

Abstract
To understand the redox-dependent arsenic partitioning, we performed batch sorption and desorption experiments using aquifer sands subjected to chemical and mineralogical characterization. Sands collected from the redox transition zone between reducing groundwater and oxic river water at the Meghna riverbank with HCl extractable Fe(III)/Fe ratio ranging from 0.32 to 0.74, are representative of the redox conditions of aquifers common in nature. One brown suboxic sediment displayed a partitioning coefficient (Kd) of 7∼8 L kg−1 at equilibrium with 100 μg L−1 As(III), while two gray reducing sediments showed Kd of 1∼2 L kg−1. Lactate amendment to aquifer sands containing 91 mg kg−1 P-extractable As resulted in the reduction of As and Fe with sediment Fe(III)/Fe decreasing from 0.54 to 0.44, and mobilized an equivalent of 64 mg kg−1 As over a month. Desorption of As from non-lactate-amended sediment was negligible with little change in sediment Fe(III)/Fe. This release of As is consistent with microbial reduction of Fe(III) oxyhydroxides and the resulting decrease in the number of surface sites on Fe(III) oxyhydroxides. Arsenic partitioning (Kd) in iron-rich, sulfur-poor aquifers with circumneutral pH is redox-dependent and can be estimated by HCL leachable sediment Fe(III)/Fe ratio with typical Fe concentrations.
Introduction
Quantification of arsenic partitioning between solute and solid phases is important to assess transport of this toxic element in aquifer. The sorption reactions of As have been extensively investigated for synthetic Fe, Mn, and Al oxyhydroxides and for clay minerals.1-4 A handful of studies examined natural soil or sediments under oxic conditions and found that sorption behavior was mostly similar to that of pure Fe(III) oxyhdyroxides.5-7 A knowledge gap exists for As partitioning in reducing aquifer conducive for As mobilization due to difficulties in sample preservation for sorption experiments and mineralogical characterization.8
Sorption isotherms of As(III) onto sands from a coastal aquifer of Waquoit Bay9 suggest a redox-dependent As partitioning,10,11 showing increasing As(III) sorption with higher Fe(III) oxides content. In the Ganges-Brahmaputra-Meghna Delta (GBMD), the As(III) sorption coefficient (Kd) at equilibrium with a typical groundwater As concentration of ∼100 μg L−1 established by batch experiment or field investigation ranged from 2 to 6 L kg−1 for Holocene reducing aquifer sediments,8, 12, 13 and were approximately 20 to 30 L kg−1 for brown sediments from deeper depths.14-16 To the best of our knowledge, batch sorption studies have not evaluated As partitioning in sediments of Bangladesh Holocene shallow aquifer, frequently enriched in groundwater arsenic under reducing conditions that are typical for many aquifers with elevated As. Because transport in aquifers is thought to influence the distribution of As in aquifers,17 improved understanding of As partitioning has implication on interpretation of the source and fate of As in the GBMD8 and elsewhere.
The objectives of this study are to quantify As partitioning by batch sorption-desorption experiments using natural sediment and porewater samples with minimal disturbance of redox condition, and to understand parameters and mechanisms controlling the mobility of As in iron-rich and sulfur-poor aquifer sediment through mineralogical characterization and geochemical modeling.
Along the Meghna Riverbank where iron and arsenic rich anoxic reducing groundwater interacts with oxic river water, a redox transition zone spanning a wide range of redox conditions within small spatial distances and with enrichment of sediment As18 up to hundreds or thousands mg kg−1 provides an ideal setting. The study is especially relevant to As transport in floodplain aquifers of South Asia where elevated groundwater As is widespread19 with similar hydrogeological settings,20-22 and has implications for As transport in aquifers.
Materials and Methods
Sediment and Porewater Samples
In addition to shallow well groundwater and river water (Table S1), sediment and porewater depth profiles (Fig. 1) were collected from surface to 6∼7 m depth near the Meghna River shore (23.6° N, 90.6° E) in Gazaria, Bangladesh (Fig. S1) using a soil probe (AMS Inc., USA) and a stainless steel drive point piezometer system (“Retract-A-Tip“, AMS Inc) at corresponding depths in Jan. 2006 and Nov. 2007. Sampling and analysis procedures are similar to Jung et al.9 and are described in the Supporting Information in detail. Sorbed sediment As was determined by N2-purged 1M sodium phosphate (Fisher) solution containing 0.1 M ascorbic acid for 36 hours in a crimp sealed amber vial to characterize sediment samples, including the sorption-desorption experiments (Tables S2, S6, and S7). A hot 1.2 N HCl extraction was conducted at 80 °C for 1 hour for the same samples to determine As, Fe, Mn, S, and P associated with Fe oxides. For the sorption experiment, three sediment samples at depths of 1.5, 3.6, and 5.4 m from core RS39, paired with porewater samples (PZ10) from the same depth interval, were collected on Nov. 4, 2007. For the desorption experiment, a section of RS19-4 (depth 1.5−1.8 m) collected on Jan. 25, 2006 was used. Sediment cores were double sealed in nitrogen-filled Mylar bags with oxygen absorbent (SorbentSystems) during transportation, and was refrigerated at 4°C prior to use.
Fig. 1.
