

Abstract
Previous structure-activity relationship studies of salvinorin A have shown that modification of the acetate functionality off the C-2 position to a methoxy methyl or methoxy ethyl ether moiety leads to increased potency at KOP receptors. However, the reason for this increase remains unclear. Here we report our efforts towards the synthesis and evaluation of C-2 constrained analogs of salvinorin A. These analogs were evaluated at opioid receptors in radioligand binding experiments as well as in the GTP-γ-S functional assay. One compound, 5, was found to have affinity and potency at κ opioid (KOP) receptors comparable to salvinorin A. In further studies, 5 was found to attenuate cocaine-induced drug seeking behavior in rats comparably to salvinorin A. This finding represents the first example of a salvinorin A analog that has demonstrated anti-addictive capabilities.
Introduction
The neoclerodane diterpene salvinorin A is the active component of Salvia divinorum L. Epling&Jativa (Lamiaceae), a widely available psychoactive plant.1 S. divinorum is a member of the sage family and is indigenous to Oaxaca, Mexico.1, 2 For centuries it has been used by the Mazatec Indians in divination ceremonies as well as to treat headaches, rheumatism, and panzón de barrego, a semi-magical disease.1 Salvinorin A was isolated from S. divinorum and determined to be a potent hallucinogen when vaporized and inhaled, with an active dose in humans of 4.5 – 21 μg/kg.3 This rivals the potency of classical hallucinogens such as lysergic acid diethylamide (LSD) and 4-bromo-2,5-dimethoxyamphetamine (DOB).4 However, unlike the classical hallucinogens, salvinorin A does not have activity at serotonin 5HT2A receptors.4–6 Instead, it was found to be a potent and selective κ opioid (KOP) receptor agonist.4 KOP agonists have received recent research focus because they may have applications as drug abuse therapeutics.7
Recently it has been reported that converting the acetate group at the C-2 position of salvinorin A to a methoxy- or ethoxymethyl ether produces ligands of greater affinity and potency at KOP receptors than salvinorin A.8, 9However, the structural basis that underlies these changes in affinity and potency is unknown. We hypothesize that the ether moieties at C-2 are relatively flexible and can adopt different conformations when interacting with KOP receptors; for example, an eclipsed or an extended conformation as shown in Figure 1. Thus, we applied the concept of conformational constraint in order to design ligands in which the flexible portions are pre-organized in the hopes that such ligands would aid in the elucidation of the structural basis for the observed changes in affinity and potency. Here, we report our synthetic efforts towards this end, as well as the biological evaluation of C-2 constrained analogs of salvinorin A.
Figure 1.

Eclipsed (upper) or extended (lower) conformations of the ethoxymethyl ether at C-2 compared to salvinorin A.
Chemistry
Compounds 3 and 4 were synthesized according to Scheme 1. Salvinorin A (1) was extracted from commercially available dried S. divinorum leaves and converted to salvinorin B (2) as previously described.10 Treatment of 2 with either chloromethyl methyl ether or chloromethyl ethyl ether in the presence of N,N-diisopropylethylamine (DIPEA) afforded the previously reported ethers 3 and 4.8
Scheme 1.
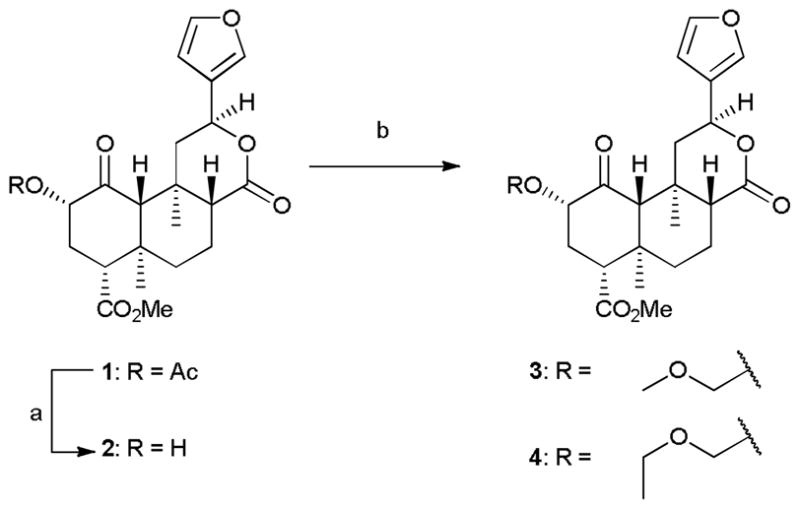
Reagents and conditions: (a) Na2CO3, MeOH; (b) appropriate chloromethyl ether, DIPEA, CH2Cl2.
