female mice, and the resulting litters were collected and genotyped at postnatal day 1 (P1)–P2 as described previously (Tang et al., 2005). The MSN cultures from YAC128 transgenic P1–P2 pups and WT littermates were established and maintained as described previously (Tang et al., 2005).
Ca2+ imaging experiments.
The 13–14 d in vitro (DIV) MSN cultures established from WT and YAC128 mice were loaded with 5 μm fura-2 AM (Invitrogen, Eugene, OR) in artificial CSF [ACSF; (in mm) 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.3] for 45 min at 37°C. The Ca2+ imaging experiments were performed using DeltaRAM illuminator (Photon Technology International, Birmingham, NJ) and IC-300 camera (Photon Technology International) and analyzed by ImageMaster Pro software (Photon Technology International) as described previously (Tang et al., 2005). Baseline (1–3 min) measurements were obtained before first pulse of glutamate. The 5 μm glutamate solution was dissolved in ACSF, and 1 min pulses of 37°C glutamate solution (SH-27B in-line solution heater) were applied using a VC-6 valve controller (Warner Instruments, Hamden, CT) driven by a square-pulse electrical waveform generator (model 148A; Willtek, IsmaningGermany) as described previously (Tang et al., 2005). For dopamine experiments, MSN cultures were incubated in the presence of 50 μm dopamine [or 25 μm (±)-1-phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrobromide (SKF38393) or 25 μm (−)-quinpirole] at 37°C for 6 min and then stimulated with 5 μm glutamate solution pulses in the continuous presence of dopamine or dopamine agonists as described above. The intracellular levels of free cytosolic Ca2+ concentrations [[Ca2+]i (nm)] in these experiments were determined from the following equation (Grynkiewicz et al., 1985): [Ca2+]i (nm) = Kd × [(R − Rmin)/(Rmax − R)] × Sf,380/Sb,380, where Kd = 285 nm is the dissociation constant of fura-2 for Ca2+ at 37°C (Groden et al., 1991), R is the experimentally determined 340/380 ratio, Rmax is the 340/380 ratio for fura-2 saturated with Ca2+, determined by application of 20 mm Ca2+ and 10 μm ionomycin, followed by washout and addition of ACSF buffer containing 10 mm EGTA to determine Rmin as 340/380 ratio for Ca2+-free fura-2. Sf,380/Sb,380 is the ratio of fluorescence intensity of the Ca2+-free and Ca2+-bond forms of fura-2 at 380 nm. Sf,380/Sb,380 was determined as 2.0 in our experiments.
In vitro apoptosis experiments.
The 14 DIV MSNs were exposed to a range of dopamine concentrations (0, 25, 50, 100, 250, and 500 μm) added to the culture medium with or without 25 μm glutamate as indicated in the text. The cells were maintained in a cell culture incubator (humidified 5% CO2 at 37°C). (5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine [(+)-MK-801], ifenprodil, 2-methyl-6-(phenylethynyl)-pyridine (MPEP), 7-(hydroxyimino) cyclopropachromen-1a-carboxylate ethyl ester (CPCCOEt), R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine [R-(+)-SCH23390], and spiperone were added to the cell culture media 30 min before the addition of dopamine and/or glutamate as indicated in the text. 2-APB and enoxaparin were added 2–3 h before dopamine and/or glutamate exposure. SKF38393 or (−)-quinpirole were added simultaneously with glutamate. Dopamine, glutamate, and all other drugs were dissolved in the culture medium as 10× concentrated solutions and prewarmed to 37°C before addition to the culture wells. Immediately after 8 h of exposure to dopamine and/or glutamate, neurons were fixed, permeabilized, and stained using the DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) according to the manufacturer's instructions. The terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining assay was performed and quantified as described previously (Tang et al., 2005). The fractions of TUNEL-positive nuclei (apoptotic cells) are presented as mean ± SD (n is the number of fields counted).
Drug delivery in mice.
The drugs were delivered to YAC128 mice by adapting an approach used previously in studies of R6/2 mice (Hickey et al., 2002). Groups of WT and YAC128 mice were fed with a mixture of 2.5 mg of l-DOPA and 0.3 mg of benserazide dissolved in PBS (l-DOPA groups) or with a mixture of 2.5 mg of l-DOPA, 0.3 mg of benserazide, and 0.125 mg of TBZ dissolved in 50 μl PBS (l-DOPA/TBZ groups). Control groups were fed with PBS. All drugs were resuspended in 2% corn flour, and mice were fed orally twice a week from 2 to 10 months of age. To determine effective concentrations of the drugs and dopamine in the brain, the wild-type mice were fed the same drugs for 2 months and anesthetized 30 min after the last drug delivery. The blood samples were collected in a heparinized tube, diluted 1:1 in water for hemolyzation, and flash frozen. The brains were immediately collected, and striatal region was dissected from one hemisphere. The other hemisphere and the striatum were flash frozen in liquid nitrogen. The blood, hemisphere, and striatum from each mouse were numbered and shipped on dry ice to Melior Discovery (Exton, PA) for analysis. Blood samples were extracted with an acetonitrile/protein precipitation method. Brain hemisphere and striatal tissue were homogenized in an equal volume of acetonitrile and clarified by centrifuge. The dopamine, l-DOPA, and TBZ levels were analyzed using HPLC and compared with a standard. Data are expressed as average of the values from five samples ± SEM (see Results).
Motor coordination assessments in mice.
The motor coordination experiments were performed as described previously for R6/2 mice and YAC128 mice (Carter et al., 1999; Slow et al., 2003). Economex rotarod apparatus (Columbus Instruments, Columbus, OH) was used to assess rotarod behavior. At each time point, the mice were trained on the accelerating rotarod (accelerated from 0 to 40 rpm over 200 s) with 3 trials/d for 3 consecutive days, by which time a steady baseline of performance was attained. The testing was executed over 1 d with 1.5 h of rest between tests. The mean latency to fall off the rotarod recorded in the two trials was used in the analysis.
The “beam walking” assay was performed using a home-built experimental setup. The 17 mm round beam, 11 mm round beam, and 5 mm square beam were used in our studies. At each time point, the mice were trained on 17 mm round beam for 3 consecutive days (4 trials/d) to traverse the beam to the enclosed box. Once the stable baseline of performance was obtained, the mice were tested in two consecutive trials on 17 and 11 mm round beams and 5 mm square beam, in each case progressing from the widest to the narrowest beam. The latency to traverse the middle section (80 cm in length) of each beam and the number of times the hind feet slipped off each beam were recorded for each trial. For each measure, the mean scores of the two trials for each beam were used in the analysis.
For the footprint test, the hindfeet and forefeet of the mice were coated with green and purple nontoxic paints, respectively. The mice were then trained to walk along a 50-cm-long, 10-cm-wide open-top runway (with 10-cm-high walls) into an enclosed box. All mice were given 3 runs/d for 3 consecutive days. A fresh sheet of white paper was placed on the floor of the runway for each run. The footprint patterns were assessed quantitatively by four measurements: stride length, hindbase width, frontbase width, and front/hind footprint overlap as described previously (Carter et al., 1999).
Neuropathological assessments in mice.
The neuropathological assessments were performed as described previously (Slow et al., 2003). At conclusion of behavioral testing (11 months time point), the mice were terminally anesthetized and perfused transcardially with 10 ml of 0.9% saline followed by 100 ml of fixative (4% paraformaldehyde in 0.1 m PBS, pH 7.4). All brains were removed from skull and weighed. The brains were postfixed overnight at 4°C in 4% paraformaldehyde and equilibrated in 20–30% (w/v) sucrose in PBS. The brains were sliced to 30-μm-thick coronal sections using a Leica (Bannockburn, IL) SM2000R sliding microtome. The coronal sections spaced 360 μm apart throughout the striatum (in the range from +1.70 mm to −2.30 mm relative to bregma) were stained with NeuN monoclonal antibody (1:1000 dilution; Millipore, Billerica, MA) and biotinylated anti-mouse secondary antibodies (1:200 dilution; Vector Laboratories, Burlington, Ontario, Canada) (MOM kit). Signal was amplified with an ABC Elite kit (Vector Laboratories) and detected with diaminobenzidine (Pierce Biotechnology, Rockford, IL). All quantitative stereological analyses were performed blindly with respect to the nature of slices (genotype and drug feeding) using Stereoinvestigator setup and software (MicroBrightField, Williston, VT). The grid size was set to 450 × 450 μm, and the counting frame was set to 50 × 50 μm. The average slice thickness after histological processing was determined to be 22 μm. The neuronal cross-sectional area was estimated by nucleator probe under the optical fractionator. Nucleator rays were set to 6.
Statistical data analysis.
For comparison between two groups, Student's unpaired t test was used to statistically analyze data. For comparison between more than two groups, ANOVA followed by post hoc Fisher's PLSD test was used.
Drugs.
Dopamine hydrobromide and 2-APB were purchased from EMD Biosciences (San Diego, CA). Glutamate, MPEP, CPCCOEt, (+)-MK801 maleate, ifenprodil, R-(+)-SCH23390 hydrochloride, spiperone hydrochloride, SKF38393, and (−)-quinpirole were purchased from Tocris Bioscience (Ellisville, MO).