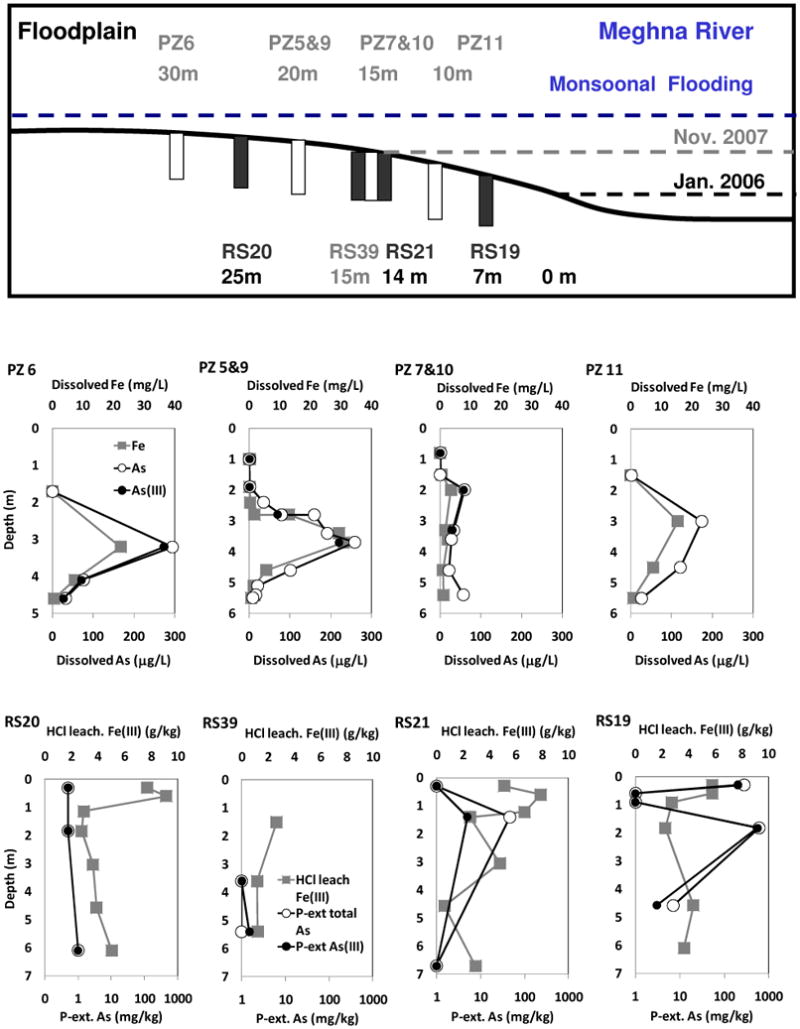
Top: Schematic diagram showing the location of porewater and sediment core sampling collected in Jan. 2006 (black font) and Nov. 2007 (gray font) with the distance from the river shore in Jan. 2006. Open bars and solid bars indicate porewater piezometers and sediment cores, respectively; Middle: Depth profiles of porewater dissolved Fe, As, and As(III) concentrations; Bottom: Depth profiles of sediment HCl leachable Fe(III) and P-extractable total As and As(III) concentrations.
X-ray Absorption Spectroscopy
The arsenic speciation and Fe mineralogy were analyzed by X-ray absorption spectroscopy (XAS) at the Stanford Synchrotron Radiation Laboratory. Iron mineralogy was obtained using extended X-ray absorption fine structure (EXAFS) spectroscopy. Arsenic speciation was determined using X-ray absorption near edge spectroscopy (XANES). Due to the very similar spectra between less crystalline goethite and ferrihydrite, which may be hard to differentiate based on linear combination fitting approaches,23 the concentrations of ferrihydrite estimated represent the sum of ferrihydrite and freshly precipitated (poorly crystalline) goethite (Supporting Information).
Batch Sorption Experiment
One suboxic brown sediment with Fe(III)/Fe of 0.58 and two reducing gray sediments with Fe(III)/Fe ratio of ∼0.37 from RS39, containing HCl-leachable or P-extractable As of ∼1 mg kg−1 (Table S2; HCl- and P-extraction were conducted using different aliquots of the same sample), were subjected to batch sorption experiment immediately after sample collection in the field in a N2 filled glove box. About 10 mL of porewater (pH 6.0∼6.5) from the same depth (PZ10, Table S3) was added to 15-mL amber serum vials containing 5∼6 g homogenized sediment spiked with As(III) and As(V) stock solutions freshly prepared using reagent grade NaAsO2 and NaHAsO4 7H2O to yield concentrations from 0 to ∼ 7000 μg L−1 As (Table S4). The vials were crimp-sealed without headspace and intermittently shaken, kept in a N2-filled chamber except during transportation in N2-filled Mylar bags, and sampled after 7 days. The supernatant was analyzed for As(III) immediately by voltammetry after filtration through a 0.45 μm membrane filter and acidification to 1% HCl and for total As by ICP-MS (Supporting Information). The pH of the supernatant likely remained circumneutral during the experiment because sediment and porewater samples were from the same depth, and thus were already at equilibrium.
Batch Desorption Experiment
To ascertain the mobility of As found at high concentrations (46-600 mg kg−1 P-extractable As), a sediment core section (1.5 to 1.8 m depth) from RS19-4 was homogenized inside a N2-filled chamber, and reacted with suboxic nanopure water (> 18 MΩ) or artificial groundwater for 1 month. Artificial groundwater and nanopure water were purged with pure nitrogen gas to reach a dissolved oxygen value of ∼0.5 mg L−1, determined by a CHEMets dissolved oxygen kit. The homogenized sample contained a P-extractable As concentration of 91 mg kg−1 with 57 mg kg−1 as As(III), HCl-leachable Fe and As concentration of 6933 and 86 mg kg−1, respectively. Artificial groundwater composition was based on that of the typical groundwater composition from adjacent tube wells at similar depths: 1 mM NaHCO3, 0.02 mM KH2PO4, 2.5 mM CaCO3, 0.16 mM MgSO4, 0.50 mM MgCl2 and 0.06 mM KCl, and was buffered to a pH of 6.8 using 20 mM PIPES (piperazine-N,N'-bis(2-ethanesulfonic acid)).
There were four types of amendment. Type I used nanopure water (pH = 6.0) with 50 mg L−1 kanamycin, an antibiotic. Type II used artificial groundwater (pH = 6.8) with 50 mg L−1 kanamycin. Type III is the same as Type II but without kanamycin, serving as a biotic control. Type IV is Type III plus 1 mM lactate, serving as an enhanced biotic control. Kanamycin was intended to limit indigenous microbial activities. Each series started in 5 replicates of 15-mL serum bottles containing 5 mL of nanopure water or artificial groundwater and ∼1 g of sediment. All glassware was autoclaved. After flushing the headspace with N2 gas and crimp sealing, bottles were shaken at 140 rpm over 1 month in a N2 filled anaerobic chamber (Coy Laboratory Products) without H2 but with oxygen absorbent (SorbentSystems). The supernatant and sediment for 4 types of incubation were sampled sacrificially from a replicate of each type on the 2nd, 5th, 10th, 17th, 30th day. Total Fe, Mn, S, P, and As in filtered supernatant and sediment extracts by hot HCL and anoxic sodium phosphate were analyzed, as well as Fe(II) and As(III) (Supporting Information).
