

Abstract
A simple mass spectrometry based method capable of examining protein structure called SNAPP (selective noncovalent adduct protein probing) is used to evaluate the structural consequences of point mutations in naturally occurring sequence variants from different species. SNAPP monitors changes in the attachment of noncovalent adducts to proteins as a function of structural state. Mutations which lead to perturbations to the electrostatic surface structure of a protein affect noncovalent attachment and are easily observed with SNAPP. Mutations which do not alter the tertiary structure or electrostatic surface structure yield similar results by SNAPP. For example, bovine, porcine, and human insulin all have very similar backbone structures and no basic or acidic residue mutations, and the SNAPP distributions for all three proteins are very similar. In contrast, four variants of cytochrome c (cytc) have varying degrees of sequence homology, which are reflected in the observed SNAPP distributions. Bovine and pigeon cytc have several basic or acidic residue substitutions relative to horse cytc, but the SNAPP distributions for all three proteins are similar. This suggests that these mutations do not significantly influence the protein surface structure. On the other hand, yeast cytc has the least sequence homology and exhibits a unique, though related, SNAPP distribution. Even greater differences are observed for lysozyme. Hen and human lysozyme have identical tertiary structures, but significant variations in the locations of numerous basic and acidic residues. The SNAPP distributions are quite distinct for the two forms of lysozyme, suggesting significant differences in the surface structures. In summary, SNAPP experiments are relatively easy to perform, require minimal sample consumption, and provide a facile route for comparison of protein surface structure between highly homologous proteins.
Introduction
Sequence variation in proteins occurs frequently in nature and has significant importance in various biological contexts. Certain proteins have similar amino acid sequences but exhibit very different behaviors which are related to structure. For example, the three proteins from the synuclein family (α-, β-, γ-synuclein) share a high level of sequence homology, but only α -synuclein is linked with fibril formation and the pathology of Parkinson's disease.1 Furthermore, single point mutations in the α -synuclein gene are known to greatly increase the rate of protein aggregation.2-4 Sickle cell anemia is another well-known disease caused by a point mutation.5 The glutamic acid residue in the sixth position of the β chain of normal hemoglobin is replaced by valine in sickle cell hemoglobin.6 This mutation shifts the hydrophobicity of the protein surface and dramatically reduces the solubility of the deoxygenated hemoglobin in the blood, which is ultimately responsible for the disease. It is clear from these examples that small changes in the primary sequence can dramatically alter protein structure and function. It is also well known that not all amino acid substitutions lead to significant structural perturbations in terms of three-dimensional backbone structure. For example, the native fold of ubiquitin can accommodate a large number of amino acid substitutions without significant perturbation.7,8 Point mutations are also regularly introduced into proteins intentionally by researchers for a variety of reasons,9 frequently with the implicit hope that the structure and behavior of the protein will not be affected.
It is possible to examine the effects of point mutations with traditional protein structure determination methods such as x-ray crystallography10 and NMR.11 In some cases this may be warranted, but frequently the time and sample consumption required for these methods will preclude their use. In contrast, mass spectrometry (MS) is well suited to examine proteins both quickly and with excellent sensitivity. Although the three dimensional structures obtained by x-ray and NMR cannot be derived from MS based experiments, there are aspects of protein structure which are most easily examined by MS. The development of soft ionization methods, which has enabled protein analysis by MS, also led to the emergence of methods that provide information about protein structure. It was recognized early on that the mere process of electrospraying a protein reveals some information about structure. For example, the charge state distribution observed for a protein that has been electrosprayed can be used to coarsely determine folding state.12-14 Subsequently, more sophisticated experiments utilizing hydrogen/deuterium exchange, irreversible covalent labeling, or crosslinking have been used to reveal more detailed information about protein conformation.15-17 These methods are the preferred manner to probe solvent accessibility and protein-protein interactions.
