

Abstract
A convenient route for the synthesis of Fmoc-protected phosphinic dipeptide building blocks is described. The protected amino acid isosteres benzyloxycarbonyl aminomethyl phosphinic acid (glycine surrogate), benzyl α-isopropyl acrylate (valine surrogate), and benzyl α-isobutyl acrylate (leucine surrogate) were synthesized starting from commercially available materials. Reaction of either the valine or leucine surrogate with bis(trimethylsilyl) phosphonite generated the pseudodipeptide bond. The synthesis concluded with an efficient one-pot three-step procedure involving a bis-deprotection of the N- and C-termini under catalytic hydrogenation conditions followed by selective capping of the N-terminus with an Fmoc group to yield either Fmoc-NHCH2PO(OAd)CH2CH(Pri)CO2H or Fmoc-NHCH2PO(OAd)CH2CH(Bui)CO2H.
Keywords: phosphinic pseudodipeptides, matrix metalloproteinase inhibitors, MMPI, peptide synthesis, phosphinates
INTRODUCTION
We are interested in developing effective matrix metalloproteinase (MMP) inhibitors as activation of these enzymes have been associated with primary and metastatic tumor growth, angiogenesis, and pathological degradation of extracellular matrix components, such as collagen and laminin.1,2 Several MMPs have been validated as targets in certain cancers (MMP-2, MMP-9, MT1-MMP), whereas others have been found to be host-beneficial and thus antitargets (MMP-3, MMP-8).3
A general approach to the design of an inhibitor of metalloproteases involves replacing a trigonal planar amide or ester bond of a substrate with a hydrolytically stable functional group possessing tetrahedral geometry in the original carbonyl position to mimic the intermediate formed during enzyme-catalyzed hydrolysis. The phosphinate functional group (Figure 1) with its pair of electronically equivalent anionic oxygens (under physiological conditions) has emerged as an effective tetrahedral substitution motif in peptides leading to potent and selective transition state inhibitors of metalloproteases such as MMPs.4–7 Indeed, due to their growing medicinal relevance, a number of elegant strategies have been developed over the past few years for the synthesis of phosphinate dipeptides. Meldal and coworkers, Yiotakis et al., and Yokomatsu and coworkers reported the synthesis of different phosphinate dipeptide building blocks by using a procedure involving the Michael addition of variously protected aminomethyl phosphinic acids to an acrylic acid ester followed by protecting group manipulation to afford the desired product.8 Using a creative variation of these methods, FmocNHCH2PO(OAd)-CH2CH(Pri)CO2H (1a, Scheme 2) was prepared and subsequently incorporated into a triple-helical sequence, resulting in a construct, which exhibited potent and selective inhibition of MMP-2 and MMP-9.7 However, this previously described synthetic route to 1a required a low yielding (35%) and problematic final step involving a Ru-catalyzed deprotection of an allyl ester.
FIGURE 1.

Common structure of phosphinic peptides.
SCHEME 2.
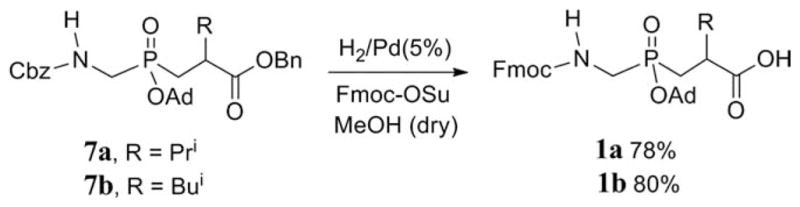
Tandem bis-deprotection and Fmoc-amine formation leading to 1.
In our continued studies of MMP inhibition involving this phosphinate dipeptide system, we required a more scale-able route to 1a. In this communication, we describe an efficient bis-deprotection strategy leading to the desired phosphinate dipeptide 1a and its Gly-Leu analog 1b.
DISCUSSION
Our general strategy for the synthesis of the Fmoc-protected phosphinic dipeptide 1 entails a Michael-type addition for the formation of the P—C bond pioneered by Regan and Yiotakis and subsequently modified by Meldal and Hammer.8–10 But our initial attempt of Michael-type addition of Fmoc-protected aminomethyl phosphinic acid and electron deficient acrylate was not successful.8,11 Thus, Cbz-protected aminomethyl phosphinic acid 3 was converted to its trivalent state by the action of HMDS and subsequently reacted with an α-isopropyl-α,β-unsaturated ester12 to give product 4 in good yield (Scheme 1). However, as will be described subsequently, the Cbz group is very efficiently exchanged for Fmoc in the final step of the synthesis.
SCHEME 1.

