

Abstract
Little is known of the environmental factors that initiate and promote disease. The aryl hydrocarbon receptor (AHR) is a key regulator of xenobiotic metabolism and plays a major role in gene/environment interactions. The AHR has also been demonstrated to carry out critical functions in development and disease. A qualitative investigation into the contribution by the AHR when stimulated to different levels of activity was undertaken to determine whether AHR-regulated gene/environment interactions are an underlying cause of cardiovascular disease. We used two congenic mouse models differing at the Ahr gene, which encodes AHRs with a 10-fold difference in signaling potencies. Benzo[a]pyrene (BaP), a pervasive environmental toxicant, atherogen, and potent agonist for the AHR, was used as the environmental agent for AHR activation. We tested the hypothesis that activation of the AHR of different signaling potencies by BaP would have differential effects on the physiology and pathology of the mouse cardiovascular system. We found that differential AHR signaling from an exposure to BaP caused lethality in mice with the low-affinity AHR, altered the growth rates of the body and several organs, induced atherosclerosis to a greater extent in mice with the high-affinity AHR, and had a huge impact on gene expression of the aorta. Our studies also demonstrated an endogenous role for AHR signaling in regulating heart size. We report a gene/environment interaction linking differential AHR signaling in the mouse to altered aorta gene expression profiles, changes in body and organ growth rates, and atherosclerosis.
Keywords: aryl hydrocarbon receptor, gene/environment interactions, Western diet, benzo[a]pyrene, atherosclerosis, body and organ growth
Activation of aryl hydrocarbon receptor (AHR) signaling is the body’s primary molecular defense following environmental toxicant exposures, and there is growing evidence linking toxicant-activated AHR signaling and some major diseases, e.g., heart disease (Bond et al., 1981; Dalton et al., 2001; Moorthy et al., 2002, 2003) and cancer (Liu et al., 2006; Ohtake et al., 2003; Schlezinger et al., 2006; Zhang et al., 2009). The AHR is a ligand-activated nuclear receptor/transcription factor that regulates genes involved in a number of biological and developmental pathways (Andreola et al., 1997; Fernandez-Salguero et al., 1995, 1997; Lahvis et al., 2000), most notably, those involved in detoxification pathways (Nebert, 1989; Nebert et al., 1993). Upon ligand binding, the AHR translocates to the nucleus where it complexes with the AHR nuclear translocator (ARNT) (Hoffman et al., 1991). The AHR/ARNT heterodimer regulates the transcription of genes in the cytochrome P450 (Cyp1) family, several phase II detoxification genes (Gonzalez et al., 1984; Hankinson, 1995), as well as hundreds if not thousands of other genes (Guo et al., 2004; Karyala et al., 2004) by binding cognate DNA sequences known as AHR, dioxin, or xenobiotic response elements (Whitlock, 1993) as well as interacting with other signaling pathways (Puga et al., 2005).
Mice with a homozygous loss of Ahr gene function (Brenner, 1996; Fernandez-Salguero et al., 1995; Mimura et al., 1997; Schmidt et al., 1996) are resistant to most chemical toxicities (Fernandez-Salguero et al., 1996). Although a definitive endogenous AHR ligand has not been identified, evidence that one or more critical intrinsic ligands exist is indirectly supported by the many defects that develop when AHR signaling is absent. Ahr-null mice have heart and vascular hypertrophy, immune system deficiencies, hepatic fibrosis (Fernandez-Salguero et al., 1995), liver retinoid accumulation, decreased retinoic acid metabolism, mineralization in the uterus, and gastric hyperplasia with progression into polyps (Andreola et al., 1997; Fernandez-Salguero et al., 1997). The most obvious developmental effects are the failure for the portosystemic shunt from the portal vein to the inferior vena cava to close, a loss of liver sinusoids, and vascular anomalies in the eye and kidney (Lahvis et al., 2000). Among the many candidates for an endogenous AHR ligand are eicosanoids and low-density lipoproteins (McMillan and Bradfield, 2007; Nebert and Karp, 2008).
The AHR is known to bind hundreds of environmental agents, including benzo[a]pyrene (BaP). BaP is pervasive in the environment; a relatively strong activator of the AHR; an atherogen, teratogen, mutagen, and carcinogen; and one of the major toxicants in tobacco smoke (Bond et al., 1979, 1980; Miller and Ramos, 2001; Nebert et al., 1969). BaP itself is nontoxic but is converted to reactive metabolites by the AHR-induced CYP1 enzyme system (Sayer et al., 1989). Previous studies showed that B6 mice possessing the high-affinity Ahrb1 allele fared much better when exposed to BaP than the mouse strain DBA/2, which possesses the low-affinity Ahrd allele (Fig. 1A) (Legraverend et al., 1984; Nebert, 1989). The DBA/2 mice suffered from numerous physical anomalies, bone marrow toxicity, and a significantly shorter life span. Later work demonstrated that CYP1A1 is protective against orally administered BaP (Uno et al., 2004), the means by which most BaP enters the body.
FIG. 1.

Mouse strains and experimental designs. The B6 and B6.D2 mouse strains possess the high- and low-affinity Ahr genes, respectively (A). Two different BaP exposure protocols were followed. Protocol 1, short-term BaP exposure: Mice were exposed to 10 mg/kg/day BaP by gavage 5 days/week for 10 weeks (B). Protocol 2, long-term BaP exposure: Mice were exposed to BaP ad libitum in corn oil–soaked Western chow (BaP dissolved at 800 mg/ml in corn oil) at an estimated dose equivalent of 10 mg/kg/day for 23 weeks (C). The experimental design for the microarray experiments (D).
In order to more clearly understand the biology of differential AHR signaling on the cardiovascular system, we used two mouse models that differ at the Ahr gene (Fig. 1A). The two strains were C57BL/6 (B6 strain), which naturally bears the high-affinity AHR encoded by the Ahrb1 allele and the congenic C57BL/6.D2 (B6.D2 strain) bearing the low-affinity AHR encoded by the Ahrd allele naturally found in the DBA/2 mouse strain. The two Ahr alleles encode AHRs that differ by approximately 10-fold in ligand-binding affinities, and in turn, gene induction and gene expression levels, including those of the Cyp1a1 and Cyp1b1 genes (Poland and Glover, 1980; Thomas et al., 2002).
A distinct advantage of using the B6 and B6.D2 mouse models is that by virtue of the integral role, the AHR plays in response to endogenous and environmental agents, any corresponding differences observed in disease states, gene expression profiles, and affected signaling pathways are due to the differing capacities of the corresponding AHRs. We tested the hypothesis that differential AHR signaling activity causes differential effects on the physiology and disease states of the cardiovascular system. Using the B6 and B6.D2 mouse models, we report that the activation of AHR signaling to different levels by unknown endogenous ligands and BaP differentially affects the onset of atherosclerosis and heart size.
MATERIALS AND METHODS
Materials.
The low-fat or regular mouse chow (Teklad catalog no. 2018; by mass: 18.8% protein, 50% carbohydrate, 6% fat) was purchased from Harlan Laboratories (Madison, WI); and the high-fat or Western mouse chow (catalog no. D12079B and the same as Teklad Western diet #88137; by mass: 20% protein, 50% carbohydrate, 21% fat) was purchased from Research Diets, Inc. (New Brunswick, NJ). Mazola corn oil (ACH Food Companies, Inc., Cordova, TN) was purchased locally. The BaP was purchased from Sigma-Aldrich (catalog #B1760; St Louis, MO).
Mice.
The mouse strains are available and maintained at The Jackson Laboratory (Bar Harbor, ME; Strain names: C57BL/6J and B6.D2N-Ahrd/J; Stock numbers: 000664 and 002921, respectively). The C57BL/6 mouse possesses the high-affinity AHR (Ahrb1 allele, B6 strain), and the congenic C57BL/6.D2 mouse strain the low-affinity AHR (Ahrd allele, B6.D2 strain) (Hofstetter et al., 2007; Poland et al., 1994) (Fig. 1A). The Ahrb1 allele is the naturally occurring allele in C57BL/6J mice. The Ahrd allele is from the DBA/2J mouse strain and was introgressed into the C57BL/6J background for more than 40 generations. The B6.D2 mice have a genomic insert on chromosome 12 from the DBA/2J mouse genome. The insert spans 35.4–41.0 Mbp and contains 15 genes. Of the 15 genes, only the Ahr and Zfp277 genes contain nonsynonymous single nucleotide polymorphisms (Hofstetter et al., 2007).
The B6.129P2-Apoetm1Unc/J mouse strain harbors an insertion of a neomycin resistance cassette that deleted part of exon 3 and intron 3 of the Apoe gene (Piedrahita et al., 1992) in a C57BL/6 genetic background (Zeiher et al., 1995) and was purchased from Jackson Laboratory. The above mouse strains all share the same C57BL/6 genetic background, and the Apoe and Ahr genes are not linked (chromosomes 7 and 6, respectively), allowing the straightforward cross of Apoe−/−;Ahrb1/b1 × Apoe+/+;Ahrd/d mice to generate the desired F1 Apoe+/−;Ahrb1/d genotype for self-crossing and breeding the Apoe−/−;Ahrd/d mouse. The reason for the inclusion of the Apoe−/− null gene was to reduce the time required for the onset of atherosclerosis (Breslow, 1996). The status of the Apoe and Ahr genes in each mouse was confirmed by genotyping (Song et al., 2004). All breeding programs began 2 weeks after arrival to allow the shipped mice to acclimate. The mouse strains were relatively hardy and fecund, and obtaining sufficient progeny numbers were not a problem.