Another MS based technique known as SNAPP (selective noncovalent adduct protein probing) has been developed to examine protein solution phase structure with noncovalent probes. 18-20 In this method, 18-crown-6 ether (18C6) is used as a recognition molecule which can noncovalently attach to basic sites (Lys/Arg side chains, N-terminus) in proteins. Whether or not a particular site is available for binding by 18C6 is determined by the presence and abundance of competitive intramolecular interactions including salt bridges and hydrogen bonds. Salt bridges with acidic groups are most effective at interfering with 18C6 binding;19 therefore, SNAPP is sensitive to the arrangement of basic, acidic, and polar groups at the protein surface (which is ultimately a function of the overall three dimensional protein structure). 18C6 adducts do not simply count the number of basic sites. Importantly, due to the relative solution and gas phase binding properties, 18C6 does not interact significantly with proteins until after droplet formation in the transition region of ESI.21,22 Adduct formation occurs within milliseconds in this region and should precede any structural rearrangements which might subsequently occur in the gas phase. The raw mass spectra provide a distribution of protein-18C6 complexes (SNAPP distribution) for each charge state. The shape and relative intensity of the distributions are very sensitive to protein surface structure as a whole; however, residue specific information is not typically obtained. If the protein is modified by ligand binding, denaturation, or point mutations which modulate the chemical environment around 18C6 binding sites, shifts in the SNAPP distributions will be observed. Therefore, SNAPP is primarily a comparison method which can determine if changes to a protein or its environment lead to structural shifts as well. It is important to note that because SNAPP provides information about the electrostatic arrangement of functional groups at the protein surface, changes which do not result in rearrangement of the tertiary fold can still yield different SNAPP distributions if the surface structure has been perturbed. There could be important consequences for this type of situation, e.g. the catalytic properties of two proteins with similar backbone structure might well be quite comparable, but protein-protein recognition could be significantly different due to changes at the surface where recognition occurs.
Herein several orthologous proteins from multiple species that share a common function and varying degrees of sequence homology are examined by SNAPP-MS. In each case where structures are known, these proteins have been determined to have nearly identical native folds by x-ray crystallography; however, the effects of sequence variation on surface structures have not been previously experimentally probed. For three variants of insulin, the effects of sequence mutations are very minor and SNAPP distributions are all very similar, suggesting no disturbance to the electrostatic surface structure. Cytochrome c (cytc) variants from four species were examined; three have known tertiary structures. Interestingly, yeast yields a distinct SNAPP distribution relative to horse, bovine and pigeon cytc. Yeast cytc contains the largest number of mutations in acidic/basic residues and also exhibits reduced stability, leading to denaturation under milder conditions than those required for the other variants. Two forms of lysozyme with significant sequence mutations, but identical backbone structures, were found to yield SNAPP distributions that are quite different. The relative importance of various types of mutations on the observed SNAPP distributions and protein surface structures is discussed.
Materials and Methods
Protein samples and purification
Recombinant Cytochrome c iso-1 from Yeast was purchased from abcam (Cambridge, MA). All other proteins used in this work were purchased from Sigma-Aldrich (St. Louis, MO). Cytochrome c from yeast and Lysozyme c from human was further purified by dialysis against water and lyophilized. Methanol (Sigma Aldrich, St. Louis, MO), acetic acid (Mallinckrodt Baker Inc. Phillipsburg, NJ) were of analytical grade and used without further purification. All protein samples were prepared using a Millipore Direct-Q (Millipore, Billerica, MA) purified water without any buffer or acid, unless otherwise noted. The concentrations for all proteins were kept in the 7∼10μM range. 18C6 (Alfa Aesar, Pelham, NH) at 10 times protein concentration was added to the sample solution prior to electrospray. For example, for a final concentration of 10 μM of cytc, the concentration of 18C6 would be 100μM. All samples were of neutral pH by litmus paper.
Mass Spectrometry
Mass spectra were obtained with a Finnigan LCQ 3D ion trap mass spectrometer equipped with a modified liquid desorption electrospray ionization (DESI) source. SNAPP experiments are very sensitive to the exact source conditions which are employed, and therefore the instrument is calibrated against a standard immediately prior to each experimental run to verify similar source conditions. For these experiments, cytochrome c from horse in 50/50 water methanol was used as the standard, which was verified to yield a reproducible spectrum. Although standard ESI can be used for SNAPP, we have found that the liquid DESI arrangement provides for easier reproducibility. The complete details of these findings will be the subject of a future publication. The DESI source was implemented by removal of the original electrospray nozzle from the source mount. The nozzle was then oriented as shown in Scheme 1 with a ring stand and clamp. The gas to the nozzle is provided directly from a gas cylinder rather than passing through the LCQ. The liquid sample is pumped from a fixed tube placed ∼90° to the mass spectrometer inlet, with the distance of 0.5-1cm as shown in Scheme 1. The sample is ionized through interaction with the charged solvent droplets generated by the electrospray nozzle. The sample flow rate is 3.0μL/min and the spray flow rate is 6.00μL/min. The sheath gas pressure is raise to >80 psi. The solvent solution for DESI source was 50/50 water/methanol with 1% acetic acid. Typical settings are as follows: capillary voltage 100V, capillary temperature 215°C, tube lens offset −65V, spray voltage 5kV. Once optimized, the instrument parameters remained unchanged for all experiments.
Scheme 1.