Compounds 5, 6, and 7 were synthesized according to Scheme 2. A solution of 2 in CH2Cl2 was treated with 3,4-dihydro-2H-pyran or 2,3-dihydrofuran in the presence of a catalytic amount of pyridinium p-toluene sulfonate (PPTS) to afford the previously reported tetrahydropyran (5 and 6) and novel tetrahydrofuran (7) C-2 constrained derivatives.8 Under these conditions, diastereomers were formed and, in the case of tetrahydropyrans 5 and 6, were separated from each other through flash column chromatography, and characterized by NMR, and X-ray crystallography (Figures 2 and 3). In the case of the diastereomers of 7, they were separated from each other through flash column chromatography and characterized by NMR.
Scheme 2.

Reagents and conditions: (a) 3,4-dihydro-2H-pyran or 2,3-dihydrofuran, PPTS, CH2Cl2; (b) PPTS, ethyl vinyl ether, reflux; (c) Br2, ethyl vinyl ether, DIPEA, CH2Cl2; (d) Ac2O/HCO2H, pyridine; (e) ethyl chloroformate, DMAP, TEA, CH2Cl2; (f) ethyl isocyanate, DMAP, pyridine.
Figure 2.

1D 13C NMR spectrum with composite pulse 1H-decoupling (blue) and gated decoupling to show JC,H (black) for the anomeric carbon of 5(A) and 6 (B).
Figure 3.
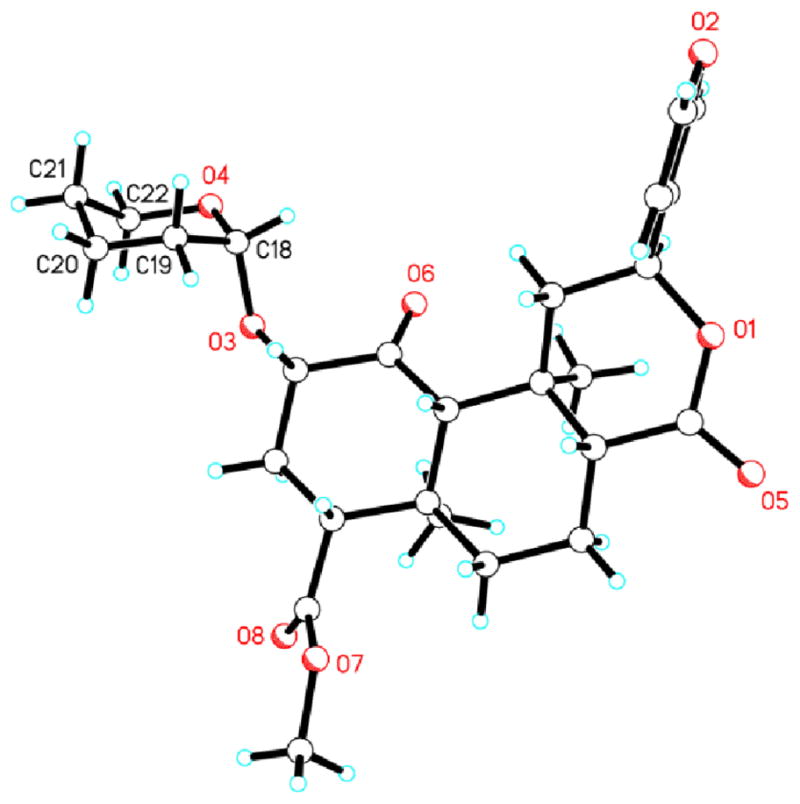
X-ray crystallographic structure of 5.