Langmuir Isotherm and Surface Complexation Modeling (SCM)
Batch sorption equilibrium data were fitted to Langmuir isotherms to determine sorption capacity (Smax) and KLa, a constant representing the binding strength, and the effective Kd values at equilibrium with 100 μg L−1 As, typical of Bangladesh groundwater.8 A semi-mechanistic SCM of sorption experimental data was conducted in PHREEQC version 2.15 with MINTEQA2 version 4.0 database, using the acidity constants and equilibrium constants (K; Table S5) for a diffuse double layer surface complexation model of hydrous ferric oxide (HFO),24 given that Fe(III) oxyhydroxides are the dominant reactive mineral phase in suboxic sediment (Table 1). Other Fe phases and primary minerals such as quartz, feldspar, and mica vary little along the depth profiles,18 and are not included. For both amorphous and crystalline Fe(III) oxide minerals, the same sorption equilibrium constants were applied because intrinsic surface complexation constants of metals and anions for both HFO and goethite are found to be similar.25 Surface complexation of Si was not considered because the K value is not available from MINTEQA2 database. Surface site density was adjusted based on HCl leachable Fe(III) concentrations, assuming that Fe minerals are goethite-like phases with ∼0.01 mol surface sites per mol Fe(III)26 to fit As data in sorption SCM modeling. Based on the composition of porewater (Table S3), pH, Ca and alkalinity were fixed to 6.0, 0.3 mM, and 0.6 mM for brown sediment, respectively, and were 6.3, 1 mM, and 2 mM for gray sediment, respectively. P concentrations of 0.005 and 0.04 mM were used for brown and gray sediments, respectively according to aqueous composition at the end of the experiment. In desorption SCM modeling for type III without lactate and type IV with lactate, all P-extractable As of 91 mg kg−1 was subjected to adsorption reactions. For each sampling time point, solid phase arsenic redox speciation was considered for modeling. Based on P-extractable sediment As data, the percentage of As(III) was initially 50%, decreasing to 40% on day 17 and 30% on day 30 in type III. In type IV, the percentage of As(III) was initially 50%, increasing to 60% on day 2, 70% on day 5, 80% on day 10, and 100% on days 17 and 30. The surface site density was estimated using HCl leachable Fe(III) concentration for each sampling point. For type III, Fe minerals were presumed to be like ferrihydrite with 0.20 mol surface sites per mol Fe(III)24 during a period of 1 month, whereas for type IV, it was assumed that Fe minerals are initially ferrihydrite-like Fe(III) mineral with 0.20 mol surface sites per mol Fe(III), and finally transformed to magnetite-like Fe(II, III) minerals with ∼0.01 mol surface sites per mol Fe(III). Based on aqueous data, pH, Ca, alkalinity and P were fixed to 6.8, 2.5, 5, and 0.01 mM, respectively.
Table 1.
Iron mineralogy of Meghna riverbank sediment collected from Gazaria, Bangladesh in Jan. 2006.
| Sampling No | Latitude | Longitude | Depth m |
HCI leach Fe(III)/Fe | HCI leach Fe(lll) mg/kg |
Fe mineralogy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| Ferrihydrite | Hematite | Goethite | Magnetite | Biotite | Siderite | FeS | Chi2 | Red_Chi2 | |||||||
| RS20−1 | 23º35.006′ | 90º35.160′ | 0.3 | 0.74 | 7715 | 45±13% | 0±3% | 0±6% | 0±4% | 21 ±5% | 34±6% | 0±3% | 145 | 0.85 | |
| RS20-4 | 25 m from the shore | 1.8 | 0.54 | 2783 | 52±8% | 0±2% | 0±4% | 0±3% | 21 ±3% | 26±4% | 0±2% | 74 | 0.43 | ||
| RS20-7 | 6.1 | 0.49 | 5029 | 49±10% | 0±2% | 0±5% | 0±3% | 20±4% | 30±5% | 0±2% | 107 | 0.63 | |||
|
| |||||||||||||||
| RS21−1 | 23º 35.008′ | 90º35.153′ | 0.3 | 0.64 | 5109 | 62±6% | 0±1% | 0±3% | 0±2% | 20±2% | 19±3% | 0±1% | 44 | 0.26 | |
| RS21-4 | 14 m from the shore | 1.4 | 0.50 | 2529 | 38±19% | 0±4% | 7±9% | 4±6% | 16±8% | 26±9% | 9±4% | 315 | 1.84 | ||
| RS21-7 | 6.7 | 0.46 | 2935 | 48±18% | 5±4% | 0±9% | 0±6% | 28±7% | 19±9% | 0±4% | 314 | 1.84 | |||
|
| |||||||||||||||
| RS19−1 | 23º 35.011′ | 90º35.149′ | 0.3 | 0.65 | 5748 | 59±8% | 0±2% | 4±4% | 0±3% | 14±3% | 21 ±4% | 1±2% | 57 | 0.34 | |
| RS19-3 | 7 m from the shore | 0.9 | 0.52 | 2741 | 45±19% | 0±4% | 0±9% | 0±6% | 23±7% | 30±9% | 1±4% | 316 | 1.85 | ||
| RS19-4 | 1.8 | 0.32 | 2246 | 39±16% | 0±4% | 0±8% | 0±6% | 33±6% | 27±8% | 1±3% | 209 | 1.22 | |||
Results and Discussion
Redox Transition Zone
A redox transition zone between approximately 0-2 m in depth is evident from depth profiles of porewater and sediment Fe and As (Fig. 1). Porewater Fe and As reach peak concentrations of 20∼30 mg L−1 and 200∼300 μg L−1 between 3 and 4 m although PZ7&10 depth profiles show less pronounced peak most likely due to deeper infiltration of oxic river water at the shore (Fig. 1). Dissolved As speciation is dominated by As(III) (90±5%; n=7) at depth ≥ 2 m but has less As(III) (57±7 %; n=3) at depth < 2 m. HCl leachable sediment Fe(III) was highest at depths < 1 m (6.3±2.7 g kg−1, n=7) and decreased rapidly (3.3±1.7 g kg−1 at depth of 1-2 m, n=6; 3.1±1.5 g kg−1 at depth > 2 m, n=10). This decrease correlates to decreases in the Fe(III)/Fe ratio (0.65±0.07 at depth < 1 m, 0.53±0.12 at depth of 1-2 m, and 0.40±0.11 at depth > 2 m). This redox transition zone develops because the river water level declines faster than the groundwater table after the monsoon season (April to October) and causes a hydraulic gradient from the aquifer towards the river,8 driving discharge of anoxic groundwater (DO < ∼0.1 mg L−1) to oxic river water (DO > 5 mg L−1). A summary of the average chemical composition of groundwater, porewater and river water is available in Supporting Information (Table S1).