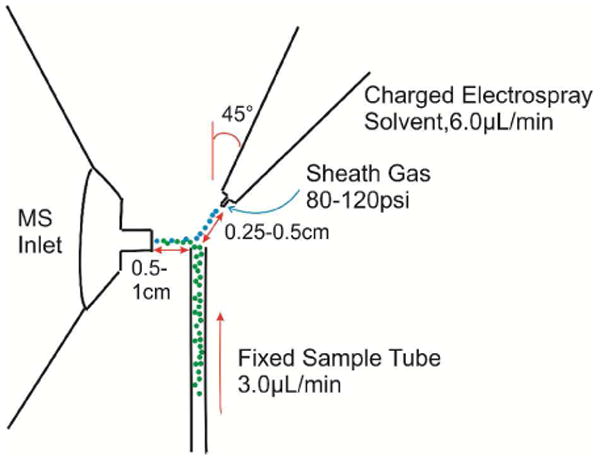
Diagram of liquid DESI source.
Protein Structures
The protein data bank (PDB) ID for the crystal structures discussed here are as follows: Porcine insulin, 2EFA; bovine insulin 9INS; human insulin 3I3Z; horse cytochrome c (cytc), 1HRC; bovine cytc, 2B4Z; yeast cytc, 1YCC; human lyzosyme, 1LZ1, hen lyzosyme, 2LYZ. The surface electrostatic maps were generated from the Maestro computing program (Schrodinger, Inc., San Diego, CA) by choosing the “molecular surface” option, and the color is based on the residue charge (positive, blue; negative, red).
Results and Discussion
Insulin
Aligned backbone structures in ribbon form obtained from the crystal structures of human, porcine, and bovine insulin are shown in Figure 1a. 23-25 The three structures are virtually identical. Sequence alignment for the same set of proteins is shown in Figure 1b. Only one residue is different between human and porcine insulin, while bovine insulin has two additional mutations (all highlighted in off-white). None of the mutations involve basic or acidic residues, which are highlighted in blue and red, respectively. Figure 1c shows the mass spectrum obtained from a solution containing bovine, porcine, human insulin and 18C6. The three proteins were examined simultaneously, which eliminates any potential effects from variations in source or solution conditions. Reassuringly, the results are almost identical to those obtained from each protein examined separately, when care is taken to ensure similar source conditions. Comparable distributions both in terms of shape and relative intensities are obtained for all three proteins. The highly aligned backbone structures and extremely similar sequences of these variants suggest that they should have virtually identical surface structures as well, which is reflected in the observed SNAPP distributions.
Figure 1.
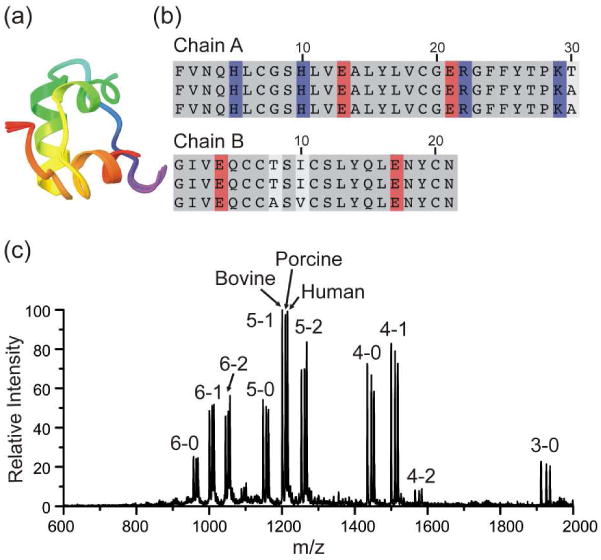
(a) Backbone structural alignment for the three variants of insulin. (b) Sequence alignment for human, porcine and bovine insulin from top to bottom, respectively. Sequence variation (white), basic residues (blue) and acidic residues (red) are highlighted. (c) ESI-MS spectrum of insulin from bovine, porcine, human and 18C6 in water. Peaks are labeled by (charge state) – (number of 18C6 adducts). The results are similar for all three proteins.