Results
Dopamine potentiates glutamate-induced Ca2+ signals in WT and YAC128 MSNs
In the previous study, we demonstrated that repetitive pulses of 20 μm glutamate resulted in a large elevation of Ca2+ levels in the YAC128 MSNs compared with a smaller Ca2+ increase in the WT or YAC18 MSNs (Tang et al., 2005). To address a potential modulatory effect of the dopaminergic signaling pathway, we compared Ca2+ signals induced by repetitive application of 5 μm glutamate to WT and YAC128 MSN primary cultures in the presence and absence of 50 μm dopamine. The MSN intracellular Ca2+ concentration in these experiments was continuously monitored by fura-2 imaging, and the data were presented as free cytosolic Ca2+ concentrations ([Ca2+]i) (Fig. 1). On average, basal Ca2+ levels before glutamate application were 131.8 ± 22.7 nm (n = 25) for WT MSNs (Fig. 1A,E) and 140.3 ± 10.0 nm (n = 11) for YAC128 MSNs (Fig. 1B,E). In the presence of 50 μm dopamine, basal Ca2+ levels were 140.2 ± 23.3 nm (n = 50) for WT MSNs (Fig. 1C,E) and 147.4 ± 27.5 nm (n = 48) for YAC128 MSNs (Fig. 1D,E). Thus, basal Ca2+ levels are not significantly different from each other for all four experimental groups (Fig. 1E). Repetitive pulses of 5 μm glutamate caused significant elevation of basal Ca2+ levels in the YAC128 MSNs (Fig. 1B), with smaller increases in Ca2+ in the WT MSNs (Fig. 1A). On average, Ca2+ levels after 20 pulses of 5 μm glutamate were equal to 164.5 ± 19.5 nm (n = 25) for WT MSNs (Fig. 1E) and 210.6 ± 22.8 nm (n = 11) for YAC128 MSNs (Fig. 1E). This is consistent with our previous findings when similar experiments were performed using 20 μm glutamate pulses (Tang et al., 2005). In the presence of 50 μm dopamine, significant Ca2+ level elevation caused by 5 μm glutamate pulses was already seen in WT MSNs (Fig. 1C), and higher Ca2+ levels were observed in YAC128 MSNs (Fig. 1D). In the presence of 50 μm dopamine, the average Ca2+ levels after 20 pulses of 5 μm glutamate were significantly elevated (p < 0.05) to 279.9 ± 46.5 nm (n = 50) for WT MSNs (Fig. 1E) and 345.2 ± 106.7 nm (n = 48) for YAC128 MSNs (Fig. 1E). In addition, the amplitudes of the first Ca2+ transient induced by 5 μm glutamate were also significantly (p < 0.05) higher in WT and YAC128 MSNs in the presence of 50 μm dopamine. On average, the amplitudes of the first Ca2+ transient were 174.4 ± 31.7 nm (n = 25) for WT MSNs (Fig. 1A), 243.6 ± 48.9 nm (n = 11) for YAC128 MSNs (Fig. 1B), 331.0 ± 94.5 nm (n = 50) for WT MSNs with dopamine (Fig. 1C), and 361.2 ± 101.8 nm (n = 48) for YAC128 MSNs with dopamine (Fig. 1D).
Figure 1.

Dopamine potentiates glutamate-induced Ca2+ signals in MSNs. A, B, Repetitive application of 5 μm glutamate induces Ca2+ signals in MSNs from the WT (A) and YAC128 (B) mice. C, D, The same experiment as in A and B was performed in the presence of 50 μm dopamine with WT (C) and YAC128 (D) MSNs. Cytosolic Ca2+ levels in A–D are presented in nanomolars. Each pulse of glutamate is shown as a black bar (1 min), and the washout step is shown as a white bar (1 min) above the Ca2+ trace. The traces shown are average traces from all MSNs for each experimental group. E, The average cytosolic Ca2+ levels (mean ± SD; n is the number of MSNs analyzed) before (open bars) and after (filled bars) 20 pulses of 5 μm glutamate are shown for WT MSNs (n = 25), WT MSNs in the presence of 50 μm dopamine (n = 50), WT MSNs in the presence of 25 μm SKF38393 (n = 17), WT MSNs in the presence of 25 μm (−)-quinpirole (n = 18), YAC128 MSNs (n = 11), YAC128 MSNs in the presence of 50 μm dopamine (n = 48), YAC128 MSNs in the presence of 25 μm SKF38393 (n = 26), and YAC128 MSNs in the presence of 25 μm (−)-quinpirole (n = 13) as indicated. After 20 pulses of 5 μm glutamate, the Ca2+ levels in WT and YAC128 MSNs are significantly (***p < 0.05) higher in the presence of dopamine or SKF38393 than in its absence. In the presence of SKF38393, Ca2+ level after 20 pulses of 5 μm glutamate was significantly higher in YAC128 MSNs than in WT MSNs (p < 0.01). ns, Not significant.
Striatal MSNs express multiple subtypes of dopamine receptors (DARs) (Surmeier et al., 1996). To determine which class of dopamine receptors is involved in potentiating glutamate-induced Ca2+ signals, we repeated glutamate pulse experiments in the presence of either specific D1-class recep-tor agonist SKF38393 (25 μm) or D2-class receptor agonist (−)-quinpirole (25 μm). We found that in the presence of 25 μm SKF38393, the basal Ca2+ levels were 123.0 ± 37.3 nm (n = 17) for WT MSNs and 151.6 ± 53.5 nm (n = 26) for YAC128 MSNs (Fig. 1E). In the presence of 25 μm (−)-quinpirole, basal Ca2+ levels were 104.3 ± 35.1 nm (n = 18) for WT MSNs and 133.0 ± 42.8 nm (n = 13) for YAC128 MSNs (Fig. 1E). The basal Ca2+ levels are not significantly different from each other for all four experimental groups (Fig. 1E). Similar to the effects of dopamine, we found that 25 μm SKF38393 caused significant (p < 0.05) potentiation of glutamate-induced Ca2+ responses in both WT and YAC128 MSNs (Fig. 1E). On average, in the presence of 25 μm SKF38393, Ca2+ levels after 20 pulses of 5 μm glutamate were elevated to 229.6 ± 42.2 nm (n = 17) for WT MSNs (Fig. 1E) and to 379.6 ± 80.0 nm (n = 26) for YAC128 MSNs (Fig. 1E). In contrast to dopamine or SKF38393, (−)-quinpirole did not have a significant effect on glutamate-induced Ca2+ signals in WT or YAC128 MSNs (Fig. 1E). From these results, we concluded that dopamine potentiates glutamate-induced Ca2+ signals in cultured WT and YAC128 MSNs via D1-class dopamine receptors. This conclusion is in agreement with our previous findings that the D1-class dopamine receptors, but not D2-class dopamine receptors, can greatly potentiate Ca2+ responses in cultured rat MSNs (Tang and Bezprozvanny, 2004). Additional analysis revealed that the glutamate-induced Ca2+ levels reached in the presence of 25 μm SKF38393 were significantly (p < 0.01) higher in YAC128 MSNs than in WT MSNs, consistent with potentiating effects of mutant Htt-128Q on intracellular Ca2+ signals in striatal MSNs (Tang et al., 2005).
Dopaminergic signaling and apoptosis of HD MSNs in vitro
To evaluate a potential role of dopaminergic signaling in the degeneration of MSNs in HD, we used the “in vitro HD model” described previously (Tang et al., 2005). In these experiments, we incubated WT and YAC128 MSN cultures with increasing concentrations of dopamine (0–500 μm) for 8 h. After incubation with dopamine, the neurons were fixed, permeabilized, and analyzed by TUNEL staining as described previously (Tang et al., 2005). At low concentrations of dopamine (0–100 μm), both WT and YAC128 MSNs display similar levels of apoptotic cell death in the range of 10–15% (Fig. 2A,C). At higher concentrations of dopamine (250 and 500 μm), the apoptosis of YAC128 MSNs was somewhat elevated compared with WT MSNs (Fig. 2A,C), but the difference did not reach a level of statistical significance. Thus, stimulation of the dopaminergic system alone is not sufficient to induce apoptosis of YAC128 MSNs in our in vitro model.
Figure 2.
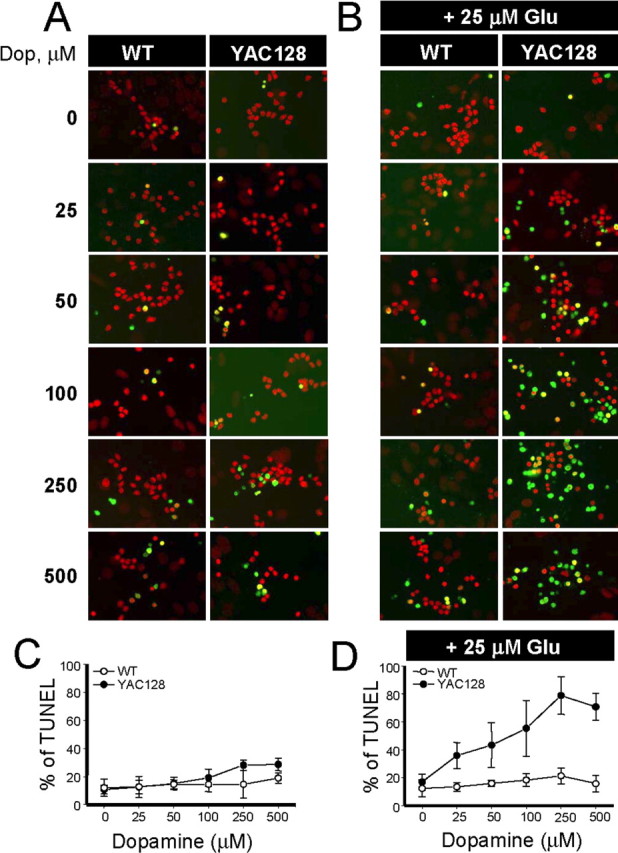
Apoptosis of YAC128 MSNs is induced by synergistic actions of dopamine and glutamate. A, MSNs (14 DIV) from the WT and YAC128 mice were exposed to a range of dopamine (Dop.) concentrations (0–500 μm) for 8 h, fixed, permeabilized, and analyzed by TUNEL staining (green) and propidium iodide counterstaining (red). B, The same experiment described in A was performed in the presence of 25 μm glutamate. C, D, The fraction of TUNEL-positive nuclei (shown in A and B) was determined and plotted against dopamine concentration for WT (open circles) and YAC128 (filled circles) MSNs. At each dopamine concentration, the data are shown as mean ± SD (n = 4–6 microscopic fields, 200–300 MSNs per field). Similar results have been obtained with six WT and YAC128 MSN cultures.