NMR spectroscopy and X-Ray crystallography were used to assign the stereochemistry of the new stereocenter in compounds 5 and 6. In carbohydrate chemistry, anomeric 1JC,H values are useful for the assignment of configuration at that position since pyranoses with an axial proton have a 1JC,H value that is approximately 10 Hz lower than the corresponding value in compounds with an equatorial anomeric proton.11, 12 Using this method, we determined that the anomeric 1JC,H values for compounds 5 and 6 are 168.55 Hz and 159.36 Hz, respectively, which are consistent with previous reports of substituted pyranoses. Given these data, we propose that 5 contains an equatorial, β-H (R) and 6 contains an axial, α-H (S) (Figure 2).13 Compound 5, the less polar diastereomer, was crystallized and we determined its structure using X-ray crystallography, confirming the orientation of the hydrogen predicted by our NMR analysis (Figure 3). In this structure, the tetrahydropyran moiety forms an α-glycosidic bond with the salvinorin A core in an axial position and the anomeric hydrogen in an equatorial, β position. Subsequently, compound 6 must be the other diastereomer.
Compounds 8 and 9 were synthesized according to Scheme 2. In the presence of a catalytic amount of PPTS, a suspension of 2 in ethyl vinyl ether was refluxed for 2 h, affording a diastereomeric mixture of previously reported ethers 8.8 These diastereomers were separated from each other by flash column chromatography, and the less polar diastereomer (8a) was isolated. The more polar diastereomer (8b) appeared to degrade during purification and could not be completely purified (purity = 81.938 %) through trituration, flash column chromatography, or HPLC. The impurity was not determined, although there was no amount of 8a present as detectable by HPLC. Both 8a and 8b were characterized by NMR. In order to produce novel ether 9, ethyl vinyl ether was added to a solution of Br2 in CH2Cl2, followed by the addition of DIPEA and finally 2. These conditions afforded a mixture of diastereomers which were subsequently separated from each other by HPLC and characterized by NMR.
Compounds 10, 11, and 12 were synthesized according to Scheme 2. Following a literature procedure, a mixture of acetic ahydride and acetic acid was added to a mixture of 2 in pyridine at 0 °C.14 The mixture was allowed to warm to room temperature and stirred for 30 min, affording formate 10. Known carbonate 119, 15 was produced by treating a solution of 2 in CH2Cl2 with ethyl chloroformate in the presence of DMAP and TEA at room temperature for 24 h. Following a literature procedure, carbamate 12 was produced by treating a mixture of 2 in pyridine with ethyl isocyanate at 35 °C overnight.16
Finally, compound 14 was synthesized according to Scheme 3. This compound was inspired by a previously published analog (15) that incorporated the piperidylamide in place of the furan ring of 1, and demonstrated retained affinity for the KOP receptor.17 Through the synthesis and evaluation of compound 14, we sought to combine both the piperidyl amide and methoxy methyl ether features into one molecule that would have high KOP receptor affinity compared to 1. Following a procedure developed in our laboratory, the furan ring of 3 was oxidatively degraded to carboxylic acid 13 using catalytic RuCl3·3H2O in the presence of NaIO4.18 With 13 in hand, standard peptide coupling conditions (as employed before in our laboratory) were used to generate amide 14.17
Scheme 3.

(a) RuCl3·3H2O, NaIO4, CCl4/CH3CN/H2O; (b) piperidine, EDCI, HOBt, TEA, CH2Cl2.
Biology
Compounds 3–12, and 14 were evaluated for affinity at human opioid receptors using methodology previously described (Table 1).19, 20 For previously reported ethers 3 and 4, the results of the radioligand binding assay are in agreement with published data.8 In past literature reports, the tetrahydropyran diastereomers 5 and 6 were not completely separated from each other before biological evaluation. For a mixture of predominantly one of the diastereomers, absolute configuration unknown, the reported binding affinity and potency were Ki = 4.0 ± 0.4 nM and EC50 = 2.8 ± 0.3 nM.8 We were able to separate and isolate each diastereomer, and the observed binding affinities for 5 and 6 in our hands reveal a eutomer (5) and a distomer (6), indicating a preference for the hydrogen of the new stereocenter to be in the β position or R configuration. Interestingly, the related tetrahydrofuran derivatives 7a and 7b have modest binding affinities at KOP receptors that are very similar to each other and show no preference for stereochemistry. One possible explanation is that by reducing the ring size by one carbon, the orientation of the new stereocenter of 7a and 7b in the receptor has been altered (relative to 5 and 6) such that neither diastereomer has a particularly unfavorable interaction with the receptor.