Aorta histology.
Perivascular fat was removed from the outer wall of the aorta, and the aorta was stained with oil red O as described (Nunnari et al., 1989). Descending aortas had been stored at 4°C in 4% paraformaldehyde, and thoracic aorta had been stored frozen at −80°C and thawed before staining. Plaque presence in the aorta was ascertained by visual assessment directly and from photos.
Ultrasound biomicroscopy.
The serial assessment of cardiac function was carried out by high-resolution echocardiography using a VisualSonics Vevo 770 in the Translational Research Animal Core at Dartmouth. Mice were anesthetized with isoflurane in oxygen (∼2.5% induction, ∼1.5% maintenance) and mounted supine on a heating pad to maintain optimal body temperature, which was measured by a rectal thermometer. Electrocardiogram (ECG) electrodes monitored heart and breathing rates throughout the procedure. Chest hair was removed with chemical hair remover, and warmed ultrasound gel was applied liberally to the area for imaging. Using the RMV 707B transducer, with an axial resolution of up to 55 μm, cine images were acquired in EKV (ECG-based kilohertz visualization) mode, which averages several thousand images across the cardiac cycle and removes respiration artifacts. Parasternal long axis EKV images of the left ventricle (LV) were obtained, ensuring the apex and aortic root were in the same plane. Measurements of the left ventricular wall dimensions (anterior and posterior) and internal cavity diameter were taken. A 90º rotation of the transducer enabled EKV images of the short axis (cross-sectional area of the LV) at the mid-papillary muscle level. Images of the aortic arch were taken, and blood velocity in the aortic root was determined 1 mm downstream from the aortic valve. Calculations of heart mass were determined from diastolic measurements using the following formula: heart mass (mg) = 1.055 × (EpiVol − LVEDV)/1000, where EpiVol (epicardial volume) is the sum of the anterior wall thickness plus the internal diameter plus the posterior wall dimensions, cubed, and LVEDV (left ventricular end diastolic volume) is the left ventricular internal dimension in diastole, cubed.
For the short-term BaP exposure (protocol 1, Fig. 1B), BaP (10 mg/kg/day, 5 days a week at the indicated durations) was given by gavage so that a more accurate dose of BaP would be administered in preparation for the comparative microarray studies. For the atherosclerosis studies (long-term BaP exposure, protocol 2, Fig. 1C), we chose to administer the BaP ad libitum (800 mg/ml BaP dissolved in corn oil–soaked chow at the indicated durations) to more accurately simulate how most BaP exposures occur.
RNA isolation.
Total RNA was isolated from aortic arches after homogenization in TRIzol reagent (Invitrogen Corp., Carlsbad, CA) according to the accompanying instructions. Total RNA from heart was isolated from formalin-fixed paraffin-embedded sections (Fedorowicz et al., 2009). Tissue pieces were removed from the paraffin blocks using a scalpel, trimmed of paraffin, and sliced finely. Xylene (1 ml) was added to each sample, the samples were vortexed, centrifuged, and incubated at 56°C for 5 min. The tissue samples were pelleted and the xylene removed. The pellet was washed several times in ethanol, dried, suspended in 150 μl of 200mM Tris-HCl, 200mM NaCl, 1.5mM MgCl2, 2% SDS, pH 7.5, and incubated with 100 μg of Proteinase K (Sigma-Aldrich) at 56°C for 15 min. The samples were incubated on ice for 3 min and centrifuged at 20,000 × g for 15 min at 4°C. The RNA was purified using RNeasy columns (Qiagen, San Diego, CA). RNA quantity and quality were determined using a NanoDrop spectrophotometer ND-1000 (Thermo Scientific, Waltham, MA) and Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA) (Wang et al., 2006).
Microarrays.
The RNA gene expression microarray experiments were carried out by the Dartmouth Genomics and Microarray Laboratory (DGML). The MouseRef-8 v2.0 Expression BeadChip array (Illumina, San Diego, CA) with approximately 25,600 annotated RefSeq transcripts covering 19,100 unique mouse genes were used for the RNA profiling. Fluorescent images were obtained with an Illumina 500GX scanner in the DGML and processed with the BeadScan software (Illumina). Following the accompanying protocol, reverse transcription was carried out using an oligo(dT) primer bearing a T7 promoter and the high-yield ArrayScript reverse transcriptase to make complementary DNA (cDNA). The cDNA was made double stranded with DNA polymerase, and the dsDNA was purified to use as a template for in vitro transcription with T7 RNA polymerase and the included biotin-NTP mix. The labeled complementary RNA was purified and 1.5 μg used for hybridization to the bead arrays for 16 h at 55°C. Following hybridization, the bead arrays were washed and stained with streptavidin-Cy3 (GE Healthcare, Piscataway, NJ). Fluorescent images were obtained with an Illumina 500GX scanner in the DGML and processed with the BeadScan software (Illumina).
Quantitative PCR.
Quantitative PCR (QPCR) analysis using SYBR Green and designed primers (Supplementary table S1) was carried out to verify the microarray results (Schwanekamp et al., 2006). Approximately 2 μg of total RNA (the same RNA used for the microarrays) was used as template for cDNA synthesis. The QPCR reactions were performed on a DNA Engine Opticon Monitor System using software version 3.1 (Bio-Rad, Hercules, CA) set at 35–40 cycles. Agarose gel electrophoresis showed that each PCR produced a single band of the predicted size. Assays to determine DNA contamination were carried out by omitting RNA from the reactions.
Data analysis.
The microarray data analysis was carried out using BRB-Array Tools Version 3.8.1, developed by the Biometric Research Branch of the Division of Cancer Treatment and Diagnosis of the National Cancer Institute under the direction of Dr Richard Simon. Raw intensity data were log2 transformed, median normalized, and filtered to remove nondetected spots as determined by the in house Illumina software program. Normalization was performed by computing a gene-by-gene difference between each array and the median (reference) array and then subtracting the median difference from the log intensities for a given array, so that the gene-by-gene difference between the normalized array and the reference array was zero. Significantly (p value ≤ 0.05) differentially expressed genes were annotated with functional assignments to help determine gene category enrichment using Gene Ontology (GO) database: Biological Process-Filtered Annotation Terms (FAT) (Ashburner et al., 2000). Significantly changed genes were also analyzed using Venn diagrams (Pirooznia et al., 2007). All the differentially expressed genes from the comparisons of the B6 to B6.D2 mice and of the 10-week BaP exposure to 0-week (no BaP) controls (p value ≤ 0.05) are listed in Supplementary tables S2–S5 for the aorta and Supplementary tables S6–S9 for the heart.
RESULTS
Our working hypothesis was that the physiological effects of BaP on the cardiovascular system would be dependent on the degree of AHR activation. We sought to determine the associations among Ahr genotype, BaP exposure, and cardiovascular disease between two C57BL/6 mouse strains that differ at the Ahr gene (Fig. 1A).
BaP Exposure
At given doses, BaP is lethal to mice possessing the allele encoding the low-affinity AHR, whereas the mortality of mice with the allele encoding the high-affinity AHR appears unaffected (Legraverend et al., 1984; Nebert, 1989). Although previous studies suggested that differential AHR signaling was the probable underlying cause for the difference in longevity because these studies compared mouse strains of different genetic backgrounds, e.g., C57Bl/6 versus DBA/2, it had not been demonstrated definitively that the different Ahr alleles were the basis for the vastly decreased longevity in mice with the low-affinity AHR. We wanted to determine (1) whether the different BaP-activated AHRs encoded by the Ahrb1 and Ahrd alleles were indeed the primary reason for the differences observed in life span and (2) if B6.D2 mice were highly susceptible to BaP, then to define a time span in which we could expose B6.D2 mice to BaP before the BaP became lethal.
B6;Apoe−/− and B6.D2;Apoe−/− mice were exposed ad libitum to Western chow and 10 mg/kg/day BaP in corn oil beginning at 4 weeks of age. The 10 mg/kg dose is known to promote atherosclerosis and other cardiac ailments when exposed to the Apoe−/− adult mouse (Curfs et al., 2004). Throughout the 28-week duration of BaP treatment, no apparent ill effects were seen in the B6 mice. However, between 12 and 23 weeks of BaP exposure (corresponding to an age range of 16–27 weeks), the B6.D2 mice had all died, as shown by the Kaplan–Meyer plot in Figure 2. We concluded that (1) the different AHRs encoded by the Ahrb1 and Ahrd alleles in B6 and B6.D2 mice exposed to BaP were the primary if not sole cause for the difference in survival and (2) at 10 mg/kg/day, the temporal effects of BaP toxicity on the physiology of B6.D2 mice can be observed from approximately 4–15 weeks of age.