In the brain, MSNs receive both glutamate and dopamine inputs simultaneously (Gerfen, 1992). To mimic this situation in the next series of experiments, we incubated WT and YAC128 MSNs in the presence of 25 μm glutamate and increasing concentrations of dopamine (0–500 μm). After 8 h of incubation with the mixture of glutamate and dopamine, the neurons were fixed, permeabilized, and analyzed by TUNEL staining. Consistent with the previous findings (Tang et al., 2005), addition of 25 μm glutamate alone did not induce apoptosis of WT or YAC128 MSNs (Fig. 2B,D). However, addition of dopamine in the presence of 25 μm glutamate induced apoptosis of YAC128 MSNs but not apoptosis of WT MSNs (Fig. 2B,D). The effects of dopamine were significant even at 25 μm concentration, and higher concentrations of dopamine further promoted apoptosis of YAC128 MSNs (Fig. 2B,D). For example, incubation with a mixture of 25 μm glutamate and 250 μm dopamine resulted in 80% apoptosis for YAC128 MSNs and only 20% apoptosis for WT MSNs (Fig. 2B,D). Thus, we concluded that dopamine and glutamate act synergistically in our in vitro HD assay to specifically induce cell death of YAC128 MSNs but not WT MSNs.
Pharmacology of dopamine/glutamate-induced apoptosis of HD MSNs
To address which DAR class is involved in dopamine/glutamate-induced apoptosis of YAC128 MSNs (Fig. 2B,D), we used specific inhibitors of D1-class DARs (SCH23390) and D2-class DARs (spiperone). In these experiments, WT and YAC128 MSNs were incubated for 8 h in the presence of 100 μm dopamine, 50 μm glutamate, or a mixture of 100 μm dopamine and 50 μm glutamate with the addition of 5 μm SCH23390 or 5 μm spiperone as indicated. Consistent with the previous findings (Fig. 2) (Tang et al., 2005), addition of 100 μm dopamine or 50 μm glutamate alone did not promote apoptosis of WT and YAC128 MSNs (Fig. 3A). Also consistent with the previous findings (Fig. 2B), the mixture of 100 μm dopamine and 50 μm glutamate increased apoptosis of YAC128 MSNs to ∼70% but had a much smaller effect on apoptosis of WT MSNs (∼30%) (Fig. 3A). The apoptosis of YAC128 MSNs induced by the dopamine/glutamate mixture was prevented by SCH23390 or by a combination of SCH23390 and spiperone but not by spiperone alone (Fig. 3A). Consistent with these findings, we found that 25 μm D1-class specific agonist SKF38393 but not 25 μm D2-class specific agonist (−)-quinpirole potentiated glutamate-induced apoptosis of YAC128 MSNs in our experiments (Fig. 3B). Thus, we concluded that in our in vitro HD model, dopamine exerts its synergistic effect on glutamate-induced apoptosis of YAC128 MSNs via activation of D1-class DARs but not via activation of D2-class DARs.
Figure 3.
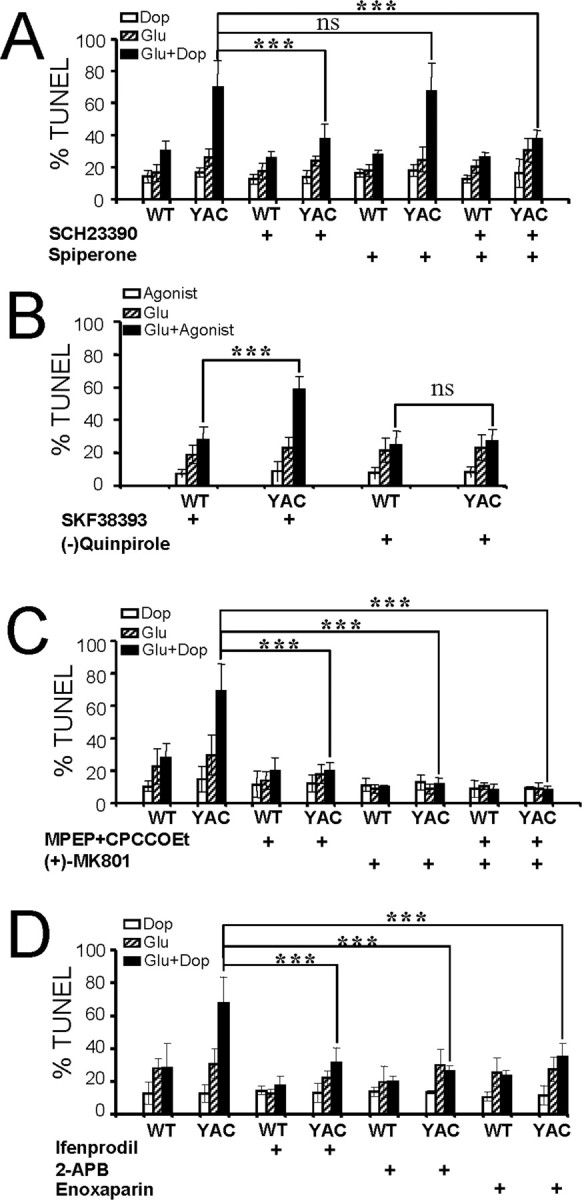
Pharmacology of dopamine/glutamate-induced apoptosis of YAC128 MSNs. A, Inhibition of D1-class DARs protects YAC128 MSNs from dopamine (Dop)/glutamate-induced apoptosis. The fraction of TUNEL-positive nuclei is shown for WT and YAC128 (YAC) MSNs incubated for 8 h in the presence of 100 μm dopamine (open bars), 50 μm glutamate (striped bars), and a mixture of 100 μm dopamine and 50 μm glutamate (filled bars). SCH23390 (5 μm) and spiperone (5 μm) were added to the culture media 30 min before addition of dopamine and glutamate as indicated. Preincubation with SCH23390 or a mixture of SCH23390 and spiperone resulted in significant (***p < 0.05) reduction of cell death in YAC128 MSNs. Preincubation with spiperone alone had no statistically significant effect on YAC128 MSN apoptosis (ns). B, D1-class DAR agonist SKF38393 promotes glutamate-induced apoptosis in YAC128 MSNs. The fraction of TUNEL-positive nuclei is shown for WT and YAC128 MSNs incubated for 8 h in the presence of DAR agonists (open bars; either 25 μm SKF38393 or 25 μm quinpirole), 50 μm glutamate (striped bars), and a mixture of DAR agonist and 50 μm glutamate (filled bars). Incubation with a mixture of SKF38393 and glutamate resulted in a significant (***p < 0.05) increase in cell death in YAC128 MSNs, whereas (−)-quinpirole had no statistically significant effect on YAC128 MSN apoptosis (ns). The apoptosis of WT MSNs was not affected. C, Inhibition of mGluR1/5 and NMDAR protects YAC128 MSNs from dopamine/glutamate-induced apoptosis. Identical experiments as described in A were performed in the presence of 20 μm MPEP and 50 μm CPCCOEt or 10 μm (+)-MK801. The inhibitors were added to the culture media 30 min before addition of dopamine and glutamate as indicated. Preincubation with MPEP/CPCCOEt or (+)-MK801 significantly (***p < 0.05) protected YAC128 MSNs from dopamine/glutamate mixture-induced cell death. D, Inhibition of NR2B NMDA receptors and InsP3R protects YAC128 MSNs from dopamine/glutamate-induced apoptosis. Identical experiments as described in A were performed in the presence of 10 μm ifenprodil, 400 μm 2-APB, or 200 μg/ml enoxaparin. Ifenprodil was added 30 min before addition of dopamine and/or glutamate, and 2-APB and enoxaparin were added 2–3 h in advance. Preincubation with ifenprodil, 2-APB, and enoxaparin significantly (***p < 0.05) protected YAC128 MSNs from dopamine/glutamate-induced cell death. A–D, Four to six microscopic fields (200–300 MSNs each) were counted for each experimental condition.