Cytochrome c
Figure 2a shows the sequence alignment for horse, bovine, pigeon and yeast cytc. Several mutations are observed among the four proteins. Horse and bovine cytc have the most similar sequences with only one basic/acidic amino acid variation, K60G. Pigeon cytc is also very similar with a total of five basic/acidic amino acid variations. In contrast, yeast cytc has numerous basic/acidic amino acid variations. It also contains 5 additional N-terminal residues (including one lysine) and is missing one C-terminal residue. Overall the yeast variant has one less basic and one less acidic residue than the horse variant. In Figure 2b, the backbone crystal structures for horse, bovine, and yeast cytc are shown from identical perspectives. Space filled atoms represent the side chains of basic and acidic residue mutations relative to horse cytc. The three proteins adopt very similar backbone structures, despite a fair number of point mutations. Electrostatic surfaces derived from the crystal structures are shown in Figure 2c from the same perspective shown in Figure 2b (red and blue areas are negatively and positively charged, respectively). Figure 2d shows the extracted SNAPP distributions for the four variants of cytc for the +9 and +10 charge states. The intensities are normalized to the non-adduct protein peak for comparison and the error bars represent the standard deviation of the mean. Although bovine cytc has one less lysine residue than the horse variant, they have nearly identical SNAPP distributions, suggesting that the additional lysine 60 in horse cytc does not contribute to the SNAPP distribution. Comparison of the two electrostatic surfaces shown in Figure 2c does not reveal significant differences in the region where Lys60 is located, in agreement with the SNAPP results. Inspection of the NMR structure26 for horse cytc also reveals a potential salt bridge interaction between Lys60 and Glu62, which would interfere with 18C6 attachment. These results confirm previous findings19 indicating that SNAPP distributions do not simply count the number of basic residues present in a protein, but rather provide information about the surface accessibility of basic residues in relation to the surrounding chemical environment. The crystal structure of pigeon cytc has not been reported yet, but based on the SNAPP distributions, it is likely similar to the known horse and bovine structures. Although SNAPP only directly probes surface structures, it is unlikely that two proteins with high sequence homology and similar surface structures would be able to simultaneously adopt two highly dissimilar backbone structures. Therefore, for homologous proteins, similar SNAPP distributions will likely imply similar backbone structures, although dissimilar SNAPP distributions may not imply dissimilar backbone structures.
Figure 2.
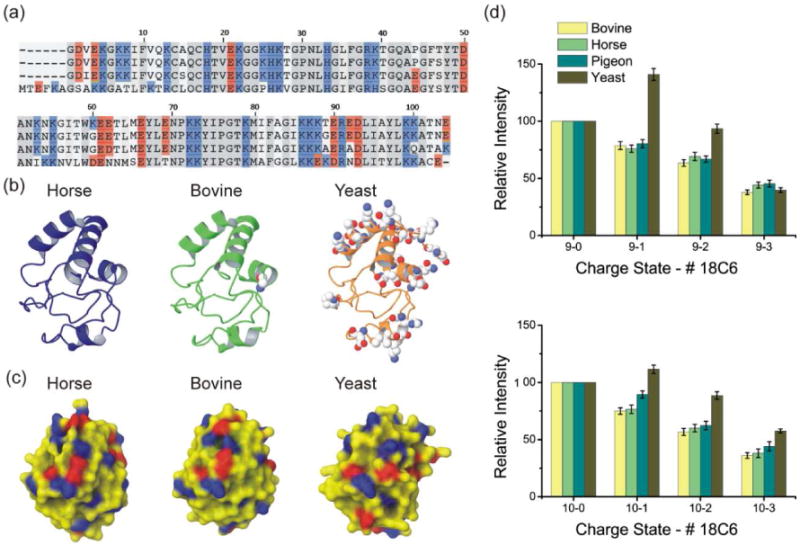
(a) Sequence alignment for horse, bovine, pigeon, and yeast cytc, respectively, from top to bottom. (b) Backbone structures of bovine, horse, and yeast cytc. Displayed atoms represent basic and acidic sequence variations relative to horse cytc. (c) Surface electrostatic distributions derived from crystal structures. The colors represent charge polarity (positive charge: blue, negative charge: red). (d) SNAPP distributions for four variants of cytc for charge state +9 and +10. Bovine, horse and pigeon cytc share similar results while yeast cytc has a distinct distribution.
This is illustrated in the SNAPP distributions for yeast cytc, which are somewhat different from the remaining proteins despite the fact that the backbone structures are the same. In Figure 2c, it is clear that more 18C6 attaches to yeast cytc than the remaining variants. It is tempting to suggest that the additional N-terminal lysine residue in yeast might account for the increased 18C6 attachment, but the comparison above between the horse and bovine variants reveals that one specific lysine residue does not necessarily affect the SNAPP distribution. In this case, the lysine residue in the additional n-terminal segment is also accompanied by an additional glutamic acid, which may mediate any increased 18C6 attachment. Other potentially important mutations involving change of polarity (basic and acidic residues), such as K7A, K8T, V11K, K25P, P44E, K53I, N54K, K60D, E62N, E69T, K88E, T89K, E92N, N103E, are also probable to contributors to the observed shift. It is most likely that the sum of these mutations leads to perturbation of the overall surface structure and a small shift in the observed SNAPP distributions. Comparison of the electrostatic surfaces in Figure 2c also confirms that yeast, while similar, has distinct features relative to the other variants, again in agreement with the data obtained by SNAPP.