FIG. 2.

BaP is lethal to B6.D2 male mice but has few discernable effects on B6 mice. Kaplan–Meyer analysis of B6;Apoe−/− and B6.D2;Apoe−/− male mice. BaP exposure was initiated at 4 weeks of age and administered ad libitum in corn oil–soaked Western chow (800 mg/ml) at an estimated dose equivalent of 10 mg/kg/day.
Effect of the Apoe Gene
The Apoe−/− mouse model is used extensively to study atherosclerosis because mice lacking a functional Apoe gene develop severe hypercholesterolemia and atherosclerotic lesions similar to those found in humans (Nakashima et al., 1994). Before undertaking the studies to determine the effects of BaP plus Western diet on the cardiovascular system of B6 and B6.D2 mice within the defined time frame of 4–15 weeks, we wanted to determine whether the Apoe gene had any apparent differential gross effects on the anatomy of the cardiovascular system among the different strains of mice.
To determine whether the Apoe gene differentially contributed to body and heart masses, B6, B6;Apoe−/−, B6.D2, and B6.D2;Apoe−/− mice were fed a Western diet in corn oil beginning at 5 weeks of age following the design shown in Figure 1C. Heart mass was determined over time by ultrasound biomicroscopy at 8, 12, 16, 20, and 28 weeks of age (corresponding to 3, 7, 11, 15, and 23 weeks of BaP exposure) (Supplementary fig. S1). We found no differences in body mass, heart mass, and heart mass:body mass ratio between Apoe+/+ and Apoe−/− mice within the B6 or B6.D2 mouse strains, which allowed us to combine the data for the B6 and B6;Apoe−/− mice and for the B6.D2 and B6.D2;Apoe−/− mice in some of the subsequent studies.
Heart and Ventricle Size
Heart mass was calculated from ultrasound biomicroscopy measurements at 8, 12, 16, 20, and 28 weeks of age (Figs. 3A–C). The heart and body masses for each mouse at each of the five time points in a given strain were summed and divided by the total number of mice for that strain (Figs. 3D–F). The results showed that relative to B6.D2 mice, B6 mice had significantly larger hearts but no significant difference in body mass. As a result, heart mass to body mass ratios were also significantly larger in B6 mice to that of B6.D2 mice. These results were confirmed by a second set of ultrasound biomicroscopy analyses of the B6 and B6.D2 mouse groups, in which the thickness of the anterior and posterior wall of the LV and the inner diameter of the LV were measured at diastolic and systolic cycles (Table 1). At the later time points, the anterior and posterior LV walls were significantly thicker in the B6 mice (values in gray). We concluded that the relatively larger heart mass and thicker ventricle walls observed in B6 mice to that of B6.D2 mice fed Western diet are probably dependent on the increased activity of endogenous ligand–induced AHR signaling.
FIG. 3.

B6.D2 male mice have inherently relatively smaller hearts and smaller heart mass to total body mass ratios than do B6 mice. Data were combined for the B6 and B6;Apoe−/− mice and B6.D2 and B6.D2;Apoe−/− mice, respectively. Heart mass was calculated from ultrasound biomicroscopy measurements for time points 8, 12, 16, 20, and 28 weeks following the long-term BaP exposure protocol 2 (Fig. 1C). Body (A and D) and heart (B and E) masses were determined to calculate heart mass/body mass ratios (C and F) over time (A, B, and C) and cumulatively, in which the heart and body masses for each mouse at each of the five time points in a given strain were summed and divided by the total number of mice for that strain (D, E, and F). Error bars represent SEM. Numbers of mice for each experimental group are shown (G).
TABLE 1.
Differential Heart Sizes of Control (No BaP) B6 Versus B6.D2 Mice
| Mouse strain (number mice per group) | Age (weeks) | Body mass (grams) | LV parameters |
|||||
| Diastolic anterior wall width (mm) | Systolic anterior wall width (mm) | Diastolic posterior wall width (mm) | Systolic posterior wall width (mm) | Diastolic inner diameter (mm) | Systolic inner diameter (mm) | |||
| B6 (4) | 8 | 30.88 | 0.785 | 1.219 | 0.760 | 1.159 | 3.875 | 2.559 |
| B6.D2 (4) | 8 | 29.70 | 0.709 | 1.044 | 0.768 | 1.126 | 3.770 | 2.451 |
| B6 (3) | 16 | 33.53 | 0.857 | 1.280 | 0.823 | 1.243 | 4.042 | 2.665 |
| B6.D2 (4) | 16 | 32.93 | 0.753 | 1.113 | 0.729 | 1.141 | 3.903 | 2.615 |
| B6 (4) | 20 | 32.45 | 0.890 | 1.221 | 0.931 | 1.304 | 3.933 | 2.608 |
| B6.D2 (4) | 20 | 33.65 | 0.734 | 0.969 | 0.743 | 1.028 | 4.061 | 2.921 |
Notes. B6, high-affinity AHR mouse strain; B6.D2, low-affinity AHR mouse strain. All mice were fed a Western diet in corn oil. All mice were male. Values shaded in gray are significantly different (p value = 0.05).
Gene Expression in the Aorta
Our observation that inherent differential AHR signaling activity resulted in larger hearts in B6 mice (Fig. 3) suggested that the regulation of muscle-specific gene expression of the cardiovascular system may be heavily dependent on AHR signaling. Inherent differences in gene expression levels of the aorta between B6;Apoe−/− and B6.D2;Apoe−/− mice were determined (Supplementary table S2) by examining control mice at 5 weeks of age following the experimental design shown in Figure 1B, i.e., no BaP exposure and regular diet. Of the top 50 genes in aorta with the greatest increase in messenger RNA (mRNA) expression levels in B6 mice, 44 of 50 were muscle-specific genes in a range spanning 3- to 25-fold increases (Supplementary table S10). Furthermore, primarily due to the increased relative mRNA expression of the muscle-specific genes in B6 mice, 9 of 20 pathways of all the differentially expressed genes from the B6 versus B6.D2 control mice were associated with muscle synthesis/metabolism as generated by the Biological Process FAT analysis in the GO program (Supplementary table S11A). The microarray results were confirmed by QPCR for selected genes (Supplementary table S12).
Early Atherosclerosis in Mice Exposed to BaP and Western Diet
Following the BaP exposure and diet regimen depicted in Figure 1B, aortas were harvested from vehicle- and BaP-exposed B6;Apoe−/− and B6.D2;Apoe−/− mice (five mice per group) at 5 weeks of age (no BaP control), 7 weeks of age (2-week BaP exposure and Western diet), and 15 weeks of age (10-week BaP exposure and Western diet). The descending and thoracic portions of each aorta were stained with oil red O and examined for atherosclerotic plaque (Fig. 4). A categorical tally of the aortic segments with plaque or no plaque revealed that relative to the B6.D2 mice, the 10-week BaP exposure not only caused B6 mice to have a greater number of aortic segments with plaque (8/10 aorta segments with plaque, Table 2) than B6.D2 mice (3/9 aorta segments with plaque) but the plaque was more extensive than that seen in the B6.D2 mice. No plaque was observed in the 5-week- and 7-week-old mice for both mouse strains (data not shown).
FIG. 4.

Male B6;Apoe−/− mice develop atherosclerotic plaque earlier than do B6.D2;Apoe−/− mice. Shown are representative descending aorta from B6;Apoe−/− mice (A) and B6.D2;Apoe−/− mice (B). Thoracic and descending aorta were harvested from control and BaP-exposed mice sacrificed at 5, 7, and 15 weeks of age and assayed for atherosclerotic plaque by staining with oil red O. The mice were exposed to BaP as depicted in BaP exposure protocol 1 (Fig. 1B). The arrows are pointing at some stained plaque.
TABLE 2.
Early Presence of Atherosclerotic Plaque in the Aorta of 15-Week-Old B6;Apoe−/− Versus B6.D2;Apoe−/− Mice Exposed to BaP
| Mouse strain | Thoracic aorta | Descending aorta | Thoracic + descending | Total aorta |
| B6 | 4/5 | 4/5 | 8/10 | 4/5 |
| B6.D2 | 3/5 | 0/4 | 3/9 | 3/5 |
Notes. B6, high-affinity AHR mouse strain; B6.D2, low-affinity AHR mouse strain. Mice (all male) were fed a Western diet beginning at 5 weeks of age as depicted in BaP exposure protocol 1 (Fig. 1B). n = 5 mice per experimental group.
Immune Response/Inflammation Gene Expression in the Aorta
A hallmark of atherosclerosis is an excessive inflammatory response in the arterial wall (Wu et al., 2011). We therefore investigated whether immune response genes were expressed at greater levels in B6 mice than those of B6.D2 mice. A third segment of the aorta, the aortic arch, was used to extract total RNA for global gene expression analysis using the Illumina bead array system. The differentially expressed genes from B6;Apoe−/− and B6.D2;Apoe−/− mice at the 10-week exposure to BaP and Western diet (Fig. 1B) showed that there were several pathways (GO Biological Process FAT) involved in immune response (the pathways blocked in gray in Supplementary table S13). The differentially expressed genes that comprised the immune response pathways were for the most part more highly expressed in B6 mice to that of B6.D2 mice. For example, a list of the genes comprising the “immune system process” pathway (marked by the black box in Supplementary table S13) shows that 36 of the 46 genes (78%) are expressed at significantly greater levels in B6 relative to B6.D2 mice (Table 3). Eight of the genes in Table 3 possess highly matched AHR response elements (Sun et al., 2004), suggesting they may be possible AHR target genes, including Hoxb4, which is involved in hematopoietic stem cell regulation (Oshima et al., 2011).