Attempted synthesis of Cbz-protected phosphinic dipeptide 6.
Phosphinic acid 3 was derived from aminomethyl phosphinate (2) (Scheme 1), which in turn was prepared from ammonium phosphinate using a previously described imine addition method.10 Other reported methods for the synthesis of 2 were less satisfactory.13 The main difficulty was in purifying highly polar fragment 2 especially on a preparative scale. The use of an ion-exchange resin described by Hammer and coworkers proved least problematic.10,14 A subsequent Cbz-protection of compound 2 followed by recrystallization from ethyl acetate/light petroleum ether7b,15 gave highly pure fragment 3 in 65% yield.
The adamantyl protection of 4 using AgNO3 and AdBr failed in our hands to provide protected dipeptide 5 (Scheme 1).8 However, esterification of 4 was achieved by treating the in situ generated phosphinic acid chloride with the sodium salt of adamantanol to give fully protected fragment 5a in 75% yield.8b,16 However, saponification of the ethyl ester unit of 5a by following a previously reported procedure8 was unsuccessful in our hands and led to a complex reaction mixture along with a significant amount of Cbz deprotection of the starting dipeptide after prolonged reaction time. Others have also noted the requirement of long reaction times for similarly substituted esters.17
This unsuccessful saponification led us to develop a new deprotection strategy. We reasoned that benzyl ester protection of the C-terminus would potentially allow for its removal under the same hydrogenation conditions required for unveiling of the amine. To test our hypothesis, we prepared benzyl ester fragment 7a (Scheme 2) following the same route as in the synthesis of 5a (Scheme 1). Catalytic hydrogenation of 7a with 5% Pd/C under hydrogen (atm. pressure) in the presence of Fmoc-OSu afforded desired pseudodipeptide 1a in good yields (Scheme 2). This one-pot reaction involved the simultaneous removal of the Cbz and benzyl protecting groups and reprotection of N-terminus with Fmoc. Optimal yields (>75%) were observed with a reaction time of 2 h, whereas prolonged exposure of the reaction mixture to hydrogenation resulted in partial Fmoc deprotection. Importantly, this method does not appear to be sensitive to the steric environment of the ester group. Following the same synthetic path as 5b, pseudo-Gly-Leu fragment 7b was also prepared and underwent efficient bis-deprotection and Fmoc addition (at a scale of 0.5 mmol) to give 1b (Scheme 2). We note that products 7a and 7b were obtained as a 1:1 mixture of diastereomers, which were easily separable by silica gel flash chromatography.
CONCLUSIONS
In conclusion, we have developed an efficient and scaleable method for the synthesis of Fmoc-protected phosphinic analogs of Gly-Val and Gly-Leu. Our modified synthetic route involves a highly abbreviated protecting group manipulation procedure that further optimizes previous work on this fragment that has found compelling applications in MMP inhibition.
Supplementary Material
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Egeblad M, Werb Z. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 2.Fingleton B. Curr Pharm Design. 2007;13:333–346. doi: 10.2174/138161207779313551. [DOI] [PubMed] [Google Scholar]
- 3.Overall CM, Kleifeld O. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 4.Vassiliou S, Mucha A, Cuniasse P, Georgiadis D, Lucet-Levannier K, Beau F, Kannan R, Murphy G, Knauper V, Rio MC, Basset P, Yiotakis A, Dive V. J Med Chem. 1999;42:2610–2620. doi: 10.1021/jm9900164. [DOI] [PubMed] [Google Scholar]
- 5.Buchardt J, Schiodt CB, Krog-Jensen C, Delaissé JM, Foged NT, Meldal M. J Comb Chem. 2000;2:624–638. doi: 10.1021/cc000031q. [DOI] [PubMed] [Google Scholar]
- 6.(a) Yiotakis A, Vassiliou S, Jiráček J, Dive V. J Org Chem. 1996;61:6601–6605. doi: 10.1021/jo9603439. [DOI] [PubMed] [Google Scholar]; (b) Buchardt J, Ferreras M, Krog-Jenson C, Delaissé JM, Foged NT, Meldal M. Chem Eur J. 1999;5:2877–2884. [Google Scholar]
- 7.Lauer-Fields J, Brew K, Whitehead JK, Li S, Hammer RP, Fields GB. J Am Chem Soc. 2007;129:10408–10417. doi: 10.1021/ja0715849.and references cited therein; Lauer-Fields J, Whitehead JK, Li S, Hammer RP, Brew K, Fields GB. J Biol Chem. 2008;283:20087–20095. doi: 10.1074/jbc.M801438200.
- 8.(a) Buchardt J, Ferreras M, Krog-Jenson C, Delaissé JM, Foged NT, Meldal M. Chem Eur J. 1999;5:2877–2884. [Google Scholar]; (b) Yamagishi T, Ichikawa H, Haruki T, Yokomatsu T. Org Lett. 2008;10:4347–4350. doi: 10.1021/ol801743d. [DOI] [PubMed] [Google Scholar]
- 9.Boyd EA, Regan AC, James K. Tetrahedron Lett. 1992;33:813–816. [Google Scholar]
- 10.Li S, Whitehead JK, Hammer RP. J Org Chem. 2007;72:3116–3118. doi: 10.1021/jo070266p. [DOI] [PubMed] [Google Scholar]
- 11.Georgiadis D, Matziari M, Yiotakis A. Tetrahedron. 2001;57:3471–3478. [Google Scholar]
- 12.Lee HS, Park JS, Kim BM, Gellman SH. J Org Chem. 2003;68:1575–1578. doi: 10.1021/jo026738b. [DOI] [PubMed] [Google Scholar]
- 13.(a) Baylis EK, Campbell CD, Dingwall JG. J Chem Soc Perkin Trans I. 1984:2845–2853. [Google Scholar]; (b) McCleery PP, Tuck Brian. J Chem Soc Perkin Trans I. 1989:1319–1329. [Google Scholar]; (c) Jiao XY, Verbruggen C, Borloo M, Bollaert W, Groot AD, Dommisse R, Haemers A. Synthesis. 1994;1:23–24. [Google Scholar]
- 14.In our hands, product 2 was eluted from the ion-exchange column using mere double-distilled water instead of ammonium hydroxide solution as previously reported.
- 15.Maligres PE, Houpis I, Rossen K, Molina A, Sager J, Upadhyay V, Wells KM, Reamer RA, Lynch JE, Askin D, Volante RP, Reider PJ. Tetrahedron. 1997;53:10983–10992. [Google Scholar]
- 16.Rogers RS. Tetrahedron Lett. 1992;33:7473–7474. [Google Scholar]
- 17.Takamura M, Sakurai M, Yamada E, Fujita S, Yachi M, Takagi T, Isobe A, Hagisawa Y, Fujiwara T, Yanagisawa H. Bioorg Med Chem. 2004;12:2419–2439. doi: 10.1016/j.bmc.2004.01.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.