Table 1.
Opioid receptor binding affinity for compounds 3–12, and 14.
| Cmpd | Ki ± SD nMa | ||
|---|---|---|---|
| [3H]DAMGO (μ) | [3H]DADLE (δ) | [3H]U69,593 (κ) | |
| 1b | EC50 = 2860 ± 980 Emax = 75 ± 8 % |
>10,000 | 7.4 ± 0.7 |
| 3 | 390 ± 20 | 2,840 ± 180 | 1.9 ± 0.2 |
| 4 | 41 ± 3 | 1,017 ± 99 | 3.13 ± 0.40 |
| 5 | >10,000 | >10,000 | 6.21 ± 0.40 |
| 6 | >10,000 | >10,000 | 300 ± 23 |
| 7a | >10,000 | >10,000 | 75 ± 3 |
| 7b | >10,000 | >10,000 | 81 ± 6 |
| 8a | 777 ± 40 | >5,000 | 6.7 ± 0.32 |
| 8b | >5,800 | >5,000 | 1,233 ± 33 |
| 9a | >3,000 | >5,000 | 5,261 ± 680 |
| 9b | 3,523 ± 389 | >5,000 | 7,438 ± 764 |
| 10c | ND | ND | 41 ± 1 |
| 11c | ND | ND | 171 ± 7 |
| 12c | 1,540 ± 140 | ND | ND |
| 14 | >3,000 | >3,000 | 59 ± 4 |
Receptor binding was performed in CHO cells which express the human MOP, DOP, or KOP receptors. All results are N = 3.
Data from reference 21.
ND indicates that Ki was found to be >10,000 nM in a range finding study.
In similar fashion to the tetrahydropyran derivatives, ethers 8a and 8b are previously reported, but the diastereomers were not completely separated from each other before biological evaluation.8 The published data show both diastereomers as having very similar binding affinities (Ki = 11 ± 1 nM and Ki =6.6 ± 0.3 nM) and potencies (10 ± 1 nM and 5.7 ±0.7 nM). We were able to separate the diastereomers and isolate the less polar one (8a). By HPLC, the more polar diastereomer (8b) appeared to degrade during purification and was not completely purified (purity = 81.938 %), although it is important to note that there was no amount of 8a present. In our hands ethers 8a and 8b also reveal a eutomer (8a) and distomer (8b), indicating a preference for the configuration of the new stereocenter, and reflecting our observations of tetrahydropyrans 5 and 6. In our NMR characterization of 8a and 8b, we observed 1JC,H values at the new stereocenter that followed the accepted trend of pyranoses as previously described, although the difference was less pronounced (data not shown). Given this observation and the pattern in the radioligand binding data in which the less polar diastereomers 5 and 8a had the highest affinity, we propose that the configuration of the new stereocenter in 8a is very likely the same as that of 5. However, this needs to be confirmed through X-ray crystallography. The related brominated ethers 9a and 9b had poor binding affinities at the KOP receptor and show no preference for stereochemistry. One explanation for this observation is that the bromine atom was too bulky and/or too lipophilic such that it precluded receptor binding altogether.
Formate 10 produced radioligand binding affinity data that was well in agreement with published reports.14, 22 However, in our hands compounds 11 and 12 behaved differently than previously reported. In two previously published radioligand binding studies, carbonate 11 showed no appreciable affinity at the KOP receptor vs. [3H]bremazocine15 or [3H]diprenorphine.9 Our data show that carbonate 11 displaces [3H]U69,593 at the KOP receptor at Ki = 171 ± 7 nM. Additionally, carbamate 12 was reported to have a KOP receptor binding affinity (vs. [3H]diprenorphine) and potency of Ki = 462 ± 20 nM and EC50 = >1000 nM.16 We found that while carbamate 12 has no appreciable affinity at the other opioid receptors, it does displace [3H]DAMGO at the μ opioid(MOP) receptor at Ki = 1540 ± 140 nM. While this particular Ki is unimpressive, it does agree with previously published reports in which alkyl carbamates at the C-2 position of salvinorin A were found to lose KOP affinity and gain MOP affinity relative to the parent compound.