TABLE 3.
Immune Response Genes Involved in Atherosclerosis Are Expressed At Greater Levels in BaP-Exposed B6 Mice Relative to B6.D2 Mice (p Value ≤ 0.05)
| Gene symbol | Fold change B6/B6.D2 | AHR response element |
| SERPINA3G | 2.27 | |
| H2-AB1 | 2.15 | |
| H2-AA | 2.11 | |
| H2-EB1 | 2.07 | |
| PSMB9 | 1.81 | ✓ |
| C1QB | 1.75 | |
| LY86 | 1.74 | |
| CXCL9 | 1.73 | |
| OAS1G | 1.69 | |
| C1QA | 1.63 | ✓ |
| C1QC | 1.55 | |
| C2 | 1.55 | |
| FCGR2B | 1.54 | |
| CD79B | 1.50 | |
| H2-M2 | 1.50 | |
| CFP | 1.43 | |
| H2-M3 | 1.43 | |
| CD86 | 1.42 | |
| LOC100044190 | 1.42 | |
| PSMB8 | 1.42 | |
| TLR7 | 1.39 | |
| CLEC4N | 1.37 | |
| CIITA | 1.35 | |
| CD34 | 1.34 | ✓ |
| CD40 | 1.27 | |
| 2610524H06RIK | 1.26 | |
| TNFAIP8L2 | 1.26 | |
| IL7 | 1.24 | |
| CCR5 | 1.18 | |
| IL18 | 1.18 | |
| TLR6 | 1.18 | |
| CD180 | 1.16 | |
| CXCL10 | 1.15 | |
| TLR1 | 1.15 | ✓ |
| BTLA | 1.14 | |
| FOXP3 | 1.11 | |
| FTH1 | 0.94 | |
| TESC | 0.82 | |
| NLRX1 | 0.79 | |
| RB1 | 0.76 | ✓ |
| G6PDX | 0.71 | |
| HOXB4 | 0.69 | ✓ |
| HDAC5 | 0.68 | |
| HSPD1 | 0.65 | ✓ |
| TXNRD2 | 0.63 | |
| DNAJA3 | 0.46 | ✓ |
Notes. B6, high-affinity AHR mouse strain; B6.D2, low-affinity AHR mouse strain. Mice (all male) were fed a Western diet. n = 4 mice per experimental group. Mice were administered 10 mg/kg/day BaP in corn oil five times a week by gavage.
Gene Expression in the Aorta of Mice Exposed to BaP and Western Diet
Differential expression of muscle-specific genes has been associated with atherosclerosis (Tabibiazar et al., 2005a,b), thus, we sought to determine whether BaP differentially affected muscle-specific gene expression and other gene sets in the B6;Apoe−/− and B6.D2;Apoe−/− mice (Supplementary tables S4 and S5). Mice were exposed to BaP and Western diet as shown in Figure 1B. There was a substantial drop in mRNA expression levels for an unexpectedly large number of muscle-specific genes in the B6 mice, in that 29 of the top 50 genes (58%) with the greatest decrease in relative mRNA expression levels were muscle-specific genes (Supplementary table S14). These results were a remarkable reversal of mRNA levels inherently expressed in the B6 mouse, in that many of the same muscle-specific genes that had shown the greatest decrease in mRNA gene expression after 10 weeks of BaP treatment and Western diet were those with the highest levels of expression in B6 mice relative to B6.D2 mice (Supplementary table S10).
Table 4 is a compilation of the results shown in Supplementary tables S10 and S14, which reveal that 23 of 36 (64%) of the top endogenously expressed muscle-specific genes in aorta of B6 versus B6.D2 (Supplementary table S10) also showed the greatest decrease in gene expression in B6 when exposed for 10 weeks to BaP and Western diet (Supplementary table S14). For example, the natriuretic peptide precursor type A (Nppa) gene product, which is involved in myocardium differentiation and the adaptive response to heart failure (Houweling et al., 2005), was expressed 25-fold greater in B6 mice relative to B6.D2 mice at 5 weeks of age (no BaP, regular diet) but at minus 30-fold in B6 mice exposed to BaP and Western diet for 10 weeks relative to control 5-week-old B6 mice. Only two of the same muscle-specific genes were differentially expressed (Myl3 and Myl1, the latter gene in the opposite direction) in the B6.D2 mouse after 10 weeks of BaP and Western diet (Supplementary table S5). Six of the differentially expressed muscle-specific genes in Supplementary table S14 contain high-stringency AHR response elements (Sun et al., 2004). The microarray results were confirmed by QPCR assays with selected genes (Supplementary table S12).
TABLE 4.
A Comparison of Muscle-Specific Genes Inherently Expressed in Aorta of B6 Versus B6.D2 Mice to Muscle-Specific Genes in Aorta of B6 Mice Exposed to BaP for 10 Weeks
| Gene symbol | Fold change |
AHR response element | |
| B6/B6.D2 no BaP | B6 BaP 10 week/0 week | ||
| Nppa | 25.64 | −30.53 | |
| Myh6 | 20.63 | −27.87 | |
| Ckm | 19.48 | −29.94 | |
| Sln | 17.86 | −24.07 | |
| Eef1a2 | 16.25 | −9.66 | |
| Ckmt2 | 15.66 | −20.91 | |
| Cox6a2 | 14.84 | −26.02 | |
| Myl4 | 14.26 | −16.48 | |
| Csrp3 | 13.85 | −17.14 | |
| Myl3 | 12.64 | −25.00 (−2.17) | |
| Tnnc1 | 12.54 | −22.37 | |
| Myl7 | 12.14 | −14.15 | |
| Mybphl | 11.70 | −15.03 | |
| Actc1 | 11.56 | −22.90 | |
| Mb | 11.49 | −11.33 | |
| Ttn | 10.67 | −16.52 | |
| Eno3 | 10.52 | −12.53 | |
| Ankrd1 | 9.54 | ||
| Actn2 | 9.36 | −7.83 | ✓ |
| Pgam2 | 7.83 | −8.69 | |
| Trim72 | 5.51 | −7.19 | |
| Hrc | 5.50 | −6.95 | |
| Mybpc3 | 5.33 | ||
| Myl1 | 5.08 | −8.33 (1.33) | |
| Atp2a2 | 4.66 | −7.68 | ✓ |
| Myoz2 | 4.25 | ||
| Casq2 | 4.04 | ||
| Tmod1 | 3.90 | ✓ | |
| Tcap | 3.41 | ||
| Gpc1 | 3.37 | ✓ | |
| Stard10 | 3.35 | ||
| Nrap | 3.34 | ||
| Corin | 3.30 | ||
| Des | 3.30 | ||
| Atp2a1 | −10.14 | ✓ | |
| Tnni2 | −14.34 | ✓ | |
Notes. B6, high-affinity AHR mouse strain; B6.D2, low-affinity AHR mouse strain. Values in parentheses are the fold change of the corresponding statistically significant differentially expressed genes in B6.D2 mice exposed to BaP for 10 weeks.
Further support that the primary biological pathways affected in the aorta by the AHR involve muscle genes is made evident in Table 5. Pathway analysis (GO Biological Process FAT) of the unique differentially expressed genes of B6 versus B6.D2 mice exposed to BaP and Western diet for 10 weeks (the checked category in the Venn diagram) revealed that the predominate pathways for B6 were involved in muscle synthesis and metabolism (blocked in gray). In contrast, most of the pathways for B6.D2 mice were involved in cell metabolism and protein synthesis (Supplementary table S15C). All the differentially expressed genes for the 10-week BaP/Western diet versus control mice are displayed for the B6 and B6.D2 mice in Supplementary tables S15A–E.
TABLE 5.