Lysozyme
The crystal structures of the hen and human variants of Lysozyme are well aligned as shown by the ribbon representations in Figure 3a. However, the predicted charge distribution on the surface of these proteins is quite different, as shown in Figure 3b. The origin of the dissimilarity can be seen in the sequence alignment for the two proteins as shown in Figure 3c. The mutated acidic residues are highlighted with red stars and the mutated basic residues are denoted by blue asterisks. In contrast to cytc, where many mutations correspond to minor shifting in location of the same residue, in lysozyme many mutations occur in regions that contain no charged residues in the other variant. It is not surprising to find that the SNAPP distributions for hen and human lysozyme are not very similar, as shown in Figure 3d for charge states +11 and +12. Neither the relative intensities nor the shapes of the distributions are similar for the two variants. Human lysozyme attaches significantly more 18C6, suggesting greater overall availability of the basic residues. The SNAPP results confirm that human and hen lysozyme have distinct surface structures, which is consistent with the picture predicted by x-ray crystallography in Figure 3b.
Figure 3.
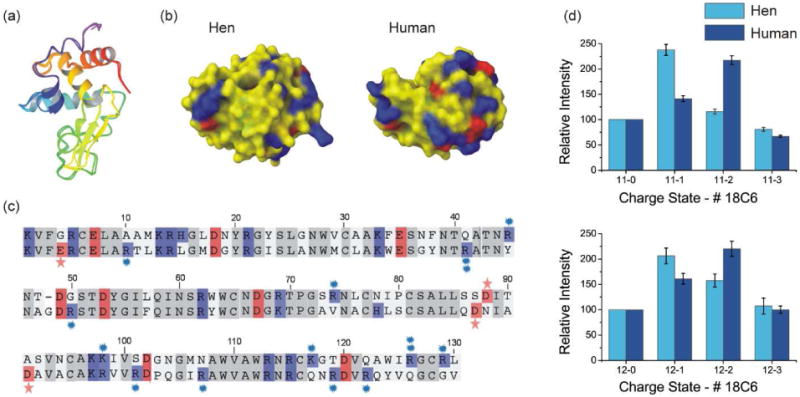
(a) Backbone structural alignment of lysozyme from hen and human. (b) Surface electrostatic distributions derived from crystal structures. The colors represent charge polarity (positive charge: blue, negative charge: red). (c) Sequence alignment for lysozyme from hen and human, from top to the bottom, respectively. (d) SNAPP distributions for lysozyme from hen and human, for charge state +11 and +12.
Comparative Analysis
The relative changes in SNAPP distributions for insulin, cytochrome c, and lysozyme are summarized in Table 1 in comparison with various changes in sequence. The ΔSNAPP column shows the percent change in the SNAPP distribution for each protein (P) as a function of charge state relative to the first variant (Pref) according to equation 1. The calculated differences reflect values between the error bars. Although it is difficult to summarize the complexity of a SNAPP distribution in a single number, the results in Table 1 do offer some interesting insight. For example, with insulin none of the ΔSNAPP scores are particularly high, but bovine insulin exhibits greater scores than porcine which does correlate with the overall sequence variation. In the case of cytochrome c, the yeast variant clearly stands out as the most distinct protein. Interestingly, the observed shift does not correlate well with changes to the sequence since yeast has more 18C6 attached but fewer basic residues. Many of the basic residues in yeast are shifted in location (see Figure 2), which may be the more important factor. For lysozyme, the two proteins are clearly distinct by ΔSNAPP and have significant sequence variation. Overall, the results from this limited data set indicate that a ΔSNAPP score >50 indicates a high probability of surface structure variation. Furthermore, the structural shifts between hen and human lysozyme and between yeast and the remaining cytochrome c proteins are comparable in magnitude. The distinct surface features of proteins with similar backbone structures led us to investigate if other properties, such as denaturation, were dissimilar as well.
Table 1. Comparison of Relative Changes in SNAPP versus Sequence.