Compound 14 is reflective of a furan ring analog of 1 that was previously produced in our lab (Scheme 3). Amide 15 had binding affinity at KOP receptors of Ki = 140 ± 10 nM, and potency and efficacy of ED50 = 5110 ± 1800 nM, Emax = 85 ± 13.17 Our hypothesis was that through the combination of the amide in place of the furan ring (from 15), and the methoxy methyl ether in place of the C-2 acetate (from 3), we would be able to observe improved (or at least retained) binding affinity relative to the parent analogs. Through the addition of the methoxy methyl ether off the C-2 position, the KOP receptor binding affinity of 14 was improved over that of 15 by approximately 2-fold to Ki = 59 ± 4 nM. However, the binding affinity of 14 is roughly 31-fold worse compared to 3. Still, the improvement in receptor binding affinity from compound 14 over compound 15 is of similar magnitude (roughly 2–4 fold) to the improvement in receptor binding affinity from compound 3 over compound 1.
Compounds 3, 4, 5, 7, 8a, and 14 were also evaluated for functional activity at KOP receptors using a [35S]GTP-γ-S functional assay (Table 2).19, 20 Compounds 3, 4, 5, and 8a were found to have potencies comparable to or better than 1 as agonists. Compounds 7a and 7b were observed to be approximately 30-and 17-fold less potent than 1 as agonists, respectively. Finally, compound 14 was 48-fold less potent as an agonist than 1, which is actually an approximately 2.5-fold improvement over the previously published furan analog 15 (approximately 125-fold less potent than 1 as an agonist).17 However, 14 was also roughly 322-fold less potent than 3 as an agonist. Thus, the incorporation of the methoxy methyl ether at C-2 improved the binding affinity and potency of compound 14 over parent compound 15 by approximately 2-fold and 2.5 fold, respectively. These improvements are of similar magnitude to the improvements in the binding affinity and potency (approximately 4-fold and 7-fold, respectively) of compound 3 over compound 1. However, the combination of furan substitution with C-2 acetate substitution in compound 14 ultimately greatly decreased the potency of the compound relative to ether 3 as well as compound 1.
Table 2.
[35S]GTP-γ-S functional assay for compounds 3, 4, 5, 7,8a, and 14.
| Cmpd | ED50 ± SDa (κ) nM | Emax ± SDb (κ) |
|---|---|---|
| 1c | 40 ±10 | 120 ± 2 |
| 3 | 6 ± 1 | 118 ± 2 |
| 4 | 0.65 ± 0.17 | 127 ± 5 |
| 5 | 60 ± 6 | 109 ± 3 |
| 7a | 1220 ± 230 | 112 ± 8 |
| 7b | 690 ± 80 | 103 ± 4 |
| 8a | 150 ± 14 | 101 ± 3 |
| 14 | 1934 ± 239 | 113 ± 5 |
ED50 = Effective dose for 50% maximal response.
Emax is % at which compound stimulates [35S]GTP-γ-Sbinding compared to (−)-U50,488 (500 nM) at KOP receptors.
Data from reference 21.
Finally, among the compounds reported, compound 5 had KOP receptor binding affinity and potency comparable to 1. Compound 5 also lacks a hydrolyzable ester at C-2 and other analogs of 1 with this particular feature have been found to have a greater duration of action than 1.23 Therefore, compound 5 met our criteria for evaluation in vivo. Using methodology previously reported, compound 5 was evaluated for its effect on cocaine-induced drug-seeking in rats at doses of 0.3 mg/kg and 1 mg/kg (Figure 4).24
Figure 4.