Iron Mineralogy and Sediment Arsenic
In most cases, the dominant Fe mineral in 9 samples analyzed was ferrihydrite-like, accounting for 49±8% of Fe minerals at depths < 2 m (Table 1, Fig. S2). Small concentrations of crystalline goethite and hematite were also detected in 2 samples (Table 1), and these concentrations probably represent minimum values given that microcrystalline Fe(III) oxides have spectra that are similar to ferrihydrite and thus may in some cases not be quantified separately. The preponderance of ferrihydrite is somewhat surprising because it commonly converts to more stable oxides.27,28 We explain this apparent discrepancy as follows. First, ferrihydrite commonly forms through the rapid oxidation of Fe(II) at neutral pH,27 and the study site is characterized by the active discharge of Fe(II)-rich groundwater. Second, co-precipitated silica, organic matter, and trace metals from groundwater may stabilize the ferrihydrite.29 Kinetics of seasonal transformation of ferrihydrite to stable Fe phases is likely affected by the seasonal transition of hydrological condition between dry season and rainy season. The prevalence of Fe(III) minerals at shallow depths and then decreasing with depth is consistent with the chemical extraction data (Fig. 1, Table 1). A high quantity of Fe(II) minerals (26±5%; fit as siderite), as well as iron silicates (22±6%; fit as biotite) are also present in the samples. The presence of biotite or other Fe silicates indicates that sediments are young because it weathers rapidly to secondary Fe(III) oxides. Although green rust may possibly be present in Bangladesh aquifer,8 green rust was not considered due to its metastable nature.27
P-extractable sediment As concentration is elevated between 0-2 m depth and within ∼15 m distance from the river, showing 46 mg kg−1 As at RS21, 284 and 600 mg kg−1 As at RS19 (Fig. 1). XRF analyses of another sediment core (RS34) collected 2 m away from the shore found that bulk sediment As concentration is 100 mg kg−1 at 1.4 m depth. Sediment As speciation determined by P-extraction and XANES (Fig. 1, Fig. S2, Table S6) suggests mixed As(III) and As(V) (Supporting Information).
Langmuir Sorption Isotherms
As(III) sorption to sediment core RS39 was equilibrated within 10 hours. Adsorption data fit well to Langmuir isotherms (Fig. 2; RMSE ≤ 0.6) that differ considerably depending on the color (redox state) of the sediment. Based on these isotherms, the calculated Kd values at equilibrium with 100 μg L−1 solute As, are 7∼8 and 1∼2 L kg−1 for brown and gray sediments respectively (Table S2). These values are comparable regardless of whether sediments were spiked with As(III) or As(V). For gray sediment, as solute As increases from 10 to 1000 μg L−1, a typical groundwater As concentration range in the GBMD aquifers, the Kd value of the gray sediment is nearly constant at 1∼2 L kg−1 similar to a relatively constant Kd of ∼ 4 L kg−1 that were proposed for reducing shallow aquifer of GBMD across a range of As concentrations of nearly 3 orders of magnitude.13
Fig. 2.
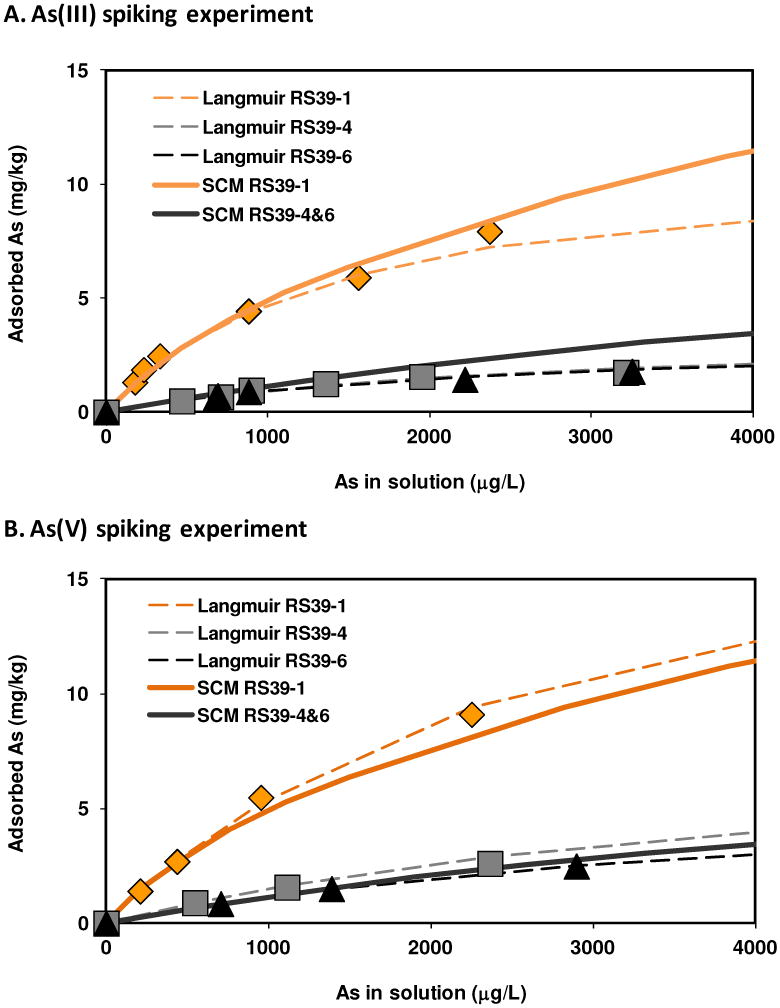
Equilibrium isjournalms of As sorption to one brown (diamond) and two gray sediment (triangle and square) samples (RS39) collected in Nov. 2007. (A) As(III) spiking experiment; (B) As(V) spiking experiment. Dashed lines represent Langmuir isjournalm fit; Solid lines represent surface complexation model fit and sorbed As species is 100% As(III) in SCM.