| ΔSNAPP | ΔSequence | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein Name | Charge State | +8 | +9 | +10 | R | K | D,E | R,K,D,E | All | ||
| Cytc | Horse | 0 | 0 | 0 | 2 | 19 | 12 | 33 | 104 | ||
| ΔR | ΔK | Δ(D,E) | |Δ(R,K,D,E)| | |ΔAll| | |||||||
| Bovine | 1.7 | 1.3 | 0.9 | 0 | -1 | 0 | 1 | 3.03% | 3 | 2.88% | |
| Pigeon | 6.7 | 0.4 | 2.6 | 0 | -1 | -1 | 2 | 6.06% | 11 | 10.6% | |
| Yeast | 20.2 | 20.0 | 10.5 | +1 | -3 | -1 | 5 | 15.2% | 45 | 43.3% | |
| Insulin | +4 | +5 | +6 | R | K | D,E | R,K,D,E | All | |||
| Human | 0 | 0 | 0 | 1 | 1 | 4 | 6 | 51 | |||
| ΔR | ΔK | Δ(D,E) | |Δ(R,K,D,E)| | |ΔAll| | |||||||
| Bovine | 2.9 | 3.6 | 1.6 | 0 | 0 | 0 | 0 | 0% | 3 | 5.88% | |
| Porcine | 2.1 | 1.5 | 0.3 | 0 | 0 | 0 | 0 | 0% | 1 | 1.96% | |
| Lysozyme | + 10 | +11 | +12 | R | K | D,E | R,K,D,E | All | |||
| Hen | 0 | 0 | 0 | 11 | 6 | 9 | 26 | 129 | |||
| ΔR | ΔK | Δ(D,E) | |Δ(R,K,D,E)| | |ΔAll| | |||||||
| Human | 26.1 | 33.1 | 9.7 | +3 | -1 | +2 | 6 | 23.1% | 52 | 40.3% | |
| Equation (1) |
Protein Denaturation
The stability of native protein structures in atypical solvent systems can vary substantially and is a property which can be easily examined with ESI-MS.27 As shown above in Figure 2, the tertiary structure of cytc contains three major α-helices and no β-sheets. There is also a structurally relevant heme group covalently linked to the protein, and there are no disulfide bonds to inhibit unfolding.28 When sufficient methanol is present, cytc adopts a compact state with native-like secondary structures but ill-defined tertiary structure.29 This state is easily detected by SNAPP,18 but is not obvious by scrutiny of charge state distributions. Lowering the pH in the presence of methanol unfolds the protein further and a distinct shift in the charge state distribution can be observed by ESI-MS.30 Figure 4a shows the DESI mass spectra of horse, bovine, pigeon and yeast cytc in 50/50 water/methanol at neutral pH. The first three variants exhibit charge state distributions similar to those observed in water (inset spectra Figure 4a), with +9 being the most intense peak. However, for yeast, the introduction of methanol alone is sufficient to unfold the protein, which can be observed by the dramatic shift in the charge state distribution. The results indicate that the tertiary structure of yeast cytc is less stable than horse, bovine, or pigeon despite the fact that the backbone structures are virtually identical. Structural stability is determined by a variety of factors, including packing of the hydrophobic core, hydrogen bonding, salt bridges, etc…31 Comparison of the cytc sequences in Figure 2a reveals that yeast has several mutations to the hydrophobic core in addition to the mutations that lead to a distinct surface structure. The SNAPP data in Figure 2c indicates more 18C6 attachment to yeast compared with the other variants, which is consistent with fewer favorable intramolecular ionic or hydrogen bonding interactions and reduced stability. It is likely that some combination of both of both surface and core mutations leads to the reduced structural stability of yeast cytc.
Figure 4.
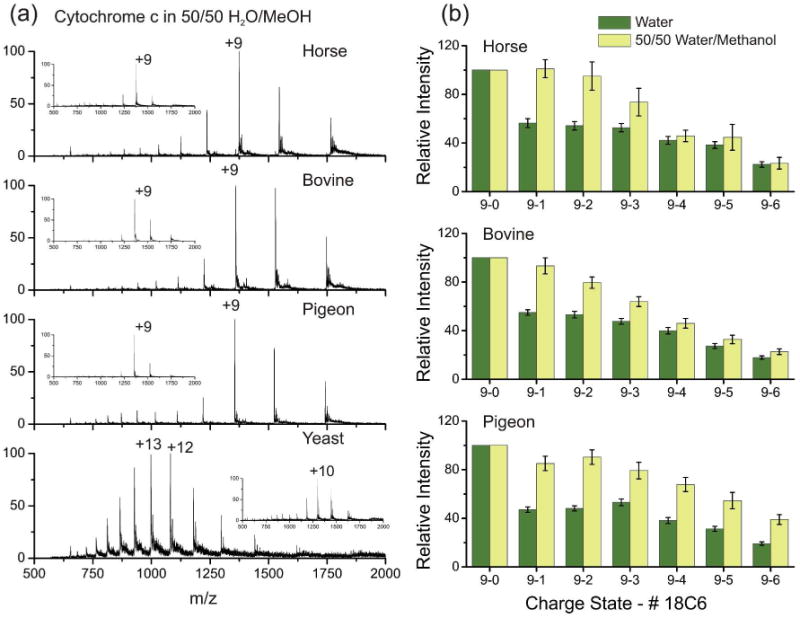
(a) DESI mass spectra of horse, bovine, pigeon, and yeast cytc in 50/50 water/methanol. Inset spectra were acquired in water. The yeast variant is significantly unfolded. (b) SNAPP distributions reveal partial denaturation in the presence of methanol when compared to water.