To examine which glutamate receptors are involved in the dopamine/glutamate-induced apoptosis of YAC128 MSNs, we repeated the same experiments in the presence of MPEP and CPCCOEt [metabotropic glutamate receptor 1/5 (mGluR1/5) blockers] or (+)-MK-801 [NMDA receptor (NMDAR) blocker]. We found that the apoptosis of YAC128 MSNs induced by the dopamine/glutamate was prevented by either the MPEP/CPCCOEt or (+)-MK-801 (Fig. 3C), suggesting that activation of both mGluR1/5 and NMDAR is necessary for dopamine/glutamate-induced apoptosis of YAC128 MSNs in our in vitro HD model. Httexp specifically facilitates activity of the NR2B subtype of NMDA receptors (Chen et al., 1999; Sun et al., 2001; Zeron et al., 2002, 2004; Song et al., 2003) and InsP3R1 (Tang et al., 2003b). The involvements of NR2B subtype of NMDAR and InsP3R were also confirmed by specific blocker of NR2B (ifenprodil) and membrane-permeable InsP3R blockers (2-APB and enoxaparin) (Fig. 3D).
Dopamine signaling and motor coordination deficits in HD mice
Our in vitro results indicated that dopamine and glutamate pathways act synergistically to cause apoptosis of HD MSNs (Figs. 2, 3). Next we aimed to examine the contribution of dopamine signaling pathway to striatal neurodegeneration in vivo with YAC128 HD mouse model. l-DOPA (100 mg/kg, twice a week) was used to elevate dopamine levels in mouse brains, and TBZ (5 mg/kg, twice a week) was used to antagonize the effects of l-dopa and to inhibit dopamine signaling (Table 1). The effectiveness of oral drug delivery (twice a week) was determined by 2 months of “drug dosage” trial. We determined that the dopamine concentration in the striatal region of control mice was 4705 ± 1158 ng/g (Table 2). Feeding l-DOPA to mice for 2 months resulted in approximately twofold elevation of striatal dopamine levels (10,656 ± 1529 ng/g), whereas TBZ could efficiently enter the brains (19 ± 2 ng/g) and prevent the increase in striatal dopamine levels resulting from l-DOPA feeding (Table 2).
Table 1.
Design of l-DOPA/TBZ trial in YAC128 mice
| Group | Group name | Number of mice | Mouse genotype | Single dose (50 μl) (two per week) | Drug dosage (mg/kg) |
|---|---|---|---|---|---|
| 1 | WT-Ctrl | 10 (5F, 5M) | WT | 50 μl PBS | 50 μl PBS |
| 2 | WT-DOPA | 10 (7F, 3M) | WT | 2.5 mg l-DOPA and 0.3 mg benserazide | 100 l-DOPA and 12 benserazide |
| 3 | WT-DOPA/TBZ | 10 (7F, 3M) | WT | 2.5 mg l-DOPA, 0.3 mg benserazide, and 0.125 mg TBZ | 100 l-DOPA, 12 benserazide, and 5 TBZ |
| 4 | YAC-Ctrl | 9 (4F, 5M) | YAC128 | 50 μl PBS | 50 μl PBS |
| 5 | YAC-DOPA | 8 (4F, 4M) | YAC128 | 2.5 mg l-DOPA and 0.3 mg benserazide | 100 l-DOPA and 12 benserazide |
| 6 | YAC-DOPA/TBZ | 8 (4F, 4M) | YAC128 | 2.5 mg l-DOPA, 0.3 mg benserazide, and 0.125 mg TBZ | 100 l-DOPA, 12 benserazide, and 5 TBZ |
Six groups of mice were tested in our experiments. At 2 months of age, WT and YAC128 mice were divided into three groups each, and drug treatment was initiated. The group number, group name, number and genotype of mice in each group, and dose of single drug treatment (two/week) are shown for each group. Also shown is the estimated drug dosage in mg/kg. Ctrl, Control; F, female; M, male; YAC, YAC128.
Table 2.
l-DOPA, dopamine and tetrabenazine levels in blood and/or brains of experimental mice (30 min after drug feeding)
| Tissue | Group | Measurements |
||
|---|---|---|---|---|
| l-DOPA | Dopamine | TBZ | ||
| Blood (ng/ml) | Vehicle | |||
| l-DOPA | 6997 ± 1908 | |||
| l-DOPA/TBZ | 15269 ± 1857* | 13 ± 5 | ||
| Striata (ng/gm) | Vehicle | 4705 ± 1158 | ||
| l-DOPA | ND | 10656 ± 1529** | ||
| l-DOPA/TBZ | ND | 5911 ± 2180* | 10 ± 3 | |
| Brain hemisphere (ng/gm) | Vehicle | 311 ± 23 | ||
| l-DOPA | 1072 ± 181 | 840 ± 77 | ||
| l-DOPA/TBZ | 1127 ± 89 | 662 ± 49 | 19 ± 2 | |
Data are expressed as their average ± SEM (n = 5). Units are ng/ml (blood) and ng/gm (brain tissue). *Statistically different (p < 0.05) from l-DOPA alone. **Statistically different (p < 0.05) from control.
Is neurodegenerative process in YAC128 mice affected by a persistent increase in striatal dopamine levels resulting from l-DOPA administration? Is it affected by the presence of TBZ? To answer these questions, we performed a behavioral analysis of six groups of WT and YAC128 mice (Table 1). In our experiments, the motor coordination of these mice was assessed by “rotarod” and beam walk assays (for details, see Materials and Methods). Basal rotarod and beam walk performance for all groups was determined before initiation of drug feeding when these mice were 2 months of age. Starting at 2 months of age, all mice were fed with drugs twice a week as described above until all mice were 10 months of age. The drug feeding was stopped when all mice were 10 months of age and was followed by a 1 month washout period. The rotarod and beam walk data were collected at 2, 5, 7, 9, 10, and 11 (washout) months of age. When results were analyzed, we found that the rotarod performance of WT and YAC128 mice was similar when these mice were 2 months of age (Fig. 4). Consistent with the previous description of YAC128 mice phenotype (Slow et al., 2003), control group of WT mice (fed with PBS) performed significantly better (p < 0.05) than control group of YAC128 mice at 5, 7, 9, 10, and 11 months of age (Fig. 4). Feeding l-DOPA or l-DOPA/TBZ to WT mice had no apparent effects on their rotarod performance at any age (Fig. 4). Feeding l-DOPA to YAC128 mice impaired their performance on the rotarod (Fig. 4), with significant (p < 0.05) reduction in the latency to fall measured at 10 and 11 months of age. In contrast, feeding YAC128 mice with l-DOPA/TBZ mixture significantly (p < 0.05) improved the rotarod performance of these mice at 9, 10, and 11 months of age compared with control YAC128 mice (Fig. 4).
Figure 4.

Rotarod performance of YAC128 mice. An average latency to fall from the accelerating rotarod is shown for the WT control (Ctrl) mice (open black circles), the YAC128 (YAC) control mice (filled black circles), the WT mice fed with l-DOPA (open red triangles), the YAC128 mice fed with l-DOPA (filled red triangles), the WT mice fed with l-DOPA/TBZ (open blue triangles), and the YAC128 mice fed with l-DOPA/TBZ (filled blue triangles). For each group of mice, the results are shown as mean ± SEM (for the number of mice in each group, see Table 1) at 2, 5, 7, 9, 10, and 11 month (washout) time points. At 5, 7, 9, 10, and 11 months of age, control WT mice performed significantly better (p < 0.05) than control YAC128 mice. YAC128 mice fed with l-DOPA (filled red triangles) performed significantly worse (***p < 0.05) than control YAC128 group at 10 and 11 months of age. YAC128 mice fed with l-DOPA/TBZ performed significantly better (***p < 0.05) than control YAC128 mice at 9, 10, and 11 months of age but significantly worse (p < 0.05) than WT mice at 5, 7, 9, 10, and 11 months of age.
In addition to rotarod experiments, balance beam experiments were performed to compare the fine motor coordination and balance capabilities among different groups of mice. In our experiments, we tested the mice on 17 mm round, 11 mm round, and 5 mm square beams to achieve different levels of task difficulty. The mice were trained to walk across these beams to reach an enclosed safety box, and the time to traverse the beam and the number of foot slips while walking on the beam were recorded for each trial. When results were analyzed, we found that control group of YAC128 mice (fed with PBS) exhibited a progressive impairment in beam-walking ability (longer beam traverse latencies and increased number of foot slips) with age and beam difficulty compared with control group of WT mice. The significant differences (p < 0.05) between beam performance of control YAC128 and control WT groups were observed at 10 and 11 months of age on 17 mm round beam (Fig. 5A,B); at 9, 10, and 11 months of age on 11 mm round beam (Fig. 5C,D); and at 7, 9, 10, and 11 months of age on 5 mm square beam (Fig. 5E,F). Similar to results obtained with rotarod assay (Fig. 4), feeding l-DOPA or l-DOPA/TBZ to WT mice had no significant effects on balance beam performance of these mice (Fig. 5). Feeding l-DOPA to YAC128 mice caused significant (p < 0.05) impairment to performance of these mice at 10 and 11 months of age on 17 mm round beam (Fig. 5A,B), at 9, 10, and 11 months of age on 11 mm round beam (Fig. 5C,D), and at 7, 9, 10, and 11 months of age on 5 mm square beam (Fig. 5E,F). In contrast, YAC128 mice fed with l-DOPA/TBZ mixture performed as well as WT mice at all ages and levels of task difficulty and significantly better (p < 0.05) than control YAC128 group at 9, 10, and 11 months of age on all three beams (Fig. 5).
Figure 5.