The sorption capacity (Smax) for brown sediment was also about 2-3 times higher than that for gray sediment (Table S2). Regardless of whether As(III) or As(V) was spiked, the dissolved As was primarily As(III) (94±11% and 89±14% for As(III) and As(V) additions respectively, Table S4) possibly due to redox processes readily reducing As(V) to As(III). This may in part explain the similar sorption behaviors in As(III) and As(V) spiked experiments because it is essentially As(III) equilibrium (Fig. 2).
Surface Complexation Model
For As(III) and As(V) spikes, the best-fitted surface site density was 0.6 and 0.2 μM g−1 for brown and gray sediments, respectively (RMSE < 0.8), assuming that all sorbed As is As(III) in SCM (Fig. 2). The three-fold higher surface site density in brown sediment than gray sediment indicates that they have different concentrations of similar assemblage of minerals controlling adsorption. This is supported by differences in the concentration of HCl-leachable Fe(III) (Table S2), with ∼0.01 mol surface sites per mol Fe(III), assuming that Fe(III) phases are principally responsible for As retention. Using a surface site density of 3.5 sites nm−2,30 an Fe mineral surface area of ∼20 m2 g−1 yields Fe concentrations that are consistent with observed HCl leachable Fe(III) concentrations of ∼2700 and ∼1200 mg kg−1 for brown and gray sediments, respectively (Table S2). This surface area is consistent with a range of microcrystalline Fe oxides such as goethite but is much lower than that is common for ferrihydrite, which was identified by XAS spectroscopy. This difference can be explained in three ways. First, it is possible or even likely that microcrystalline goethite present in these sediments anomalously fit with ferrihydrite due to spectral similarities. Second, ferrihydrite, which often has measured surface areas in excess of 200 m2 g−1,26 may form aggregates or have blocked surfaces that make them less reactive. Indeed, the mole ratio of As effectively scavenged by fresh ferrihydrite is about 1:50,31 consistent with a surface area of about ∼60 m2 g−1, and more aged forms could be less reactive. Third, the XAS data were collected in the middle of dry season (January) during groundwater discharge. Fresh precipitates formed in such environments could indeed contain ferrihydrite that could be converted to more stable forms, catalyzed by their partial reduction28 during the following rainy season. Other sediments collected in this area have also exhibited widely different sorption capacities, possibly due to different Fe mineral composition.4 The As sorption capacity of 13-23 mg kg−1 established by the batch sorption experiment using suboxic brown sediment collected in Nov. 2007 is considerably lower than that observed for suboxic sediment collected in Jan. 200318 or Jan. 2006 (Fig. 1), which had P-extractable sediment As concentrations as high as 100−1000 mg kg−1; this difference is also attributed to the seasonal transformation of Fe minerals. The extent of such seasonal transformations needs to be assessed in the future.
Desorption of Arsenic and Iron Mineralogy
Desorption experiments performed in the absence of lactate (types I, II, and III) released measurable As into solution within 2 days, its concentration reaching 604±57 μg L−1, primarily as As(III) (Fig. 3, Table S7). This As release was decoupled from a rise of Fe concentration, with 1237, <10, and 18 μg L−1 for types I, II, and III, respectively. Over the next 28 days, dissolved As concentrations decreased by 15∼31% and was oxidized to nearly 100% As(V) (Table S7). The oxidation of As is also observed in the sediment, with P-extractable As(III) decreasing from 57 mg kg−1 to finally 15, 32 and 22 mg kg−1 for types I, II, and III, respectively, while the total pool of adsorbed As remained constant, with the nearly unchanged total P-extractable (80∼90 mg/kg) and HCl leachable As (∼70 mg/kg) concentrations (Fig. 3B, Table S7). While we are unsure of the responsible oxidants, limited introduction of oxygen (∼0.5 mg L−1 dissolved O2) at the beginning of the experiment, or another oxidant such as Mn(IV) oxides in the sediments are possibilities.14 The HCl leachable Mn concentration is ∼100 mg kg−1, but the mineralogy of Mn phase has not been confirmed in these sediments (Table S7). The overall oxidation state of sediment Fe was constant as evidenced by a constant Fe(III)/Fe ratio throughout the experiment (0.5-0.55), while the HCl leachable Fe(III) decreased from its initial concentration of 3500 mg kg−1 to ∼2700 mg kg−1 over the course of experiment (Fig. 3B). This could take place when some of Fe(III) oxyhydroxides are transformed to more crystalline Fe(III) phases (e.g. goethite, hematite), resulting in the decreased leachability by HCl with little change in overall oxidation state.
Fig. 3.
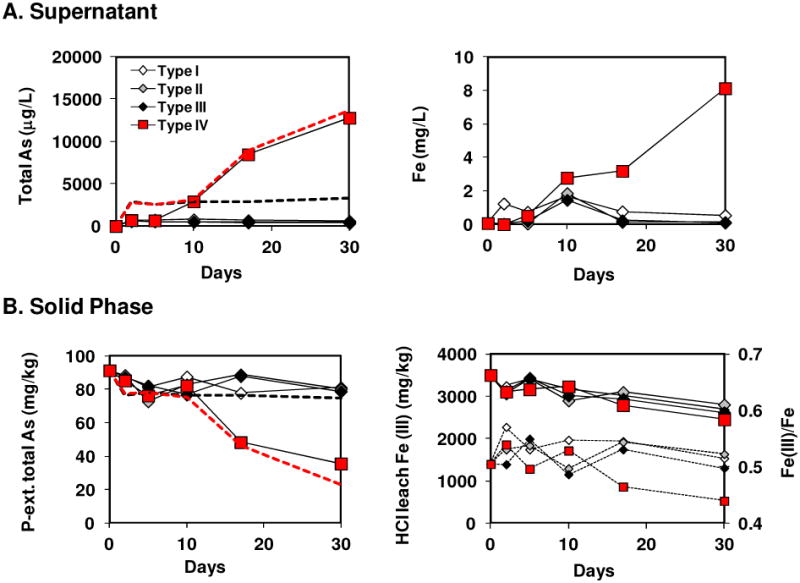
Fe and As in aqueous phase (A) and solid phase (B) during desorption experiment using RS19-4 sediment core collected in Jan. 2006. Type I: nanopure water + kanamycin (50 mg/L), Type II: artificial groundwater + kanamycin (50 mg/L), Type III: artificial groundwater, Type IV: artificial groundwater + lactate (1 mM); In the panel showing HCl leachable Fe (B right), solid lines with large symbols represent Fe(III) concentration, and dotted lines with small symbols are Fe(III)/Fe ratio. Surface complexation modeling of As desorption in type III without lactate and type IV with lactate are indicated in black dashed line and red dashed line, respectively.