In Figure 4b, the SNAPP distributions for horse, bovine, and pigeon cytc in 50/50 water/MeOH are shown in comparison to those obtained in water. The SNAPP distributions clearly indicate a structural shift has occurred for each of these proteins as well, even though complete denaturation is not observed as in the case of yeast cytc. The SNAPP distributions further suggest that the structure of pigeon cytc has changed more than horse or bovine (this is particularly evident by comparison of the higher crown adduct peaks). The native structures of all three variants were very similar by SNAPP (Figure 2); however, pigeon cytc contains a P44E mutation in a turn region that may enable pigeon to adopt a different partially denatured state. Following the addition of acid to yield complete denaturation, the SNAPP distributions for the denatured states of all four proteins are comparable. Furthermore, the addition of acid does not significantly change the yeast SNAPP distribution relative to that obtained with just the addition of methanol.
Conclusions
SNAPP-MS is demonstrated to be a useful probe of surface structure for proteins with highly homologous sequences and nearly identical three dimensional backbone structures. For insulin, very minor sequence variation leads to very similar SNAPP distributions for three variants. In the case of cytochrome c, yeast exhibits the greatest change in surface structure and also denatures more easily in the presence of organic solvent. These changes do not correlate well solely with differences in the number of 18C6 binding sites, suggesting that sequence shifts which alter the surface environment of basic residues are also important. This hypothesis is further supported by results with lysozyme where significant sequence shifting yields quite disparate SNAPP distributions. These findings are consistent with previous results where SNAPP distributions have been shown to be sensitive to the availability of charged basic side chains. Changes to the arrangement of charged groups on the protein surface lead to changes in the observed SNAPP distributions. These results clearly demonstrate that proteins which adopt nearly identical tertiary structure may have substantially different electrostatic surfaces which could easily modulate protein-protein or small molecule recognition responses. The SNAPP-MS method is easy to carry out and requires minimal sample consumption, which should make it ideal for structure validation for proteins which have been subjected to site directed mutagenesis. Furthermore, SNAPP can assess variations in structurally ill-defined or highly dynamic states, such as proteins which have been partially or fully denatured. The effects of mutations can therefore be tracked from completely folded to completely unfolded structures by the same method.
Abbreviations
- SNAPP
Selective noncovalent adduct protein probing
- MS
mass spectrometry
- cytc
Cytochrome c
- ESI
electrospray ionization
- DESI
desorption electrospray ionization
- NMR
nuclear magnetic resonance
Footnotes
The authors gratefully acknowledge funding from the NIH (R01GM084106).
The authors declare no competing financial interest.
References
- 1.Sung YH, Eliezer D. Residual structure, backbone dynamics, and interactions within the synuclein family. J Mol Biol. 2007;372:689–707. doi: 10.1016/j.jmb.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nature Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 3.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 4.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 5.Ingram VM. Gene mutations in human haemoglobin- chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326–328. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- 6.Harrington DJ, Adachi K, Royer WE. Crystal structure of deoxy-human hemoglobin beta 6 Glu -> Trp - Implications for the structure and formation of the sickle cell fiber. J Biol Chem. 1998;273:32690–32696. doi: 10.1074/jbc.273.49.32690. [DOI] [PubMed] [Google Scholar]
- 7.Rao-Naik C, delaCruz W, Laplaza JM, Tan S, Callis J, Fisher AJ. The rub family of ubiquitin-like proteins - Crystal structure of arabidopsis Rub1 and expression of multiple rubs in arabidopsis. J Biol Chem. 1998;273:34976–34982. doi: 10.1074/jbc.273.52.34976. [DOI] [PubMed] [Google Scholar]
- 8.Finley D, Varshavsky A. The Ubiquitin System – Functions and Mechanisms. Trends Biochem Sci. 1985;10:343–347. [Google Scholar]