The effect of 5 on cocaine-induced drug-seeking. In the first phase (cocaine baseline), the active lever delivered an infusion of cocaine. Recorded active lever responses consisted of access to cocaine (0.5 mg/kg/inf) for a period of 2 h in operant chambers. At the beginning of the second phase (extinction), cocaine was replaced with sterile heparinized saline and responses were recorded for 2 h, until active lever responses were <20 for a 2 h daily session. At the beginning of the third phase, animals received an I.P. injection of vehicle (75% DMSO, N = 7), 5 (0.3 mg/kg, N = 3), 5 (1 mg/kg, N = 6), or 1 (0.3 mg/kg, N = 7) 5–10 min prior to a priming injection of cocaine (20 mg/kg). Responding in phase 3 was measured for 2 h (± SEM).
Briefly, rats were trained on a fixed ratio 5 schedule of reinforcement for cocaine self-administration where a single cocaine infusion was received (0.5 mg/kg/infusion, paired with a light cue) for every 5 correct active lever responses. The first phase involved daily 2 h sessions until responding showed <20% variation across three consecutive days (cocaine baseline). In phase 2, cocaine was replaced with heparinised saline and daily 2 h sessions continued in the same operant chambers until active lever responses were <20 for a session (typically 2–3 days). When the extinction criteria had been achieved, reinstatement of cocaine-induced cocaine-seeking was tested the following day. Animals received IP injections of vehicle (75% DMSO), compound 5 (0.3 mg/kg or 1 mg/mg), or compound 1 (0.3 mg/kg) 5–10 min prior to a priming injection of cocaine (IP, 20 mg/kg). Immediately following the cocaine injection, animals were placed into the operant chamber and responding was recorded for a further 2 h. Results show a statistically significant reduction in responding between vehicle and the 1 mg/kg dose of 5. In fact, this dose appeared to attenuate responding comparably to 0.3 mg/kg salvinorin A (1), our positive control, which we have previously shown to attenuate cocaine-primed reinstatement of drug seeking at a dose of 0.3 mg/kg or greater.24
Conclusions
In summary, we have evaluated C-2 modified analogs of 1 for opioid receptor affinity and efficacy. We have isolated and confirmed the binding affinities and potencies of two agonists, compounds 5 and 8a. In comparison to their diastereomers (6 and 8b), these analogs show preferential interactions with the KOP receptor. Since our NMR-based assignment of the absolute configuration of compound 5 was confirmed through X-ray crystallography, and given our observation that the less polar diastereomers appear to have the highest binding affinities, we propose that the new stereocenter in ether 8a shares the same absolute configuration as that of tetrahydropyran 5. This will need to be further investigated through X-ray crystallography. Interestingly, we did not observe a stereochemical influence in the other related diastereomeric compounds (7a, 7b, 9a, and 9b). This may be accounted for in steric considerations: the smaller ring size of 7a and 7b has changed their orientation in the receptor such that neither diastereomer interacts particularly unfavourably; and 9a and 9b are too large and/or lipophilic to allow for receptor interaction at all. Additionally, we have shown that contrary to previous reports, compound 11 does show affinity for the KOP receptor in radioligand binding assays. We have also demonstrated that compound 12, in agreement with previous reports, has very little affinity at KOP receptors, but does have some affinity for the MOP receptor, in agreement with past radioligand binding studies of similar compounds. We also found that compound 14, which is reflective of a previously published furan ring analog (15), demonstrated improved binding affinity over the parent compound upon the incorporation of the methoxy methyl ether moiety off C-2. However, the potency of 14 relative to compounds 3 and 1 was greatly diminished. Finally through further evaluation, it was found that compound 5 attenuated cocaine-induced drug seeking behavior comparably to salvinorin A (1). This finding is consistent with other reported KOP receptor agonists, and represents the first example of a salvinorin A analog that has demonstrated anti-addictive capabilities. The identification of differences in chirality and receptor tolerances for these ring constrained analogs should be of great value for the design of new compounds and or pharmacophore models of the binding site of 1.