Sediments incubated with 1 mM lactate (type IV) released similar quantities of As into solution (∼ 630 μg L−1, Fig 3A) on the 2nd day and also without Fe (< 10 μg L−1), but As release was much more appreciable between the 5th and the 30th day, at which point the dissolved As reached 12,081 μg L−1. This high As concentration is equivalent to mobilization of 64 mg kg−1 sediment As, or 70% of the initial P-extractable As. Concurrent decreases in P-extractable and HCl-leachable As concentrations were also observed, as was the nearly complete conversion of dissolved As to As(III) (Fig. 3B, Table S7). The release of dissolved Fe resembled that of As except for the 17th day. In the first 5 days, Fe release was low and comparable to type I, with 482 μg L−1 dissolved Fe, equivalent to 2.5 mg kg−1 sediment Fe. It then increased to 3 mg L−1, and finally to 8 mg L−1, equivalent to 40 mg kg−1 sediment Fe. The HCL leachable Fe(III) decreased from 3500 mg kg−1 to ∼2450 mg kg−1 and sediment Fe(III)/Fe ratio displayed a measurable and consistent decrease from 0.54 to 0.44 between the 10th and the 30th day (Fig. 3). The change in extractable Fe(III)/Fe in the solid phase indicates that a significant portion of the Fe(III) was reduced (and not just changes in leachability by HCl), and that much of the Fe(II) produced was conserved in the solid-phase. The reduction of Fe and As in both solution and sediment, as well as subsequent desorption of As appear to have been caused by microbial activity32 stimulated with addition of labile organic carbon to sediment preserved at 4 °C for 11 months.15 This is different from the naturally occurring reduction of As during sorption experiment without addition of labile organic carbon perhaps because sorption experiment was conducted using fresh sediment containing active microbial community immediately after sample collection.
Decrease in surface site density due to microbial transformation of Fe minerals stimulated by lactate was likely a major cause for extensive As desorption. Although no XAS data were available to ascertain the nature of iron mineralogy change during desorption experiment, SCM modeling based on surface sites derived from HCl leachable Fe(III) concentration simulated the evolution of solute (Fig. 3A) and solid As (Fig. 3B) reasonably well, although dissolved As is overestimated (Fig. 3A). The overestimation may be due to surface site density of natural Fe(III) oxyhydroxides in sediment being higher than that of typical synthetic ferrihydrite (0.2 mol surface sites per mol Fe), or some As co-precipitated with Fe in sediment could not be mobilized during the experiment. The sediment Fe(III)/Fe decrease with lactate over 1 month was interpreted as a redox transformation of Fe minerals from an initially ferrihydrite-like Fe(III) mineral with 0.20 mol surface sites per mol Fe(III) to finally magnetite-like Fe(II, III) minerals with 0.01 mol surface sites per mol Fe(III) (same as the surface site concentration from sorption experiment with RS39 sediments collected in Nov. 2007), while the surface site concentrations for day 5, 10, and 17 were adjusted to 0.20, 0.15, and 0.04 mol per mol Fe(III), respectively for the best fit. For types without lactate, 0.20 mol surface sites per mol Fe(III) was used throughout. It is worth noting that color change of Bangladesh Holocene aquifer sediment from brown to gray corresponds to a threshold Fe(III)/Fe ratio of ∼0.6.33 Therefore, the SCM modeling results suggest that ferrihydrite-like Fe(III) mineral or poorly crystalline goethite with higher surface site density is initially the dominant Fe mineral in RS19 sediment collected in Jan. 2006 and is supported by XAS data (Table 1), whereas microcrystalline goethite with lower surface site density is possibly dominant in RS39 sediment collected in Nov. 2007. The results also suggest that Meghna Riverbank sediment As will remain reasonably immobile in the absence of labile organic carbon.
Redox-dependent As Partitioning in Iron Dominant System
Arsenic partitioning to aquifer sands appears to vary systematically with sediment redox state. The Kd values for As(III) at equilibrium with 100 μg L−1 As(III) observed in batch sorption experiments using sands from the redox transition zones of Meghna Riverbank in this study compare favorably with those determined within the Waquoit Bay aquifer,9 as well as Bangladesh Pleistocene orange sands.14,15 Arsenic adsorption is considerably greater in orange sands than gray sands. Interestingly, adsorption (as indicated by the effective Kd) is not clearly proportional to sediment Fe(III) concentration (Fig. S4) probably due to mineralogical differences such as crystallinity and morphology, which affects the leachability of Fe minerals by HCl. Instead, the Kd and sediment Fe(III)/Fe ratio can be fitted with an exponential function (R2 =0.97, n=7; Fig. 4) excluding a black reducing sediment from Waquoit Bay with Fe(III)/Fe of 0.26.9 The higher than expected Kd value of the black sediment from Waquoit Bay is attributed to As-sulfide precipitation11 during sorption experiment because of high sulfur and low iron concentration in porewater (Fe/S molar ratio < l).9,11,34 This relationship can be used to estimate Kd for aquifer sands where sulfur content is low (e.g. Fe/S > 1), sediment Fe(III)/Fe ratio is between 0.3∼0.9 and As mobility is governed by interaction with Fe minerals. The nearly constant Kd values of 2∼6 reported for reducing GBMD aquifer sediments with Fe(III)/Fe < 0.58, 12, 13 is thus not surprising.
Fig. 4.