Biological Pathways Associated With Differentially Expressed Genes Unique to B6 Compared With B6.D2 Mice Exposed to BaP
| Biological pathway/term | p Value | FDR |
| GO:0009987∼cellular process | 1.70 × 10−14 | 3.08 × 10−11 |
| GO:0007517∼muscle organ development | 2.57 × 10−6 | 4.67 × 10−2 |
| GO:0003012∼muscle system process | 1.25 × 10−5 | 2.27 × 10−2 |
| GO:0001944∼vasculature development | 1.40 × 10−5 | 2.54 × 10−2 |
| GO:0001568∼blood vessel development | 1.83 × 10−5 | 3.32 × 10−2 |
| GO:0044237∼cellular metabolic process | 4.71 × 10−5 | 8.54 × 10−2 |
| GO:0006936∼muscle contraction | 4.85 × 10−5 | 8.79 × 10−2 |
| GO:0014706∼striated muscle tissue development | 5.09 × 10−5 | 9.22 × 10−2 |
| GO:0060537∼muscle tissue development | 5.77 × 10−5 | 1.05 × 10−1 |
| GO:0044238∼primary metabolic process | 6.81 × 10−5 | 1.23 × 10−1 |
| GO:0042592∼homeostatic process | 9.89 × 10−5 | 1.79 × 10−1 |
| GO:0048514∼blood vessel morphogenesis | 1.88 × 10−5 | 3.40 × 10−1 |
| GO:0008152∼metabolic process | 1.91 × 10−4 | 3.47 × 10−1 |
| GO:0055001∼muscle cell development | 2.30 × 10−4 | 4.16 × 10−1 |
| GO:0006066∼alcohol metabolic process | 2.41 × 10−4 | 4.36 × 10−1 |
| GO:0006006∼glucose metabolic process | 2.46 × 10−4 | 4.45 × 10−1 |
| GO:0006937∼regulation of muscle contraction | 2.50 × 10−4 | 4.52 × 10−1 |
| GO:0030239∼myofibril assembly | 4.11 × 10−4 | 7.43 × 10−1 |
| GO:0031032∼actomyosin structure organization | 4.78 × 10−4 | 8.64 × 10−1 |
| GO:0005996∼monosaccharide metabolic process | 5.03 × 10−4 | 9.08 × 10−1 |
Notes. B6, high-affinity AHR mouse strain; B6.D2, low-affinity AHR mouse strain; FDR, false discovery rate. Mice (all male) were fed a Western diet. n = 5 mice per experimental group. Mice were administered 10 mg/kg/day BaP in corn oil five times a week by gavage.
Gene Expression of the Heart Exposed to BaP and Western Diet
Heart size and aorta RNA expression levels of muscle-specific genes were so greatly affected in B6 versus B6.D2 mice exposed BaP and different diets; hence, we sought to determine the gene expression levels of the heart itself. To our surprise, relatively few genes in the heart were affected by BaP and diet, and those for the most part were affected only slightly (Supplementary tables S6–S9). None of the differentially expressed genes pointed convincingly to a biological pathway to lend insight into the underlying cause for the difference in heart and ventricle wall sizes. However, there were several differentially expressed genes of potential interest, including three major urinary protein genes (Mup1, Mup2, and Mup3) (Supplementary table S16). MUP1 in mice regulates systemic glucose metabolism via hepatic gluconeogenic and/or lipogenic programs (Zhou et al., 2009). The Mup2 gene is regulated by the toxicant-activated AHR, in which MUP2 protein levels in mice declined when exposed to 3-methylcholanthrene, a potent AHR inducer (Nukaya et al., 2004). The authors concluded that the toxicant-activated AHR indirectly suppressed Mup2 gene expression by inhibiting expression of the growth hormone receptor and Janus kinase 2 (Jak2) directed pathways, both of which regulate Mup2 expression. Jak2 RNA levels were also affected (declined) in B6 mice exposed to BaP.
Body and Organ Mass in Mice Exposed to BaP and Western Diet
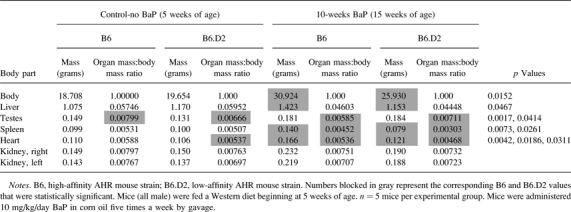
We observed in the process of necropsy inherent differences not only in body and heart masses but also in the mass of several other organs of B6;Apoe−/− mice versus B6.D2;Apoe−/− mice (Supplementary table S17) subjected to the short-term BaP exposure regimen (Fig. 1B). At the 5-week time point, at which the mice had not been exposed to BaP and had been fed only regular chow, B6 mice had significantly larger testes and a greater heart mass:body mass ratio than did B6.D2 mice, in which the latter results replicated earlier findings shown in Figure 3C. After 10-week exposure to BaP and Western diet, body mass, liver mass, spleen mass, heart mass, spleen mass:body mass ratio, and heart mass:body mass ratio were significantly greater in B6 mice relative to B6.D2 mice. In contrast, B6 mouse testes mass:body mass ratio went from significantly larger at 5 weeks to significantly smaller to that observed in B6.D2 mice after 10 weeks of BaP and Western diet. There were no significant differences in B6 mice exposed to BaP plus Western diet at 20 weeks (which would have been lethal in B6.D2 mice) versus B6 mice exposed to Western diet alone (data not shown).
DISCUSSION
B6 and B6.D2 Mice as Model Systems to Study Gene/Environment Interactions in the Context of Disease
BaP is one of the more potent toxicants in cigarette smoke, and cigarette smoking combined with obesity can be deadly. The life expectancy of obese males after age 40 who smoke is lowered by 14 years compared with normal weight nonsmokers (Peeters et al., 2003). Obese individuals who smoke are more likely to develop type 2 diabetes and cancer, as well as heart disease. How smoking and obesity interact to cause disease and reduce life expectancy is not clear. Thus, there is a real need to develop model systems that take into account the dual effects of harmful toxicants and obesity, and the Ahr mouse models used here may be very useful in this regard.
In humans, differential responses to drugs and toxicants are usually due to allelic differences rather than to gene loss (Weinshilboum, 2003). We speculate that in our mouse model system, in an unstressed environment, the two Ahr alleles are the underlying basis of relatively negligible physical differences. However, in a stressed environment, an exposure to BaP for instance, the two alleles can be the source of large phenotypic variations. Hence, we suggest that the use of mouse models with allelic differences more closely exemplify the human condition than a gene knockout mouse model. Knocking out a gene often has unforeseen secondary effects, as is the case for the Ahr gene (Lahvis et al., 2000), which can confound interpretation.
The Role of BaP and the AHR in Atherosclerosis
In the work reported here, in which atherosclerosis was considerably more prevalent in B6 mice to that of B6.D2 mice (Fig. 4, Table 2), the Cyp1a1 gene was not differentially expressed in the aorta of both BaP-treated mouse strains, and the Cyp1b1 gene (Supplementary tables S3 and S4) was differentially expressed to approximately the same levels in both (approximately threefold). Thus, a differential activity of the Cyp1 genes between the mouse strains does not appear to be playing a large role in the differential effects produced by BaP in the aorta.
However, our findings are in contrast to studies showing that induction of the Cyp1 genes by the BaP-activated AHR contributes to atherosclerosis. CYP1-dependent bioactivation of BaP by chicken aortic microsome homogenates indicated that oxidized BaP metabolites formed to generate reactive intermediates that covalently bound DNA in vitro (Bond et al., 1980). Similar results were observed using human thoracic aortic tissue (Juchau et al., 1976) and cultured human fetal aortic smooth muscle cells (SMCs) (Bond et al., 1979). Other evidence supporting the involvement of the CYP1 family in atherosclerosis comes from studies showing that atherosclerosis-susceptible White Carneque Pigeons have greater inducible CYP1A1 activity than atherosclerosis-resistant Show Racer Pigeons (St Clair, 1983), consistent with reports that 3-methylcholanthrene causes more severe vascular lesions in the high-affinity AHR B6 mice than in the low-affinity AHR B6.D2 mice (Paigen et al., 1987).
The activation of AHR signaling by BaP and other toxicants is known to promote atherosclerosis in mice (Iwano et al., 2005a,b; Oesterling et al., 2008). Although in vivo studies with mice and other rodents have linked different disease states to specific polymorphic forms of the AHR (Watanabe et al., 2000; Williams and Phillips, 2000), none has examined the effects of different AHR polymorphic forms on atherosclerosis. For humans, epidemiological studies have shown an association between some cancers and different forms of the AHR but no linkage to cardiac disease (Harper et al., 2002; Van den Berg et al., 2006; Wong et al., 2001); however, the human AHR polymorphic studies have been relatively small and focused on cancer. Nevertheless, the results from the Ahr mouse model studies are translatable to humans because the affinity and responsiveness of the AHR encoded by the mouse lower affinity Ahrd gene are similar to that of the human Ahr gene (Ema et al., 1994).
Differential Gene Expression in Atherosclerosis
It is well established that atherosclerosis involves the inflammatory response (Wu et al., 2011), and candidate genes that may contribute to the earlier atherosclerotic state in B6 mice are the immune response genes with elevated expression shown in Table 5 and Supplementary table S13. It is also well established that vessel walls respond to injury and disease by undergoing SMC dedifferentiation, which requires the repression of smooth muscle gene expression, prior to proliferation and migration (Owens et al., 2004). We found mRNA levels for a considerable number of muscle-specific genes dropped steeply when B6 mice were exposed to BaP (Supplementary tables S4 and S14). Others have shown that the expression of muscle-specific genes is inhibited in the aorta by the toxicant-activated AHR in B6; Apoe−/− mice (Curfs et al., 2004; Nakashima et al., 1994). The transcriptional regulation of many smooth muscle–specific genes requires serum response factor (SRF) and myocardin (MYOCD), a powerful SRF cofactor that activates smooth muscle–specific gene expression (Pipes et al., 2006). Srf mRNA was expressed at significantly lower levels in B6 mice exposed to BaP for 10 weeks (Supplementary table S3), whereas B6.D2 mice exposed to BaP for 10 weeks showed an increase in Srf mRNA levels of over twofold (Supplementary table S5), which may account for the observed differential expression levels of muscle-specific genes in B6 mice relative to B6.D2 mice when the strains are exposed to BaP. Myocd mRNA was expressed at significantly lower levels in B6 mice relative to B6.D2 mice in both control mice (no BaP, regular diet, Supplementary table S2) and mice exposed to BaP and Western diet for 10 weeks (Supplementary table S3). Both the Srf and Myocd genes have lower stringency AHR response elements (Sun et al., 2004) suggesting a possible direct link to the AHR.