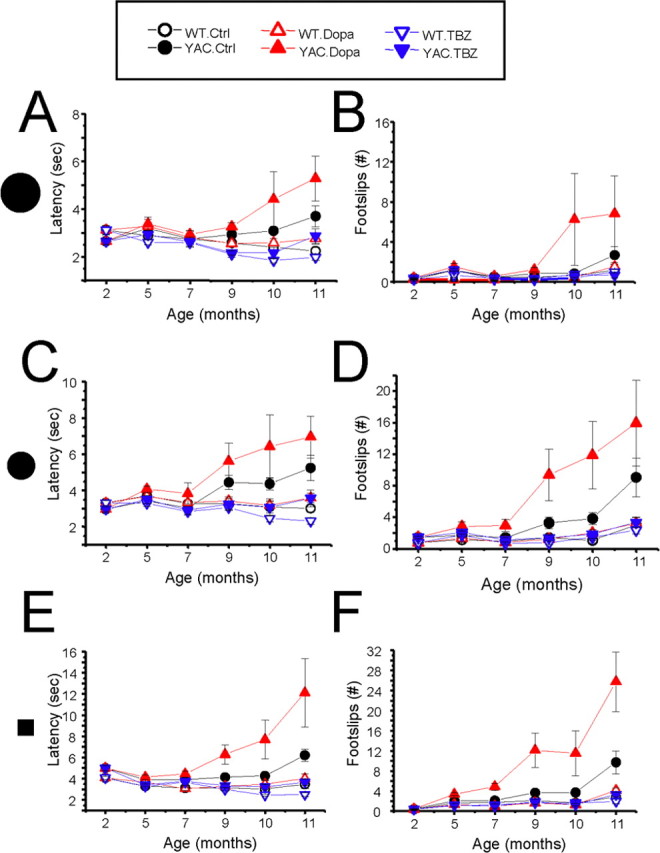
Beam walk performance of YAC128 mice. The average time to cross the beam (A, C, E) and the average number of foot slips on the beam (B, D, F) are shown for beam walk experiments performed with 17 mm round beam (A, B), 11 mm round beam (C, D), and 5 mm square beam (E, F). The data for WT control (Ctrl) mice (open black circles), YAC128 (YAC) control mice (filled black circles), WT mice fed with l-DOPA (open red triangles), YAC128 mice fed with l-DOPA (filled red triangles), WT mice fed with l-DOPA/TBZ (WT. TBZ; open blue triangles), and YAC128 mice fed with l-DOPA/TBZ (YAC. TBZ; filled blue triangles) are shown as mean ± SEM (for the number of mice in each group, see Table 1) at 2, 5, 7, 9, 10, and 11 month (washout) time points.
While conducting beam walk experiments, we noticed that some aging mice exhibited periods of “crawling behavior” (defined as prolonged contact between the thorax and abdomen of the mice and beam surface, with the mice using forelimbs to drag themselves along the beam). Two mice in YAC128 control group crawled on 11 mm round and 5 mm square beams at 11 months time point, two mice in YAC128/l-DOPA group crawled on 11 mm round beam and 5 mm square beam at 9, 10, and 11 months of age, and one mouse in YAC128/l-DOPA group crawled on 17 mm round beam at 10 and 11 months of age. Furthermore, 1 mouse in YAC128/l-DOPA group fell off 5 mm square beam when tested at 11 months time point. In contrast, none of the mice in WT groups or in YAC128 TBZ group exhibited crawling behavior or fell off the beams at any age tested. The frequency of crawling behavior was consistent with a general performance of these mice in balance beam assay, with YAC128/l-DOPA group performing the worst, YAC128 control group performing better, and YAC128/TBZ group performing as well as WT groups.
At the conclusion of rotarod and beam walking behavioral experiments (11 months of age), we also assessed gait abnormalities in all six groups of mice by using footprint pattern analysis (Fig. 6A). The footprint patterns were assessed quantitatively by four measurements: stride length, hindbase width, frontbase width, and front/hind footprint overlap (for details, see Materials and Methods). We found that 11-month-old control WT and YAC128 mice walked in similar stride lengths (Fig. 6B). Consistent with rotarod and beam walk experiments, feeding l-DOPA and l-DOPA/TBZ to WT mice had no significant effects on any of the gait measurements (Fig. 6B). Our analysis further revealed that the hindbase width and frontbase width were not significantly different between any of the six groups of mice (Fig. 6B). However, we found that feeding l-DOPA to YAC128 mice caused significantly (p < 0.05) shorter stride lengths and increased front/hind paw overlap compared with control group of YAC128 mice (Fig. 6B). In contrast, YAC128 mice fed with l-DOPA/TBZ showed similar stride lengths and front/hind paw overlap compared with control YAC128 mice and all WT groups of mice (Fig. 6B).
Figure 6.
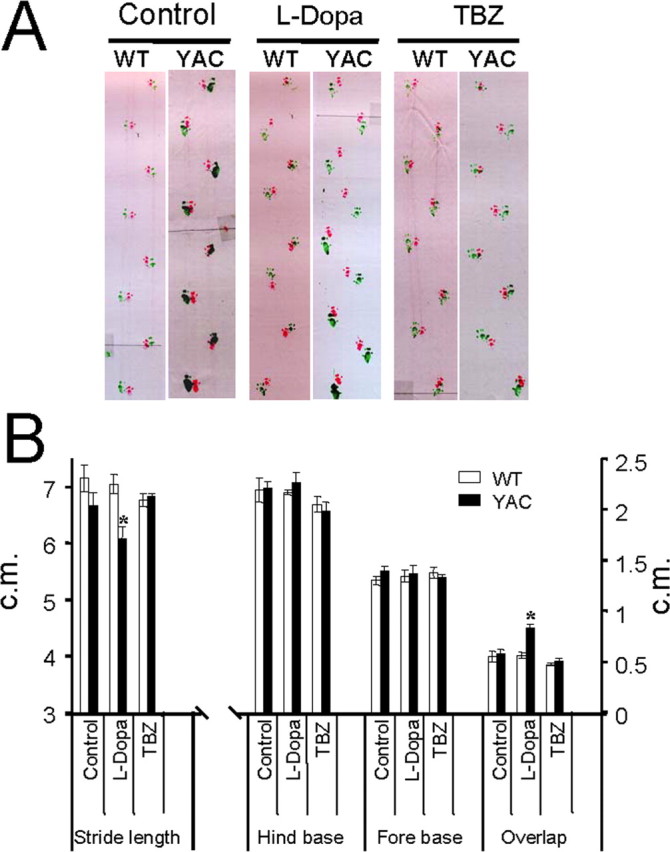
Gait analysis of YAC128 mice. A, The footprint patterns of 11-month-old YAC128 (YAC) and WT mice. The footprints for control, l-DOPA, and TBZ groups are shown for both WT and YAC128 mice. B, The footprint patterns were assessed quantitatively by four measurements: stride length, hindbase width, frontbase width, and front/hind footprint overlap. The hindbase and frontbase width measurements are similar for all six groups of mice. Feeding l-DOPA to YAC128 mice caused significantly shorter stride lengths and greater front/hind paw overlap than observed for control YAC128 mice. The YAC128 mice fed with l-DOPA/TBZ exhibited stride lengths and front/hind paw overlap similar to those from control WT and control YAC128 mice. Data for each measure are presented as mean ± SEM (for the number of mice in each group, see Table 1). *p < 0.05, significantly different from control YAC128 group.
The combined results from rotarod (Fig. 4), beam walk (Fig. 5), and “gait walk” (Fig. 6) behavioral analysis lead us to conclude that persistent elevation of dopamine levels has a detrimental effect on motor coordination of aging YAC128 mice. We further concluded that TBZ alleviates motor deficits developed by aging YAC128 mice.
Dopamine signaling and striatal cell loss in HD mice
Selective loss of striatal MSNs is a major neuropathological hallmark of HD. Previous analysis of YAC128 mice revealed strong correlation between impaired motor coordination and striatal MSN loss in these mice (Slow et al., 2003). To determine the contribution of dopamine signaling pathway to neuronal loss in YAC128 mice, at conclusion of behavioral analysis (11 months time point) the brains from all six groups of experimental mice were removed from skull and weighed after transcardial perfusion (Table 3). Consistent with the previous report (Slow et al., 2003), control YAC128 mice (fed by PBS) showed significant brain weight reduction (p < 0.05) compared with control WT mice group (Fig. 7A, Table 3). Feeding l-DOPA or l-DOPA/TBZ to WT mice had no significant effect on brain weight of these mice (Fig. 7A, Table 3). There was some reduction in brain weight of YAC128 mice fed with l-DOPA, but the difference did not reach a level of statistical significance compared with control YAC128 group (Fig. 7A, Table 3). However, feeding l-DOPA/TBZ to YAC128 mice resulted in significant increase (p < 0.05) in brain weight of these mice compared with control YAC128 group (Fig. 7A, Table 3).
Table 3.
Neuropathological analysis of brains from l-DOPA/TBZ trial mice
| Group number | Group name | Number of mice | Brain weight (g) | Neuron counts (×106 cells) | MSN cross-sectional area (μm2) |
|---|---|---|---|---|---|
| 1 | WT-Ctrl | 9 (5F, 4M) | 0.508 ± 0.025 | 1.264 ± 0.035 | 85.75 ± 2.33 |
| 2 | WT-DOPA | 7 (5F, 2M) | 0.501 ± 0.020 | 1.225 ± 0.016 | 87.51 ± 1.98 |
| 3 | WT- DOPA/TBZ | 8 (5F, 3M) | 0.501 ± 0.019 | 1.258 ± 0.020 | 91.34 ± 2.57 |
| 4 | YAC-Ctrl | 9 (4F, 5M) | 0.432 ± 0.007 | 1.090 ± 0.020 | 71.99 ± 1.48 |
| 5 | YAC-DOPA | 8 (4F, 4M) | 0.423 ± 0.008 | 1.014 ± 0.028 | 75.53 ± 1.16 |
| 6 | YAC- DOPA/TBZ | 7 (4F, 3M) | 0.464 ± 0.010 | 1.182 ± 0.026 | 77.21 ± 1.15 |
The average brain weight, striatal neuronal counts, and striatal neuron cross-sectional area were measured in 11-month-old WT and YAC128 mice at the conclusion of the l-DOPA/TBZ trial. Several mice died during the behavioral stage of the trial and were not used in neuropathological analysis. The data are shown as mean ± SEM.