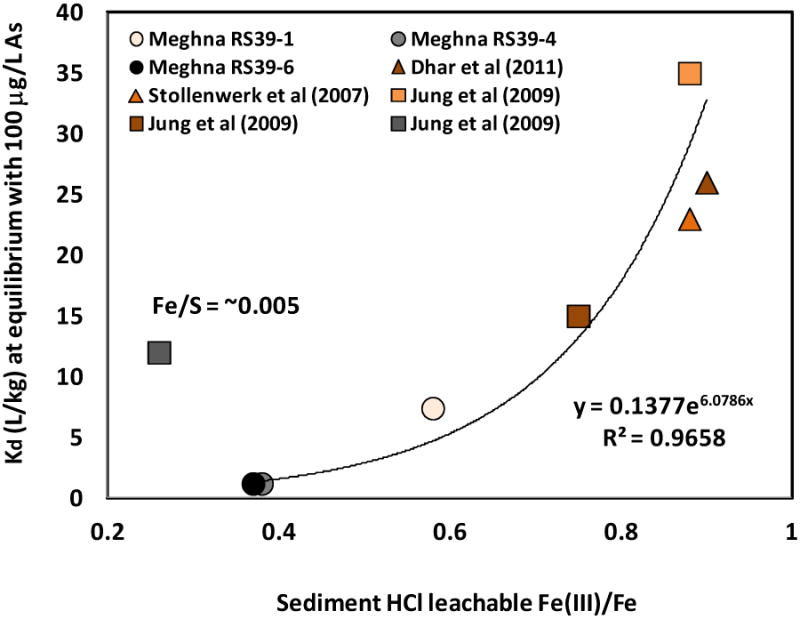
Correlation between HCl leachable Fe(III)/Fe ratio and As partitioning coefficient (Kd) for a solute As of 100 μg L−1 at circumneutral pH. Data are from the Meghna riverbank (this study) and the Waquoit Bay coastal aquifer (9), and Pleistocene aquifer sands from Bangladesh (14, 15). Langmuir isjournalm fit was used to obtain Kd, except for Stollenwerk et al (14), in which Kd was estimated based on SCM fit. The exponential fit excludes the Kd for a black Waquoit Bay sediment with porewater molar Fe/S of ∼0.005 (Fe/S < 1, a sulfur rich system). In Bangladesh aquifer, the Fe(III)/Fe ratio and Kd range from 0.37 to 0.90 and from 1 to 26 L kg−1, respectively.
Despite a number of limitations such as unavailable Fe mineralogy data for desorption experiment, spatially and seasonally heterogeneous sediment composition, and lack of differentiation between adsorption and co-precipitation, the results from this coupled field, experimental and modeling study underscore the importance of characterizing the mineralogy, redox state and surface site density of sediments of aquifers that are affected by seasonal and spatial redox transition. Recharge and discharge zones of the floodplain aquifer of South Asia, where seasonal and spatial redox transition is considerable due to groundwater-surface water interaction,18, 20, 22 are likely most susceptible to redox-dependent As partitioning through precipitation/dissolution and transformation of Fe minerals causing the change of the surface sorption site density. The sediment Fe(III)/Fe ratio is a useful parameter to assess the mobility and transport of groundwater As in iron-rich and sulfur-poor aquifers with circumneutral pH, but will not apply in the iron-poor environment where sulfides dominate the mobility of As.11,34
Supplementary Material
Acknowledgments
Funding is provided by NIEHS SBRP 2 P42 ES10349 to YZ. HBJ received a University Fellowship and Mina Rees Dissertation Fellowship from the Graduate Center, CUNY. We are grateful to Mohammad W. Rahman, Mohammad M. Rahman, and Mohammad Rezaul Huq for assistance in the field.
Footnotes
Supporting Information Available: Detailed sampling and analysis methods, chemical composition of water and sediment, database for surface complexation modeling, batch sorption and desorption data, sampling location map. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Raven KP, Jain A, Loeppert RH. Arsenite and arsenate adsorption on ferrihydrite: Kinetics, equilibrium, and adsorption envelopes. Environ Sci Technol. 1998;32:344–349. [Google Scholar]
- 2.Goldberg S. Competitive adsorption of arsenate and arsenite on oxides and clay minerals. Soil Sci Soc Am J. 2002;66:413–421. [Google Scholar]
- 3.Manning BA, Fendorf SE, Bostick B, Suarez DL. Arsenic(III) oxidation and arsenic(V) adsorption reactions on synthetic birnessite. Environ Sci Technol. 2002;36:976–981. doi: 10.1021/es0110170. [DOI] [PubMed] [Google Scholar]
- 4.Dixit S, Hering JG. Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: Implications for arsenic mobility. Environ Sci Technol. 2003;37:4182–4189. doi: 10.1021/es030309t. [DOI] [PubMed] [Google Scholar]
- 5.Manning BA, Goldberg S. Arsenic(III) and arsenic(V) adsorption on three California soils. Soil Sci. 1997;162:886–895. [Google Scholar]
- 6.Smith E, Naidu R, Alston AM. Chemistry of arsenic in soils: I. Sorption of arsenate and arsenite by four Australian soils. J Environ Qual. 1999;28:1719–1726. [Google Scholar]
- 7.Zhang H, Selim HM. Kinetics of arsenate adsorption-desorption in soils. Environ Sci Technol. 2005;39:6101–6108. doi: 10.1021/es050334u. [DOI] [PubMed] [Google Scholar]
- 8.Kinniburgh DG, Smedley PL, editors. BGS and DPHE. Arsenic contamination of groundwater in Bangladesh, Final Report British Geological Survey Report WC/00/19. British Geological Survey: Keyworth; 2001. [Google Scholar]
- 9.Jung HB, Charette MA, Zheng Y. Field, Laboratory, and Modeling Study of ReactiveTransport of Groundwater Arsenic in a Coastal Aquifer. Environ Sci Technol. 2009;43:5333–5338. doi: 10.1021/es900080q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ford RG, Wilkin RT, Hernandez G. Arsenic cycling within the water column of a small lake receiving contaminated ground-water discharge. Chem Geol. 2006;228:137–155. [Google Scholar]
- 11.O'Day PA, Vlassopoulos D, Root RA, Rivera NA. The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions. Proc Natl Acad Sci U S A. 2004;101:13703–13708. doi: 10.1073/pnas.0402775101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harvey CF, Swartz CH, Badruzzaman ABM, Keon-Blute N, Yu W, Ali MA, Jay J, Beckie R, Niedan V, Brabander D, Oates PM, Ashfaque KN, Islam S, Hemond HF, Ahmed MF. Arsenic mobility and groundwater extraction in Bangladesh. Science. 2002;298:1602–1606. doi: 10.1126/science.1076978. [DOI] [PubMed] [Google Scholar]