Endogenous AHR Signaling and Heart Growth
Although we asserted above that in an unstressed environment the two Ahr alleles generate relatively negligible physical differences, our studies suggest that the AHR may play key roles in the development and ongoing physiology of the mouse heart. AHR signaling is involved in numerous developmental and homeostatic programs that seemingly rely on as yet unidentified endogenous ligands, or for a more accurate characterization, exposure to a known exogenous ligand is not required for many apparent AHR-dependent functions. This contention is supported by many studies that have shown that developmental programs in the mouse are profoundly affected in the Ahr-null mouse (Fernandez-Salguero et al., 1995; Schmidt et al., 1996). In regard to the cardiovascular system, there are several distinct cardiovascular phenotypes in the Ahr-null mouse, including hypertrophic remodeling of the left ventricular wall, a thickening of the arterial media wall in the aorta, increased numbers of SMCs in the aorta, and extensive fibrosis in the heart (Sauzeau et al., 2011). Thus, the AHR appears to play critical cardiovascular roles not only in development and xenobiotic metabolism but also in homeostasis, remodeling, and disease.
Few if any studies have examined the effects of differential AHR signaling derived from functional alleles of the Ahr gene, whether by intrinsic or exogenous ligand activation. In an attempt to determine how intrinsic differential AHR signaling may influence mouse physiology, i.e., the mice were not exposed to an exogenous environmental agent, we carried out studies to examine the anatomy of two strains of adult male mice that possessed high- and low-affinity alleles of the Ahr gene. What we found was contrary to what would have been predicted from the Ahr-null mouse studies. Instead of a larger heart in the presumably less potent AHR signaling output in the cardiovascular tissues of the B6.D2 mouse, the hearts were significantly smaller by mass (Fig. 3) than those of B6 mice, and the ultrasound biomicroscopy results revealed in real time that B6.D2 mice relative to B6 mice had significantly thinner anterior and posterior LV walls (Table 1). In addition, the microarray data of the 5-week-old control mice (Supplementary table S10) showed that muscle-specific genes of the aorta (but not of the heart) were expressed at much greater levels in B6 mice to that of B6.D2 mice. Thus, the more massive hearts, thicker LV walls, and relatively more highly expressed muscle-specific genes in aorta observed in B6 mice all point to a role for endogenous AHR signaling in the regulation of heart growth, such that B6 mouse hearts grow larger and more muscular than B6.D2 mouse hearts. Again, there were several muscle-specific genes that were potential transcriptional targets of the AHR (based on the presence of AHR response elements, Table 3) that may contribute to an AHR-based regulation of gene expression dominated by muscle-specific genes.
We showed a relatively small response by the heart to BaP exposure to that by the aorta. There has been little work on the toxicant-activated AHR on adult heart gene expression; however, it was shown that AHR signaling has a large impact on RNA expression of the fetal mouse heart (Aragon et al., 2008; Thackaberry et al., 2005). For our work, the RNA from aorta was isolated directly from tissue, whereas the RNA from heart was isolated from formalin-fixed paraffin-embedded sections. Although all internal controls on the microarrays were positive, the degree of degradation for the RNA isolated from the heart tissue sections may have been sufficiently severe to skew the results.
How Can BaP Accelerate Death in B6.D2 Mice but Still Be Atheroprotective?
B6 mouse heart and body mass were significantly larger than B6.D2 mice when exposed to BaP; however, liver and spleen growth were also significantly larger in B6 mice (Table 6). The underlying differential effect of BaP on B6.D2 body and organ growth may be that BaP is more injurious to mice harboring the low-affinity AHR than those with the high-affinity AHR (Galván et al., 2003; Legraverend et al., 1984; Najai et al., 2011), and the lower body and organ mass of B6.D2 mice may be a reflection of a dying state. When BaP is given orally, a 15 mg/kg dose of BaP has a half-life of 31 min in B6 mice (Uno et al., 2004). However, the half-life of BaP in mice with the Ahrd/d genotype must be considerably longer. BaP and BaP metabolites can be 20- to 80-fold in higher bone marrow, 10- to 20-fold higher in spleen, and approximately 2-fold higher in small intestine and liver for the initial 1–2 days postexposure in mice with the Ahrd genes relative to that of B6 mice (Nebert et al., 1980). As a result, BaP-exposed mice carrying the low-affinity Ahrd gene when compared with BaP-exposed B6 mice carrying the high-affinity Ahrb1 gene are much more sickly and die of bone marrow toxicity (Galván et al., 2003; Najai et al., 2011; Nebert et al., 1977). The B6.D2 mice in our study are also very likely dying primarily of bone marrow toxicity, but to be certain, histopathology of the bone marrow will need to be carried out.
TABLE 6.
Effect of BaP on the Mass and Mass to Body Mass Ratio of Different Organs in B6 Versus B6.D2 Mice
 |
It has been shown that CYP1A1 is protective against BaP when administered orally (Uno et al., 2004) (the means by which most BaP exposures enter the body). Generally, these findings were contrary to previous studies, which had used cell lines or live mice that were administered BaP via routes other than orally, e.g., ip. It was speculated that BaP given orally must pass through the small intestine and liver where the BaP is metabolized to an extent by CYP1A1 before entering the bloodstream and distributed to other organs to damage more susceptible tissues (Uno et al., 2004). An ip administration of BaP, however, bypasses the small intestine and liver in large part because it is absorbed via the mesenteric veins and lymphatic system and distributed at much higher concentrations throughout the body to cause damage in other more susceptible tissues and organs. As a result, B6 mice possessing the Ahrb1 allele fare better than mice possessing the Ahrd allele because of the greater capacity of the Ahrb1 gene product to induce CYP1A1 activity in the liver (Legraverend et al., 1984; Nebert, 1989; Robinson et al., 1975). These findings explain how BaP may be lethal for mice possessing the low-affinity AHR but not how reduced AHR signaling may be atheroprotective. Our results suggest that atherosclerosis in the aorta may be more dependent on the regulation of inflammation and muscle-specific gene expression via AHR signaling than on tissue damage caused by enhanced BaP metabolism.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
The National Institutes of Health (NIH)/National Institute of Environmental Health Sciences (NIEHS) award R21ES013827, NIH/National Center for Research Resources (NCRR) Centers of Biomedical Research Excellence (COBRE) award RR028309, Norris Cotton Cancer Center Prouty award P30CA023108, NIH/National Cancer Institute (NCI) R25 CA134286 Training Program for Quantitative Population Sciences in Cancer (JSK-H) and a grant from the Dartmouth Hitchcock Foundation.
Supplementary Material
Acknowledgments
We thank the DGML (Genomics Shared Resource) for the microarray studies, the Biostatistics and Bioinformatics Shared Resource for the microarray data analyses, and the reviewers for their thoughtful comments.