Figure 7.
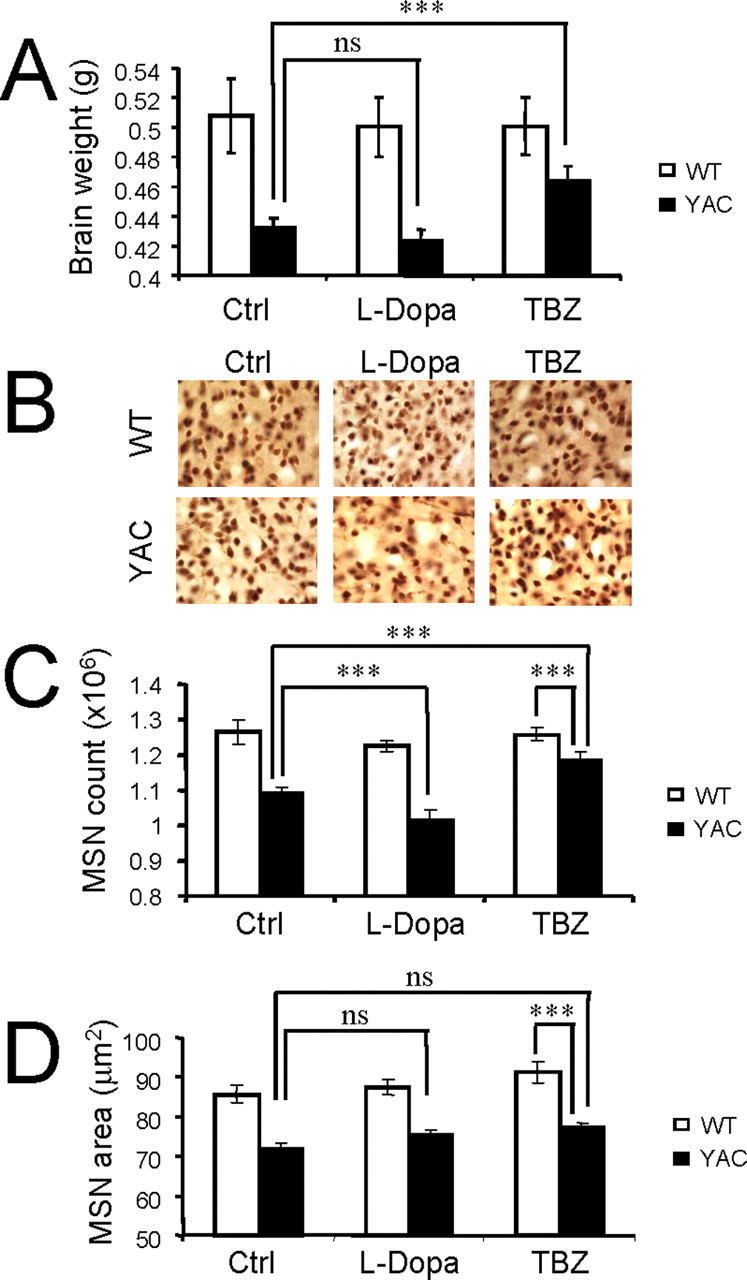
Effect of dopamine signaling on neuronal loss in YAC128 mice. A, Average brain weight of 11-month-old WT and YAC128 (4AC) mice. The brain weight of control YAC128 is significantly reduced compared with control WT group (p < 0.05). The brain weight of YAC128 mice fed with l-DOPA/TBZ is significantly increased compared with brain weight of control (Ctrl) YAC128 mice (***p < 0.05). B, Representative NeuN staining of striatal sections from 11-month-old WT and YAC128 mice. C, Average striatal neuronal counts of 11-month-old WT and YAC128 mice. Control YAC128 mice showed significant striatal neuronal loss (p < 0.05) compared with control WT mice. YAC128 mice fed with l-DOPA exhibited significant neuronal loss (***p < 0.05) compared with control YAC128 mice. YAC128 mice fed with l-DOPA/TBZ display significantly increased striatal neuronal counts compared with control YAC128 mice (***p < 0.05) but significantly reduced neuronal counts compared with WT mice (***p < 0.05). D, Average cross-sectional area of striatal neurons of 11-month-old WT and YAC128 mice. A, C, D, For each group of mice, the results are shown as mean ± SEM (for the number of mice in each group, see Table 3). Ctrl, Control; ns, not significant.
To obtain quantitative information about striatal neuronal loss in these mice, the brains were fixed, frozen, and sliced with the microtome, and coronal sections corresponding to striatal region were stained with monoclonal antibodies against NeuN protein (Fig. 7B). The number of NeuN-positive neurons in the striatum of each mouse was determined by stereology (for details, see Materials and Methods). In agreement with the previous report (Slow et al., 2003), control YAC128 mice (fed by PBS) showed significant striatal neuronal loss (p < 0.05) compared with control WT mice (Fig. 7C, Table 3). Feeding l-DOPA and l-DOPA/TBZ to WT mice did not cause significant changes in striatal neuronal counts in these mice (Fig. 7C, Table 3). In contrast, YAC128 mice fed with l-DOPA exhibited significant additional neuronal loss (p < 0.05) compared with control YAC128 mice (Fig. 7C, Table 3). Interestingly, feeding l-DOPA/TBZ to YAC128 mice significantly increased the striatal neuronal counts (p < 0.05) compared with control YAC128 mice (Fig. 7C, Table 3), indicating that TBZ protects YAC128 MSNs from cell death. Additional stereological analysis revealed significant reduction in average neuronal cross-sectional areas in all three groups of YAC128 mice compared with all three groups of WT mice (Fig. 7D, Table 3). The reduction in MSN cross-sectional area of YAC128 mice observed in our experiments (Fig. 7D, Table 3) is in quantitative agreement with the recent analysis of MSN cross-sectional area in 12-month-old YAC128 mice performed on 129 strain background (Van Raamsdonk et al., 2007). We did not observe significant difference in MSN cross-section area between l-DOPA/TBZ and control groups of YAC128 mice (Fig. 7D, Table 3), suggesting that TBZ does not prevent shrinkage of YAC128 MSNs observed in our experiments.
The results of our neuropathological analysis demonstrated that the persistent elevation of dopamine levels by feeding l-DOPA promoted loss of striatal neurons in 11-month-old YAC128 mice. We further concluded that TBZ offers significant protection from striatal neuronal loss in aging YAC128 mice but does not protect striatal neurons in these mice from shrinkage.
Discussion
Dopaminergic signaling and neurodegeneration of HD MSNs
The connection between dopamine and HD was first suspected when over one-third of asymptomatic relatives of individuals with HD developed dyskinesis in response to l-DOPA (Klawans et al., 1970). Additional biochemical analysis revealed progressive loss of striatal D1 and D2 receptors in postmortem HD brains (Joyce et al., 1988; Filloux et al., 1990; Richfield et al., 1991; Glass et al., 2000; Suzuki et al., 2001). Imaging studies also reported reduction of striatal D1 and D2 receptors in HD patients (Sedvall et al., 1994; Turjanski et al., 1995) and asymptomatic HD mutation carriers (Antonini et al., 1996; Weeks et al., 1996; Backman et al., 1997; Ginovart et al., 1997). In agreement with the analysis of human HD patients, significant reduction of striatal D1 and D2 receptor density (Cha et al., 1998; Ariano et al., 2002) and deficiencies in dopamine signaling (Bibb et al., 2000; Petersen et al., 2002) were observed in the R6/2 and R6/1 HD mouse models. To determine whether the deficient dopamine signaling plays a role in HD phenotype, R6/2 mice were chronically treated with l-DOPA (Hickey et al., 2002). Although short-term improvements in activity and rearing behavior were initially observed, long-term treatment with l-DOPA had deleterious effects on survival of R6/2 HD mice (Hickey et al., 2002). The HD-like motor dysfunction and selective MSN degeneration have been reported for the dopamine transporter knock-out mice (Cyr et al., 2003), and hyperdopaminergic transmission has been shown to accelerate the formation of Httexp aggregates in 92Q knock-in HD mouse model (Cyr et al., 2006). All of these results provided circumstantial evidence suggesting an important role of dopaminergic signaling in HD pathogenesis (Jakel and Maragos, 2000), but the mechanistic basis for these observations is poorly understood.