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. David Vandervelde at the University of Kansas for valuable insight and discussions regarding NMR spectroscopy. The authors also thank the National Institute on Drug Abuse (DA018151) and the NIH Dynamic Aspects of Chemical Biology training grant (GM008545) for financial support of ongoing research, and the National Science Foundation (CHE-0923449) and the University of Kansas for funds to purchase the x-ray instrumentation and computers. Portions of this work were supported by the Intramural Research Program, National Institute on Drug Abuse, NIH, DHHS and the Health Research Council of New Zealand. The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse, National Institutes of Health, or the National Science Foundation.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
References
- 1.Valdes LJ, 3rd, Diaz JL, Paul AG. J Ethnopharmacol. 1983;7:287–312. doi: 10.1016/0378-8741(83)90004-1. [DOI] [PubMed] [Google Scholar]
- 2.Valdes LJ, Butler WM, Hatfield GM, Paul AG, Koreeda M. J Org Chem. 1984;49:4716–4720. [Google Scholar]
- 3.Johnson MW, MacLean KA, Reissig CJ, Prisinzano TE, Griffiths RR. Drug Alcohol Depend. 2011;115:150–155. doi: 10.1016/j.drugalcdep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc Nat Acad Sci USA. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valdes LJ., 3rd J Psychoactive Drugs. 1994;26:277–283. doi: 10.1080/02791072.1994.10472441. [DOI] [PubMed] [Google Scholar]
- 6.Siebert DJ. J Ethnopharmacol. 1994;43:53–56. doi: 10.1016/0378-8741(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 7.Prisinzano TE, Tidgewell K, Harding WW. The AAPS Journal. 2005;7:E592–E599. doi: 10.1208/aapsj070361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munro TA, Duncan KK, Xu W, Wang Y, Liu-Chen L-Y, Carlezon WA, Jr, Cohen BM, Béguin C. Bioorg Med Chem. 2008;16:1279–1286. doi: 10.1016/j.bmc.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DYW, Karnati VVR, He M, Liu-Chen LY, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C, Carlezon JWA, Cohen B. Bioorg Med Chem Lett. 2005;15:3744–3747. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 10.Tidgewell K, Harding WW, Schmidt M, Holden KG, Murry DJ, Prisinzano TE. Bioorg Med Chem Lett. 2004;14:5099–5102. doi: 10.1016/j.bmcl.2004.07.081. [DOI] [PubMed] [Google Scholar]
- 11.Bock K, Lundt I, Pedersen C. Tetrahedron Lett. 1973:1037–1040. [Google Scholar]
- 12.Haellgren C. J Carbohydr Chem. 1992;11:527–530. [Google Scholar]
- 13.Horton D, editor. Advances in Carbohydrate Chemistry and Biochemistry. Academic Press, Inc; San Diego, CA: 1995. [Google Scholar]
- 14.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J Med Chem. 2005;48:345–348. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. J Pharmacol Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- 16.Béguin C, Richards MR, Wang Y, Chen Y, Liu-Chen LY, Ma Z, Lee DYW, Carlezon WA, Cohen BM. Bioorg Med Chem Lett. 2005;15:2761–2765. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- 17.Simpson DS, Lovell KM, Lozama A, Han N, Day VW, Dersch CM, Rothman RB, Prisinzano TE. Org Biomol Chem. 2009;7:3748–3756. doi: 10.1039/b905148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Gilmour B, Navarro H, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:107–112. doi: 10.1021/np050398i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fontana G, Savona G, Rodríguez B, Dersch CM, Rothman RB, Prisinzano TE. Tetrahedron. 2008;64:10041–10048. doi: 10.1016/j.tet.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu H, Hashimoto A, Rice KC, Jacobson AE, Thomas JB, Carroll FI, Lai J, Rothman RB. Synapse (N Y) 2001;39:64–69. doi: 10.1002/1098-2396(20010101)39:1<64::AID-SYN9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 21.Lozama A, Cunningham CW, Caspers MJ, Douglas JT, Dersch CM, Rothman RB, Prisinzano TE. J Nat Prod. 2011;74:718–726. doi: 10.1021/np1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee DYW, Yang L, Xu W, Deng G, Guo L, Liu-Chen LY. Bioorg Med Chem Lett. 2010;20:5749–5752. doi: 10.1016/j.bmcl.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Chen Y, Xu W, Lee DYW, Ma Z, Rawls SM, Cowan A, Liu-Chen LY. J Pharmacol Exp Ther. 2008;324:1073–1083. doi: 10.1124/jpet.107.132142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morani AS, Kivell B, Prisinzano TE, Schenk S. Pharmacol Biochem Behav. 2009;94:244–249. doi: 10.1016/j.pbb.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.