References
- Andreola F, Fernandez-Salguero PM, Chiantore MV, Petkovich MP, Gonzalez FJ, De Luca LM. Aryl hydrocarbon receptor knockout mice (AHR-/-) exhibit liver retinoid accumulation and reduced retinoic acid metabolism. Cancer Res. 1997;57:2835–2838. [PubMed] [Google Scholar]
- Aragon AC, Kopf PG, Campen MJ, Huwe JK, Walker MK. In utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure: Effects on fetal and adult cardiac gene expression and adult cardiac and renal morphology. Toxicol. Sci. 2008;101:321–330. doi: 10.1093/toxsci/kfm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond JA, Gown AM, Yang HL, Benditt EP, Juchau MR. Further investigations of the capacity of polynuclear aromatic hydrocarbons to elicit atherosclerotic lesions. J. Toxicol. Environ. Health. 1981;7:327–335. doi: 10.1080/15287398109529983. [DOI] [PubMed] [Google Scholar]
- Bond JA, Kocan RM, Benditt EP, Juchau MR. Metabolism of benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene in cultured human fetal aortic smooth muscle cells. Life Sci. 1979;25:425–430. doi: 10.1016/0024-3205(79)90574-5. [DOI] [PubMed] [Google Scholar]
- Bond JA, Yang HY, Majesky MW, Benditt EP, Juchau MR. Metabolism of benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene in chicken aortas: Monooxygenation, bioactivation to mutagens, and covalent binding to DNA in vitro. Toxicol. Appl. Pharmacol. 1980;52:323–335. doi: 10.1016/0041-008x(80)90119-2. [DOI] [PubMed] [Google Scholar]
- Brenner DA. New functions for the aryl hydrocarbon receptor. Hepatology. 1996;23:379–380. doi: 10.1002/hep.510230228. [DOI] [PubMed] [Google Scholar]
- Breslow JL. Mouse models of atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- Curfs DM, Lutgens E, Gijbels MJ, Kockx MM, Daemen MJ, van Schooten FJ. Chronic exposure to the carcinogenic compound benzo[a]pyrene induces larger and phenotypically different atherosclerotic plaques in ApoE-knockout mice. Am. J. Pathol. 2004;164:101–108. doi: 10.1016/S0002-9440(10)63101-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton TP, Kerzee JK, Wang B, Miller M, Dieter MZ, Lorenz JN, Shertzer HG, Nerbert DW, Puga A. Dioxin exposure is an environmental risk factor for ischemic heart disease. Cardiovasc. Toxicol. 2001;1:285–298. doi: 10.1385/ct:1:4:285. [DOI] [PubMed] [Google Scholar]
- Ema M, Ohe N, Suzuki M, Mimura J, Sogawa K, Ikawa S, Fujii-Kuriyama Y. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J. Biol. Chem. 1994;269:27337–27343. [PubMed] [Google Scholar]
- Fedorowicz G, Guerrero S, Wu TD, Modrusan Z. Microarray analysis of RNA extracted from formalin-fixed, paraffin-embedded and matched fresh-frozen ovarian adenocarcinomas. BMC Med. Genomics. 2009;2:23. doi: 10.1186/1755-8794-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet. Pathol. 1997;34:605–614. doi: 10.1177/030098589703400609. [DOI] [PubMed] [Google Scholar]
- Galván N, Jaskula-Sztul R, MacWilliams PS, Czuprynski CJ, Jefcoate CR. Bone marrow cytotoxicity of benzo[a]pyrene is dependent on CYP1B1 but is diminished by Ah receptor-mediated induction of CYP1A1 in liver. Toxicol. Appl. Pharmacol. 2003;193:84–96. doi: 10.1016/s0041-008x(03)00338-7. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Tukey RH, Nebert DW. Structural gene products of the Ah locus. Transcriptional regulation of cytochrome P1-450 and P3-450 mRNA levels by 3-methylcholanthrene. Mol. Pharmacol. 1984;26:117–121. [PubMed] [Google Scholar]
- Guo J, Sartor M, Karyala S, Medvedovic M, Kann S, Puga A, Ryan P, Tomlinson CR. Expression of genes in the TGF-beta signaling pathway is significantly deregulated in smooth muscle cells from aorta of aryl hydrocarbon receptor knockout mice. Toxicol. Appl. Pharmacol. 2004;194:79–89. doi: 10.1016/j.taap.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Hankinson O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- Harper PA, Wong JY, Lam MS, Okey AB. Polymorphisms in the human AH receptor. Chem. Biol. Interact. 2002;141:161–187. doi: 10.1016/s0009-2797(02)00071-6. [DOI] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Hofstetter JR, Svihla-Jones DA, Mayeda AR. A QTL on mouse chromosome 12 for the genetic variance in free-running circadian period between inbred strains of mice. J. Circadian Rhythms. 2007;5:7. doi: 10.1186/1740-3391-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houweling AC, van Borren MM, Moorman AFM, Christoffels VM. Expression and regulation of the atrial natriuretic factor encoding gene Nppa during development and disease. Cardiovasc. Res. 2005;67:583–593. doi: 10.1016/j.cardiores.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Iwano S, Asanuma F, Nukaya M, Saito T, Kamataki T. CYP1A1-mediated mechanism for atherosclerosis induced by polycyclic aromatic hydrocarbons. Biochem. Biophys. Res. Commun. 2005a;337:708–712. doi: 10.1016/j.bbrc.2005.09.109. [DOI] [PubMed] [Google Scholar]
- Iwano S, Nukaya M, Saito T, Asanuma F, Kamataki T. A possible mechanism for atherosclerosis induced by polycyclic aromatic hydrocarbons. Biochem. Biophys. Res. Commun. 2005b;335:220–226. doi: 10.1016/j.bbrc.2005.07.062. [DOI] [PubMed] [Google Scholar]
- Juchau MR, Bond JA, Benditt EP. Aryl 4-monooxygenase and cytochrome P-450 in the aorta: Possible role in atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 1976;73:3723–3725. doi: 10.1073/pnas.73.10.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karyala S, Guo J, Sartor M, Medvedovic M, Kann S, Puga A, Ryan P, Tomlinson CR. Different global gene expression profiles in benzo[a]pyrene- and dioxin-treated vascular smooth muscle cells of AHR-knockout and wild-type mice. Cardiovasc. Toxicol. 2004;4:47–73. doi: 10.1385/ct:4:1:47. [DOI] [PubMed] [Google Scholar]
- Lahvis GP, Lindell SL, Thomas RS, McCuskey RS, Murphy C, Glover E, Bentz M, Southard J, Bradfield CA. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10442–10447. doi: 10.1073/pnas.190256997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legraverend C, Guenthner TM, Nebert DW. Importance of the route of administration for genetic differences in benzo[a]pyrene-induced in utero toxicity and teratogenicity. Teratology. 1984;29:35–47. doi: 10.1002/tera.1420290106. [DOI] [PubMed] [Google Scholar]
- Liu S, Abdelrahim M, Khan S, Ariazi E, Jordan VC, Safe S. Aryl hydrocarbon receptor agonists directly activate estrogen receptor alpha in MCF-7 breast cancer cells. Biol. Chem. 2006;387:1209–1213. doi: 10.1515/BC.2006.149. [DOI] [PubMed] [Google Scholar]
- McMillan BJ, Bradfield CA. The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 2007;104:1412–1417. doi: 10.1073/pnas.0607296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KP, Ramos KS. Impact of cellular metabolism on the biological effects of benzo[a]pyrene and related hydrocarbons. Drug Metab. Rev. 2001;33:1–35. doi: 10.1081/dmr-100000138. [DOI] [PubMed] [Google Scholar]
- Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M, et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- Moorthy B, Miller KP, Jiang W, Ramos KS. The atherogen 3-methylcholanthrene induces multiple DNA adducts in mouse aortic smooth muscle cells: Role of cytochrome P4501B1. Cardiovasc. Res. 2002;53:1002–1009. doi: 10.1016/s0008-6363(01)00536-3. [DOI] [PubMed] [Google Scholar]
- Moorthy B, Miller KP, Jiang W, Williams ES, Kondraganti SR, Ramos KS. Role of cytochrome P4501B1 in benzo[a]pyrene bioactivation to DNA-binding metabolites in mouse vascular smooth muscle cells: Evidence from 32P-postlabeling for formation of 3-hydroxybenzo[a]pyrene and benzo[a]pyrene-3,6-quinone as major proximate genotoxic intermediates. J. Pharmacol. Exp. Ther. 2003;305:394–401. doi: 10.1124/jpet.102.044271. [DOI] [PubMed] [Google Scholar]
- Najai AU, Larsen MC, Bushkofsky JR, Czuprynski CJ, Jefcoate CR. Acute disruption of bone marrow hematopoiesis by benzo(a)pyrene is selectively reversed by aryl hydrocarbon receptor-mediated processes. Mol. Pharmacol. 2011;79:724–734. doi: 10.1124/mol.110.070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- Nebert DW. The Ah locus: Genetic differences in toxicity, cancer, mutation, and birth defects. Crit. Rev. Toxicol. 1989;20:153–174. doi: 10.3109/10408448909017908. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Jensen NM, Levitt RC, Felton JS. Toxic chemical depression of the bone marrow and possible aplastic anemia explainable on a genetic basis. Clin. Toxicol. 1980;16:99–122. doi: 10.3109/15563658008989927. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Karp CL. Endogenous functions of the aryl hydrocarbon receptor (AHR): Intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J. Biol. Chem. 2008;283:36061–36065. doi: 10.1074/jbc.R800053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Levitt RC, Jensen NM, Lambert GH, Felton JS. Birth defects and aplastic anemia: Differences in polycyclic hydrocarbon toxicity associated with the Ah locus. Arch. Toxicol. 1977;39:109–132. doi: 10.1007/BF00343280. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Puga A, Vasiliou V. Role of the Ah receptor and the dioxin-inducible [Ah] gene battery in toxicity, cancer, and signal transduction. Ann. N. Y. Acad. Sci. 1993;685:624–640. doi: 10.1111/j.1749-6632.1993.tb35928.x. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Winker J, Gelboin HV. Aryl hydrocarbon hydroxylase activity in human placenta from cigarette smoking and nonsmoking women. Cancer Res. 1969;29:1763–1769. [PubMed] [Google Scholar]