One possibility that has been considered previously is direct damage to HD striatal neurons from exposure to dopamine (Jakel and Maragos, 2000). High doses of dopamine are able to directly induce cell death of striatal neurons in vitro and in vivo (Cheng et al., 1996; Hastings et al., 1996; McLaughlin et al., 1998). The mechanism of direct dopamine neurotoxicity most likely involves oxidative stress and generation of reactive oxygen species (Jakel and Maragos, 2000). It has been reported that MSN cultures from R6/2 HD fragment model are sensitized to dopamine-induced oxidative stress, leading to neuronal autophagy (Petersen et al., 2001). In a more recent study, it has been demonstrated that the cultured MSNs transiently transfected with a GFP-tagged first exon Htt-103Q protein are sensitized to apoptosis induced by high concentrations of dopamine (Charvin et al., 2005). Interestingly, the latter study indicated an important role played by D2 receptors, but not by D1 receptors, in dopamine-induced toxicity (Charvin et al., 2005). In contrast to these findings, we found that dopamine alone was not sufficient to induce apoptosis of cultured YAC128 MSNs in our experiments (Fig. 2A,C), but instead we found that YAC128 MSNs were strongly sensitized to apoptosis induced by a mixture of glutamate and dopamine (Fig. 2B,D). We explained these results by the synergy between glutamate and dopamine signaling pathways resulting in Ca2+ overload (Fig. 1). Furthermore, in our experiments dopamine acted via D1 class of dopamine receptors and not via D2 class of dopamine receptors (Fig. 1E, 3A,B). Most likely, the difference between our findings and the previous results originates from using a mild YAC128 HD mouse model in our experiments in contrast to more severe first Httexp exon overexpression animal and cellular models used in the previous studies (Petersen et al., 2001; Charvin et al., 2005).
Our results lead us to conclude that dopamine and glutamate signaling pathways act synergistically to elevate Ca2+ levels and induce degeneration of MSNs in HD (Fig. 8). We propose that dopamine released from midbrain dopaminergic neurons (Gerfen, 1992) stimulates D1-class and D2-class DARs, which are abundantly expressed in MSNs (Surmeier et al., 1996; Cha et al., 1998; Missale et al., 1998; Bibb et al., 2000; Glass et al., 2000; Vallone et al., 2000). D1-class DARs are coupled to Gs/olf, activation of adenyl cyclase, and cAMP production (Missale et al., 1998). PKA activation modulates the glutamate-related Ca2+ signaling pathway by facilitating the activity of NMDAR (Levine et al., 1996; Flores-Hernandez et al., 2002), AMPA receptor (Yan et al., 1999), and InsP3R1 (Tang et al., 2003a; Tang and Bezprozvanny, 2004). As a result of the convergence of cAMP/PKA and NMDAR/InsP3R1-mediated signaling pathways on Ca2+ (Fig. 8), activation of D1-class DARs potentiates glutamate-induced Ca2+ signals in MSNs (Fig. 1). However, in HD MSNs, mutant Httexp further potentiates the activity of NR2B NMDAR (Chen et al., 1999; Sun et al., 2001; Zeron et al., 2002, 2004; Song et al., 2003) and InsP3R1 (Tang et al., 2003b). Thus, convergence of dopamine and glutamate signaling pathways promotes cytosolic and mitochondrial Ca2+ overload and apoptosis of HD MSNs (Fig. 8). The proposed model is supported by accentuated behavioral phenotype (Figs. 4–6) and increased striatal cell loss (Fig. 7C, Table 3) in response to l-DOPA feeding to YAC128 mice.
Figure 8.

Glutamate and dopamine signaling pathways act synergistically to induce Ca2+ overload and apoptosis of HD MSNs. Dopamine (Dop) released from midbrain dopaminergic neurons (Gerfen, 1992) stimulates D1-class and D2-class DARs, which are abundantly expressed in MSNs (Surmeier et al., 1996). D1-class DARs are coupled to Gs/olf, activation of adenyl cyclase, and cAMP production (Missale et al., 1998). PKA potentiates glutamate-induced Ca2+ signals by facilitating activity of NMDAR (Levine et al., 1996; Flores-Hernandez et al., 2002) and InsP3R1 (Tang et al., 2003a; Tang and Bezprozvanny, 2004). In HD MSNs, NR2B- and InsP3R1-mediated Ca2+ signaling is further facilitated by Httexp (Zeron et al., 2002, 2004; Tang et al., 2003b, 2005). As a result, simultaneous activation of glutamate and dopamine receptors leads to cytosolic and mitochondrial Ca2+ overload in HD MSNs. The mitochondrial Ca2+ handling is further destabilized by direct association of Httexp with mitochondria (Mito) (Panov et al., 2002; Choo et al., 2004). Once mitochondrial Ca2+ storage capacity is exceeded, mitochondrial permeability transition pore (MPTP) opens, leading to release of cytochrome c into the cytosol and activation of caspases 9 and 3, which leads to progression of apoptosis, MSN degeneration, and HD. The model is supported by the ability of blockers (shown in red) to protect YAC128 MSNs from dopamine/glutamate-induced apoptosis in our in vitro experiments. The blockers that were effective in in vitro experiments are as follows: D1-class DAR blocker SCH23390; NMDAR blocker (+)-MK801 and NR2B-specific blocker ifenprodil; mGluR1/5-specific blockers MPEP and CPCCOEt; and membrane-permeable InsP3R1 blockers 2-APB and enoxaparin (Enox). Also shown is a clinically relevant NMDAR antagonist, memantine (MMT), which protects YAC128 MSNs from glutamate-induced apoptosis (Wu et al., 2006), and a clinically relevant dopamine pathway inhibitor, TBZ, which demonstrated efficacy in the whole-animal experiments with a YAC128 HD mouse model as described in Results. PLC, Phospholipase C.
Dopamine pathway inhibitors and treatment of HD
The proposed model (Fig. 8) has direct implications for approaches to treatment of HD. A recent short-term clinical trial demonstrated that dopamine pathway antagonist TBZ efficiently reduced chorea in HD patients compared with placebo group (Huntington Study Group, 2006). Our results suggest that dopamine pathway inhibitors should be considered not only for symptomatic treatment of HD patients but also as neuroprotective agents. We demonstrated that the long-term treatment of YAC128 mice with tetrabenazine had significant beneficial effects on behavioral phenotype of these mice in motor coordination assays (Figs. 4, 5) and significantly reduced neuronal cell loss in these mice (Fig. 7A,C), even in the presence of l-DOPA.
Tetrabenazine is a reserpine-like drug that acts by blocking the vesicular monoamine transporter type 2 (VMAT-2) and depleting dopamine storage vesicles (Scherman et al., 1983). This mode of action was confirmed in our studies, because tetrabenazine prevented an increase in striatal dopamine content resulting from l-DOPA feeding to mice in our in vivo experiments (Table 2). Tetrabenazine also directly binds to and inhibits dopamine receptors in the brain (Login et al., 1982, 1983; Reches et al., 1983). The brain concentrations of tetrabenazine achieved in our experiments were in the range 30–60 nm (Table 2), which is sufficient to block VMAT-2 but not likely to inhibit striatal dopamine receptors in vivo [for related discussion, see Savani and Login (2007)].
Results in our study suggest that inhibitors of dopamine signaling pathway that act by decreasing the striatal dopamine concentration (similar to TBZ) or direct dopamine receptors antagonists should exert a neuroprotective effect in HD. Our results also suggest that TBZ exerted only a partial neuroprotective effect in vivo. Although YAC128 mice treated with TBZ performed similar to WT mice in beam walking assay (Fig. 5), the performance of these mice on rotarod was significantly impaired compared with WT mice (but also significantly improved compared with YAC128 control group) (Fig. 4). The striatal neuronal counts of YAC128 treated with TBZ were significantly reduced compared with WT mice (but also significantly improved compared with YAC128 control group) (Fig. 7C). Moreover, TBZ had no significant effect on cross-sectional area of striatal neurons in YAC128 mice (Fig. 7D), indicating that striatal cell shrinkage in these mice cannot be prevented by TBZ.
Partial neuroprotective effects and potentially severe side effects of TBZ (Huntington Study Group, 2006) indicate that dopamine antagonists would be most beneficial when delivered together with other neuroprotective drugs as a part of combination therapy. In a recent study with cultured YAC128 MSN neurons, we demonstrated that clinically relevant NMDAR antagonist memantine (Namenda) protects these neurons from glutamate-induced apoptosis (Wu et al., 2006) (Fig. 8). Based on the previous (Wu et al., 2006) and current results, we propose that a combination composed of NMDAR blocker (such as memantine) and dopamine pathway inhibitors (such as tetrabenazine) is worthwhile for assessment of clinical benefit in HD patients.
Footnotes
I.B. was supported by the Robert A. Welch Foundation, Huntington's Disease Society of America, the Hereditary Disease Foundation, the HighQ Foundation, and National Institute of Neurological Disorders and Stroke Grants R01 NS38082 and NS056224. We thank Elizabeth Slow, Mahmoud Pouladi, and Michael R. Hayden (University of British Columbia, Vancouver, British Columbia, Canada) for generously sharing YAC128 mice with our laboratory and for facilitating our work with clinically relevant dopamine antagonists; Tianhua Lei and Xiangmei Kong for help with maintaining the YAC128 mouse colony and with behavioral experiments; Janet Young for administrative assistance; Lisa Monteggia, Jenny Hsieh, Malu Tansey, and Keith Tansey for help and advice with behavioral and stereological experiments; Melior Discovery and David Howland (HighQ Foundation) for assistance with the measurements of dopamine and TBZ concentrations; and Mary Heng (University of Michigan, Ann Arbor, MI) for advice on balance beam experiments.