- 9.Taylor SV, Kast P, Hilvert D. Investigating and engineering enzymes by genetic selection. Angew Chem-Int Edit. 2001;40:3310–3335. doi: 10.1002/1521-3773(20010917)40:18<3310::aid-anie3310>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 10.Mirkin N, Jaconcic J, Stojanoff V, Moreno A. High resolution X-ray crystallographic structure of bovine heart cytochrome c and its application to the design of an electron transfer biosensor. Proteins. 2008;70:83–92. doi: 10.1002/prot.21452. [DOI] [PubMed] [Google Scholar]
- 11.Ferentz AE, Wagner G. NMR spectroscopy: a multifaceted approach to macromolecular structure. Q Rev Biophys. 2000;33:29–65. doi: 10.1017/s0033583500003589. [DOI] [PubMed] [Google Scholar]
- 12.Chowdhury SK, Katta V, Chait BT. Probing Conformational-Changes in Proteins by Mass-Spectrometry. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 13.Grandori R. Origin of the conformation dependence of protein charge-state distributions in electrospray ionization mass spectrometry. J Mass Spectrom. 2003;38:11–15. doi: 10.1002/jms.390. [DOI] [PubMed] [Google Scholar]
- 14.Kaltashov IA, Mohimen A. Estimates of protein surface areas in solution by electrospray ionization mass spectrometry. Anal Chem. 2005;77:5370–5379. doi: 10.1021/ac050511+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 16.Mendoza VL, Vachet RW. Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Anal Chem. 2008;80:2895–2904. doi: 10.1021/ac701999b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 18.Ly T, Julian RR. Using ESI-MS to probe protein structure by site-specific noncovalent attachment of 18-crown-6. J Am Soc Mass Spectrom. 2006;17:1209–1215. doi: 10.1016/j.jasms.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Liu ZJ, Cheng SJ, Gailie DR, Julian RR. Exploring the mechanism of selective noncovalent adduct protein probing mass spectrometry utilizing site-directed mutagenesis to examine ubiquitin. Anal Chem. 2008;80:3846–3852. doi: 10.1021/ac800176u. [DOI] [PubMed] [Google Scholar]
- 20.Ly T, Julian RR. Protein-Metal Interactions of Calmodulin and alpha-Synuclein Monitored by Selective Noncovalent Adduct Protein Probing Mass Spectrometry. J Am Soc Mass Spectrom. 2008;19:1663–1672. doi: 10.1016/j.jasms.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Sun QY, Tyler RC, Volkman BF, Julian RR. Dynamic Interchanging Native States of Lymphotactin Examined by SNAPP-MS. J Am Soc Mass Spectrom. 2011;22:399–407. doi: 10.1007/s13361-010-0042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamdy O, Julian RR. Reflections on Charge State Distributions, Protein Structure, and the Mystical Mechanism of Electrospray Ionization. J Am Soc Mass Spectrom. 2012;23:1–6. doi: 10.1007/s13361-011-0284-8. [DOI] [PubMed] [Google Scholar]
- 23.Timofeev VI, Chuprov-Netochin RN, Samigina VR, Bezuglov VV, Miroshnikov KA, Kuranova IP. X-ray investigation of gene-engineered human insulin crystallized from a solution containing polysialic acid. Acta Crystallogr F-Struct Biol Cryst Commun. 2010;66:259–263. doi: 10.1107/S1744309110000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishikawa T, Chatake T, Ohnishi Y, Tanaka I, Kurihara K, Kuroki R, et al. A neutron crystallographic analysis of a cubic porcine insulin at pD 6.6. Chem Phys. 2008;345:152–158. [Google Scholar]
- 25.Gursky O, Li YL, Badger J, Caspar DLD. Monovalent Cation Binding to Cubic Insulin Crystals. Biophys J. 1992;61:604–611. doi: 10.1016/S0006-3495(92)81865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banci L, Bertini I, Huber JG, Spyroulias GA, Turano P. Solution structure of reduced horse heart cytochrome c. J Biol Inorg Chem. 1999;4:21–31. doi: 10.1007/s007750050285. [DOI] [PubMed] [Google Scholar]
- 27.Kaltashov IA, Eyles SJ. Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrom Rev. 2002;21:37–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- 28.Fisher WR, Taniuchi H, Anfinsen CB. Role of heme in formation of structure of cytochrome-c. J Biol Chem. 1973;248:3188–3195. [PubMed] [Google Scholar]
- 29.Kamatari YO, Konno T, Kataoka M, Akasaka K. The methanol-induced globular and expanded denatured states of cytochrome c: A study by CD fluorescence, NMR and small-angle X-ray scattering. J Mol Biol. 1996;259:512–523. doi: 10.1006/jmbi.1996.0336. [DOI] [PubMed] [Google Scholar]
- 30.Konermann L, Douglas DJ. Acid-induced unfolding of cytochrome c at different methanol concentrations: Electrospray ionization mass spectrometry specifically monitors changes in the tertiary structure. Biochemistry. 1997;36:12296–12302. doi: 10.1021/bi971266u. [DOI] [PubMed] [Google Scholar]
- 31.Dyson HJ, Wright PE, Scheraga HA. The role of hydrophobic interactions in initiation and propagation of protein folding. Proc Natl Acad Sci. 2006;103:13057–13061. doi: 10.1073/pnas.0605504103. [DOI] [PMC free article] [PubMed] [Google Scholar]