- Nukaya M, Takahashi Y, Gonzalez FJ, Kamataki T. Aryl hydrocarbon receptor-mediated suppression of GH receptor and Janus kinase 2 expression in mice. FEBS Lett. 2004;558:96–100. doi: 10.1016/S0014-5793(03)01528-X. [DOI] [PubMed] [Google Scholar]
- Nunnari JJ, Zand T, Joris I, Majno G. Quantitation of oil red O staining of the aorta in hypercholesterolemic rats. Exp. Mol. Pathol. 1989;51:1–8. doi: 10.1016/0014-4800(89)90002-6. [DOI] [PubMed] [Google Scholar]
- Oesterling E, Toborek M, Hennig B. Benzo[a]pyrene induces intercellular adhesion molecule-1 through a caveolae and aryl hydrocarbon receptor mediated pathway. Toxicol. Appl. Pharmacol. 2008;232:309–316. doi: 10.1016/j.taap.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423:545–550. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- Oshima M, Endoh M, Endo TA, Toyoda T, Nakajima-Takagi Y, Sugiyama F, Koseki H, Kyba M, Iwama A, Osawa M. Genome-wide analysis of target genes regulated by HoxB4 in hematopoietic stem and progenitor cells developing from embryonic stem cells. Blood. 2011;117:e142–e150. doi: 10.1182/blood-2010-12-323212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- Peeters A, Barendregt JJ, Willekens F, Mackenbach JP, Al Mamun A, Bonneux L. Obesity in adulthood and its consequences for life expectancy: A life-table analysis. Ann. Intern. Med. 2003;138:24–32. doi: 10.7326/0003-4819-138-1-200301070-00008. [DOI] [PubMed] [Google Scholar]
- Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipes GCT, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: Versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- Pirooznia M, Nagarajan V, Deng Y. GeneVenn–A web application for comparing gene lists using Venn diagrams. Bioinformation. 2007;1:420–422. doi: 10.6026/97320630001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland A, Glover E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: Segregation of toxicity with the Ah locus. Mol. Pharmacol. 1980;17:86–94. [PubMed] [Google Scholar]
- Poland A, Palen D, Glover E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 1994;46:915–921. [PubMed] [Google Scholar]
- Puga A, Tomlinson CR, Xia Y. Ah receptor signals cross-talk with multiple developmental pathways. Biochem. Pharmacol. 2005;69:199–207. doi: 10.1016/j.bcp.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Ramos KS, Parrish AR. Growth-related signaling as a target of toxic insult in vascular smooth muscle cells: Implications in atherogenesis. Life Sci. 1995;57:627–635. doi: 10.1016/0024-3205(95)00314-v. [DOI] [PubMed] [Google Scholar]
- Robinson JR, Felton JS, Levitt RC, Thorgeirsson SS, Nebert DW. Relationship between “aromatic hydrocarbon responsiveness” and the survival times in mice treated with various drugs and environmental compounds. Mol. Pharmacol. 1975;11:850–865. [PubMed] [Google Scholar]
- Sauzeau V, Carvajal-Gonzalez JM, Riolobos AS, Sevilla MA, Menacho-Marquez M, Roman ÃC, Abad A, Montero MJ, Fernandez-Salguero P, Bustelo XR. Transcriptional factor aryl hydrocarbon receptor (Ahr) controls cardiovascular and respiratory functions by regulating the expression of the Vav3 proto-oncogene. J. Biol. Chem. 2011;286:2896–2909. doi: 10.1074/jbc.M110.187534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer JM, Whalen DL, Jerina DM. Chemical strategies for the inactivation of bay-region diol epoxides, ultimate carcinogens derived from polycyclic aromatic hydrocarbons. Drug Metab. Rev. 1989;20:155–182. doi: 10.3109/03602538909103534. [DOI] [PubMed] [Google Scholar]
- Schlezinger JJ, Liu D, Farago M, Seldin DC, Belguise K, Sonenshein GE, Sherr DH. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol. Chem. 2006;387:1175–1187. doi: 10.1515/BC.2006.145. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanekamp JA, Sartor MA, Karyala S, Halbleib D, Medvedovic M, Tomlinson CR. Genome-wide analyses show that nuclear and cytoplasmic RNA levels are differentially affected by dioxin. Biochim. Biophys. Acta. 2006;1759:388–402. doi: 10.1016/j.bbaexp.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Song Y, Sonawane ND, Salinas D, Qian L, Pedemonte N, Galietta LJ, Verkman AS. Evidence against the rescue of defective DeltaF508-CFTR cellular processing by curcumin in cell culture and mouse models. J. Biol. Chem. 2004;279:40629–40633. doi: 10.1074/jbc.M407308200. [DOI] [PubMed] [Google Scholar]
- St Clair RW. Metabolic changes in the arterial wall associated with atherosclerosis in the pigeon. Fed. Proc. 1983;42:2480–2485. [PubMed] [Google Scholar]
- Sun YV, Boverhof DR, Burgoon LD, Fielden MR, Zacharewski TR. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 2004;32:4512–4523. doi: 10.1093/nar/gkh782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabibiazar R, Wagner RA, Ashley EA, King JY, Ferrara R, Spin JM, Sanan DA, Narasimhan B, Tibshirani R, Tsao PS, et al. Signature patterns of gene expression in mouse atherosclerosis and their correlation to human coronary disease. Physiol. Genomics. 2005a;22:213–226. doi: 10.1152/physiolgenomics.00001.2005. [DOI] [PubMed] [Google Scholar]
- Tabibiazar R, Wagner RA, Spin JM, Ashley EA, Narasimhan B, Rubin EM, Efron B, Tsao PS, Tibshirani R, Quertermous T. Mouse strain-specific differences in vascular wall gene expression and their relationship to vascular disease. Arterioscler. Thromb. Vasc. Biol. 2005b;25:302–308. doi: 10.1161/01.ATV.0000151372.86863.a5. [DOI] [PubMed] [Google Scholar]
- Thackaberry EA, Jiang Z, Johnson CD, Ramos KS, Walker MK. Toxicogenomic profile of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the murine fetal heart: Modulation of cell cycle and extracellular matrix genes. Toxicol. Sci. 2005;88:231–241. doi: 10.1093/toxsci/kfi301. [DOI] [PubMed] [Google Scholar]
- Thomas RS, Penn SG, Holden K, Bradfield CA, Rank DR. Sequence variation and phylogenetic history of the mouse Ahr gene. Pharmacogenetics. 2002;12:151–163. doi: 10.1097/00008571-200203000-00009. [DOI] [PubMed] [Google Scholar]
- Uno S, Dalton TP, Derkenne S, Curran CP, Miller ML, Shertzer HG, Nebert DW. Oral exposure to benzo[a]pyrene in the mouse: Detoxication by inducible cytochrome P450 is more important than metabolic activation. Mol. Pharmacol. 2004;65:1225–1237. doi: 10.1124/mol.65.5.1225. [DOI] [PubMed] [Google Scholar]
- Van den Berg M, Birnbaum LS, Denison M, De Vito M, Farland W, Feeley M, Fiedler H, Hakansson H, Hanberg A, Haws L, et al. The 2005 World Health Organization reevaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006;93:223–241. doi: 10.1093/toxsci/kfl055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu W, Levy DE. Nuclear and cytoplasmic mRNA quantification by SYBR Green based real-time RT-PCR. Methods. 2006;39:356–362. doi: 10.1016/j.ymeth.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Watanabe J, Shimada T, Gillam EM, Ikuta T, Suemasu K, Higashi Y, Gotoh O, Kawajiri K. Association of CYP1B1 genetic polymorphism with incidence to breast and lung cancer. Pharmacogenetics. 2000;10:25–33. doi: 10.1097/00008571-200002000-00004. [DOI] [PubMed] [Google Scholar]
- Weinshilboum R. Inheritance and drug response. N. Engl. J. Med. 2003;348:529–537. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- Whitlock JP., Jr Mechanistic aspects of dioxin action. Chem. Res. Toxicol. 1993;6:754–763. doi: 10.1021/tx00036a003. [DOI] [PubMed] [Google Scholar]
- Williams JA, Phillips DH. Mammary expression of xenobiotic metabolizing enzymes and their potential role in breast cancer. Cancer Res. 2000;60:4667–4677. [PubMed] [Google Scholar]
- Wong JM, Okey AB, Harper PA. Human aryl hydrocarbon receptor polymorphisms that result in loss of CYP1A1 induction. Biochem. Biophys. Res. Commun. 2001;288:990–996. doi: 10.1006/bbrc.2001.5861. [DOI] [PubMed] [Google Scholar]
- Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz S, Van Winkle L, Matsumura F, Vogel C. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E-/- mice. Arterioscler. Thromb. Vasc. Biol. 2011;31:1260–1267. doi: 10.1161/ATVBAHA.110.220202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Jr, Capecchi MR, Welsh MJ, Thomas KR. A mouse model for the delta F508 allele of cystic fibrosis. J. Clin. Investig. 1995;96:2051–2064. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Lei P, Liu X, Li X, Walker K, Kotha L, Rowlands C, Safe S. The aryl hydrocarbon receptor as a target for estrogen receptor-negative breast cancer chemotherapy. Endocr. Relat. Cancer. 2009;16:835–844. doi: 10.1677/ERC-09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Jiang L, Rui L. Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J. Biol. Chem. 2009;284:11152–11159. doi: 10.1074/jbc.M900754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.