References
- Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, Sanchez-Pernaute R, de Yebenez JG, Boesiger P, Weindl A, Maguire RP. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain. 1996;119:2085–2095. doi: 10.1093/brain/119.6.2085. [DOI] [PubMed] [Google Scholar]
- Ariano MA, Aronin N, Difiglia M, Tagle DA, Sibley DR, Leavitt BR, Hayden MR, Levine MS. Striatal neurochemical changes in transgenic models of Huntington's disease. J Neurosci Res. 2002;68:716–729. doi: 10.1002/jnr.10272. [DOI] [PubMed] [Google Scholar]
- Backman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L. Cognitive deficits in Huntington's disease are predicted by dopaminergic PET markers and brain volumes. Brain. 1997;120:2207–2217. doi: 10.1093/brain/120.12.2207. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P. Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci USA. 2000;97:6809–6814. doi: 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charvin D, Vanhoutte P, Pages C, Borrelli E, Caboche J. Unraveling a role for dopamine in Huntington's disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc Natl Acad Sci USA. 2005;102:12218–12223. doi: 10.1073/pnas.0502698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem. 1999;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- Cheng N, Maeda T, Kume T, Kaneko S, Kochiyama H, Akaike A, Goshima Y, Misu Y. Differential neurotoxicity induced by L-DOPA and dopamine in cultured striatal neurons. Brain Res. 1996;743:278–283. doi: 10.1016/s0006-8993(96)01056-6. [DOI] [PubMed] [Google Scholar]
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- Cyr M, Beaulieu JM, Laakso A, Sotnikova TD, Yao WD, Bohn LM, Gainetdinov RR, Caron MG. Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc Natl Acad Sci USA. 2003;100:11035–11040. doi: 10.1073/pnas.1831768100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr M, Sotnikova TD, Gainetdinov RR, Caron MG. Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. FASEB J. 2006;20:2541–2543. doi: 10.1096/fj.06-6533fje. [DOI] [PubMed] [Google Scholar]
- Filloux F, Wagster MV, Folstein S, Price DL, Hedreen JC, Dawson TM, Wamsley JK. Nigral dopamine type-1 receptors are reduced in Huntington's disease: a postmortem autoradiographic study using [3H]SCH 23390 and correlation with [3H]forskolin binding. Exp Neurol. 1990;110:219–227. doi: 10.1016/0014-4886(90)90033-o. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Lundin A, Farde L, Halldin C, Backman L, Swahn CG, Pauli S, Sedvall G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington's disease. Brain. 1997;120:503–514. doi: 10.1093/brain/120.3.503. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RL. The pattern of neurodegeneration in Huntington's disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience. 2000;97:505–519. doi: 10.1016/s0306-4522(00)00008-7. [DOI] [PubMed] [Google Scholar]
- Groden DL, Guan Z, Stokes BT. Determination of Fura-2 dissociation constants following adjustment of the apparent Ca-EGTA association constant for temperature and ionic strength. Cell Calcium. 1991;12:279–287. doi: 10.1016/0143-4160(91)90002-v. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MA, Reynolds GP, Morton AJ. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington's disease. J Neurochem. 2002;81:46–59. doi: 10.1046/j.1471-4159.2002.00804.x. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2006;66:366–372. doi: 10.1212/01.wnl.0000198586.85250.13. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- Joyce JN, Lexow N, Bird E, Winokur A. Organization of dopamine D1 and D2 receptors in human striatum: receptor autoradiographic studies in Huntington's disease and schizophrenia. Synapse. 1988;2:546–557. doi: 10.1002/syn.890020511. [DOI] [PubMed] [Google Scholar]
- Klawans HC, Paulson GW, Barbeau A. Predictive test for Huntington's chorea. Lancet. 1970;2:1185–1186. doi: 10.1016/s0140-6736(70)90367-3. [DOI] [PubMed] [Google Scholar]
- Levine MS, Altemus KL, Cepeda C, Cromwell HC, Crawford C, Ariano MA, Drago J, Sibley DR, Westphal H. Modulatory actions of dopamine on NMDA receptor-mediated responses are reduced in D1A-deficient mutant mice. J Neurosci. 1996;16:5870–5882. doi: 10.1523/JNEUROSCI.16-18-05870.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Login IS, Cronin MJ, MacLeod RM. Tetrabenazine has properties of a dopamine receptor antagonist. Ann Neurol. 1982;12:257–262. doi: 10.1002/ana.410120308. [DOI] [PubMed] [Google Scholar]
- Login IS, Cronin MJ, Harcus CT, MacLeod RM. Neuroendocrine evidence that tetrabenazine is a dopamine antagonist. Proc Soc Exp Biol Med. 1983;172:225–231. doi: 10.3181/00379727-172-41550. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Nelson D, Erecinska M, Chesselet MF. Toxicity of dopamine to striatal neurons in vitro and potentiation of cell death by a mitochondrial inhibitor. J Neurochem. 1998;70:2406–2415. doi: 10.1046/j.1471-4159.1998.70062406.x. [DOI] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, Sulzer D. Expanded CAG repeats in exon 1 of the Huntington's disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet. 2001;10:1243–1254. doi: 10.1093/hmg/10.12.1243. [DOI] [PubMed] [Google Scholar]
- Petersen A, Puschban Z, Lotharius J, NicNiocaill B, Wiekop P, O'Connor WT, Brundin P. Evidence for dysfunction of the nigrostriatal pathway in the R6/1 line of transgenic Huntington's disease mice. Neurobiol Dis. 2002;11:134–146. doi: 10.1006/nbdi.2002.0534. [DOI] [PubMed] [Google Scholar]
- Reches A, Burke RE, Kuhn CM, Hassan MN, Jackson VR, Fahn S. Tetrabenazine, an amine-depleting drug, also blocks dopamine receptors in rat brain. J Pharmacol Exp Ther. 1983;225:515–521. [PubMed] [Google Scholar]
- Richfield EK, O'Brien CF, Eskin T, Shoulson I. Heterogeneous dopamine receptor changes in early and late Huntington's disease. Neurosci Lett. 1991;132:121–126. doi: 10.1016/0304-3940(91)90448-3. [DOI] [PubMed] [Google Scholar]
- Savani AA, Login IS. Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology. 2007;68:797. doi: 10.1212/01.wnl.0000259143.52138.5c. author reply 797. [DOI] [PubMed] [Google Scholar]
- Scherman D, Jaudon P, Henry JP. Characterization of the monoamine carrier of chromaffin granule membrane by binding of [2–3H]dihydrotetrabenazine. Proc Natl Acad Sci USA. 1983;80:584–588. doi: 10.1073/pnas.80.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedvall G, Karlsson P, Lundin A, Anvret M, Suhara T, Halldin C, Farde L. Dopamine D1 receptor number—a sensitive PET marker for early brain degeneration in Huntington's disease. Eur Arch Psychiatry Clin Neurosci. 1994;243:249–255. doi: 10.1007/BF02191583. [DOI] [PubMed] [Google Scholar]
- Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Song C, Zhang Y, Parsons CG, Liu YF. Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-d-aspartate receptors. J Biol Chem. 2003;278:33364–33369. doi: 10.1074/jbc.M304240200. [DOI] [PubMed] [Google Scholar]
- Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-d- aspartate receptors via post-synaptic density 95. J Biol Chem. 2001;276:24713–24718. doi: 10.1074/jbc.M103501200. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Desmond TJ, Albin RL, Frey KA. Vesicular neurotransmitter transporters in Huntington's disease: initial observations and comparison with traditional synaptic markers. Synapse. 2001;41:329–336. doi: 10.1002/syn.1089. [DOI] [PubMed] [Google Scholar]
- Tang TS, Bezprozvanny I. Dopamine receptor-mediated Ca(2+) signaling in striatal medium spiny neurons. J Biol Chem. 2004;279:42082–42094. doi: 10.1074/jbc.M407389200. [DOI] [PubMed] [Google Scholar]
- Tang TS, Tu H, Wang Z, Bezprozvanny I. Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase A and protein phosphatase 1α. J Neurosci. 2003a;23:403–415. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003b;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Slow EJ, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci USA. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Tobin AJ, Signer ER. Huntington's disease: the challenge for cell biologists. Trends Cell Biol. 2000;10:531–536. doi: 10.1016/s0962-8924(00)01853-5. [DOI] [PubMed] [Google Scholar]
- Turjanski N, Weeks R, Dolan R, Harding AE, Brooks DJ. Striatal D1 and D2 receptor binding in patients with Huntington's disease and other choreas. A PET study. Brain. 1995;118:689–696. doi: 10.1093/brain/118.3.689. [DOI] [PubMed] [Google Scholar]
- Vallone D, Picetti R, Borrelli E. Structure and function of dopamine receptors. Neurosci Biobehav Rev. 2000;24:125–132. doi: 10.1016/s0149-7634(99)00063-9. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Metzler M, Slow E, Pearson J, Schwab C, Carroll J, Graham RK, Leavitt BR, Hayden MR. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol Dis. 2007;26:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Weeks RA, Piccini P, Harding AE, Brooks DJ. Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington's disease. Ann Neurol. 1996;40:49–54. doi: 10.1002/ana.410400110. [DOI] [PubMed] [Google Scholar]
- Wu J, Tang T-S, Bezprozvanny I. Evaluation of clinically-relevant glutamate pathway inhibitors in in vitro model of Huntington's disease. Neurosci Lett. 2006;407:219–223. doi: 10.1016/j.neulet.2006.08.036. [DOI] [PubMed] [Google Scholar]
- Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2004;25:469–479. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]