- 13.van Geen A, Zheng Y, Goodbred S, Horneman A, Aziz Z, Cheng Z, Stute M, Mailloux B, Weinman B, Hoque MA, Seddique AA, Hossain MS, Chowdhury SH, Ahmed KM. Flushing history as a hydrogeological control on the regional distribution of arsenic in shallow groundwater of the Bengal Basin. Environ Sci Technol. 2008;42:2283–2288. doi: 10.1021/es702316k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stollenwerk KG, Breit GN, Welch AH, Yount JC, Whitney JW, Foster AL, Uddin MN, Majumder RK, Ahmed N. Arsenic attenuation by oxidized aquifer sediments in Bangladesh. Sci Total Environ. 2007;379:133–150. doi: 10.1016/j.scitotenv.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 15.Dhar RK, Zheng Y, Saltikov CW, Radloff KA, Mailloux BJ, Ahmed KM, van Geen A. Microbes Enhance Mobility of Arsenic in Pleistocene Aquifer Sand from Bangladesh. Environ Sci Technol. 2011;45:2648–2654. doi: 10.1021/es1022015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radloff KA, Zheng Y, Michael HA, Stute M, Bostick BC, Mihajlov I, Bounds M, Huq MR, Choudhury I, Rahman MW, Schlosser P, Ahmed KM, van Geen A. Arsenic migration to deep groundwater in Bangladesh influenced by adsorption and water demand. Nat Geosci. 2011;4:793–798. doi: 10.1038/ngeo1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann RB, Ashfaque KN, Badruzzaman ABM, Ali MA, Shoemaker JK, Harvey CF. Anthropogenic influences on groundwater arsenic concentrations in Bangladesh. Nat Geosci. 2010;3:46–52. [Google Scholar]
- 18.Datta S, Mailloux B, Jung HB, Hoque MA, Stute M, Ahmed KM, Zheng Y. Redox trapping of arsenic during groundwater discharge in sediments from the Meghna riverbank in Bangladesh. Proc Natl Acad Sci U S A. 2009;106:16930–16935. doi: 10.1073/pnas.0908168106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fendorf S, Michael HA, van Geen A. Spatial and Temporal Variations of Groundwater Arsenic in South and Southeast Asia. Science. 2010;328:1123–1127. doi: 10.1126/science.1172974. [DOI] [PubMed] [Google Scholar]
- 20.Polizzotto ML, Harvey CF, Sutton SR, Fendorf S. Processes conducive to the release and transport of arsenic into aquifers of Bangladesh. Proc Natl Acad Sci U S A. 2005;102:18819–18823. doi: 10.1073/pnas.0509539103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Postma D, Larsen F, Minh Hue NT, Due MT, Viet PH, Nhan PQ, Jessen S. Arsenic in groundwater of the Red River floodplain, Vietnam: Controlling geochemical processes and reactive transport modeling. Geochim Cosmochim Acta. 2007;71:5054–5071. [Google Scholar]
- 22.Polizzotto ML, Kocar BD, Benner SG, Sampson M, Fendorf S. Near-surface wetland sediments as a source of arsenic release to ground water in Asia. Nature. 2008;454:505–508. doi: 10.1038/nature07093. [DOI] [PubMed] [Google Scholar]
- 23.O'Day PA, Rivera N, Root R, Carroll SA. X-ray absorption spectroscopic study of Fe reference compounds for the analysis of natural sediments. Am Miner. 2004;89:572–585. [Google Scholar]
- 24.Dzombak DA, Morel FMM. Surface Complexation Modeling: Hydrous Ferric Oxide. Wiley-Interscience; New York: 1990. [Google Scholar]
- 25.Mathur SS, Dzombak DA. Surface complexation modeling: goethite. In: Lutzenkirchen J, editor. Surface Complexation Modeling. Elsevier; Amsterdam: 2006. [Google Scholar]
- 26.Appelo CAJ, Postma D. Geochemistry, groundwater and pollution. A.A. Balkema Publishers; Leiden: 2005. [Google Scholar]
- 27.Zachara JM, Kukkadapu RK, Fredrickson JK, Gorby YA, Smith SC. Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiol J. 2002;19:179–207. [Google Scholar]
- 28.Hansel CM, Benner SG, Neiss J, Dohnalkova A, Kukkadapu RK, Fendorf S. Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow. Geochim Cosmochim Acta. 2003;67:2977–2992. [Google Scholar]
- 29.Cornell RM, Schwertmann U. The Fe oxides: structure, properties, reactions, occurrences, and uses. VCH Weinheim; Federal Republic of Germany: 1996. [Google Scholar]
- 30.Sverjensky DA, Fukushi K. A predictive model (ETLM) for As(III) adsorption and surface speciation on oxides consistent with spectroscopic data. Geochim Cosmochim Acta. 2006;70:3778–3802. [Google Scholar]
- 31.Berg M, Luzi S, Trang PTK, Viet PH, Giger W, Stuben D. Arsenic Removal from Groundwater by Household Sand Filters: Comparative Field study, Model Calculations, and Health Benefits. Environ Sci Technol. 2006;40:5567–5573. doi: 10.1021/es060144z. [DOI] [PubMed] [Google Scholar]
- 32.Islam FS, Gault AG, Boothman C, Polya DA, Charnock JM, Chatterjee D, Lloyd JR. Role of metal-reducing bacteria in arsenic release from Bengal delta sediments. Nature. 2004;430:68–71. doi: 10.1038/nature02638. [DOI] [PubMed] [Google Scholar]
- 33.Horneman A, van Geen A, Kent DV, Mathe PE, Zheng Y, Dhar RK, O'Connell SO, Hoque MA, Aziz A, Shamsuddhua M, Seddique AA, Ahmed KM. Decoupling of As and Fe release to Bangladesh groundwater under reducing conditions. Part I: Evidence from sediment profiles. Geochim Cosmochim Acta. 2004;68:3459–3473. [Google Scholar]
- 34.Keimowitz AR, Zheng Y, Chillrud SN, Mailloux B, Jung HB, Stute M, Simpson HJ. Arsenic Redistribution between Sedimetns and Water near a Highly Contaminated Source. Environ Sci Technol. 2005;39:8606–8613. doi: 10.1021/es050727t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.