

Abstract
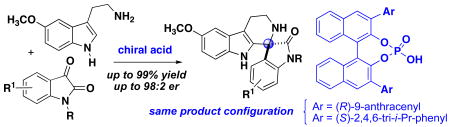
The condensation cyclization between isatins and 5-methoxy tryptamine catalyzed by chiral phosphoric acids provides spirooxindole tetrahydro-β-carboline products in excellent yields (up to 99%) and enantioselectivity (up to 98:2 er). A comparison of catalysts provides insight for the substrate scope and factors responsible for efficient catalytic activity and selectivity in the spirocyclization. Chiral phosphoric acids with different 3,3′-substitution on the binaphthyl system and opposite axial chirality afford the spiroindolone product with the same absolute configuration.
Keywords: Isatin, Pictet-Spengler, Chiral BrØnsted Acid Catalysis, Spirooxindole
Spirocyclic oxindoles are attractive targets for synthetic organic research due to their biological activity and occurrence as the core structure in natural products.1 This multicyclic scaffold is frequently utilized in medicinal chemistry where the chiral center and the spirocyclic fused core of these structures complements the flat heterocyclic compounds typically encountered in drug discovery programs. Thus, the use of catalytic asymmetric methods for the efficient synthesis of enantioenriched spirocycles provides an important synthetic challenge. Recent advances have provided efficient synthetic routes to access spirocyclic oxindoles containing both 5- and 6-membered ring-fused spirocyclic oxindoles.2 Several classes of spirooxindoles have been shown to exhibit stereospecific biological activity, e.g. MI-63, an MDM2 inhibitor,3 and NITD609, which has recently been identified as a potential treatment for Malaria based on in-vivo activity, having single dose efficacy in a rodent malaria model (Figure 1).4 Current methods often utilize expensive chiral column separations to obtain single enantiomer compounds, so asymmetric catalytic methods can provide an attractive alternative.
Figure 1. Examples of Biologically Active Spirooxindoles.

Here we report a chiral phosphoric acid-catalyzed enantioselective synthesis of functionalized spirooxindole tetrahydro-β-carbolines (spiroindolones) upon addition of tryptamines to isatins in a Pictet-Spengler type reaction.5 Several examples of enantioselective Pictet-Spengler reactions have been previously reported,6,7 but there is only one other example with isatins, recently reported while this research was in progress.8 This study provides a comparison for several catalyst systems to determine the factors responsible for efficient catalytic activity and selectivity in this reaction, and we have observed that the 3,3′-substitution on the binaphthyl chiral catalyst directs the enantioinduction for the spirocyclization.
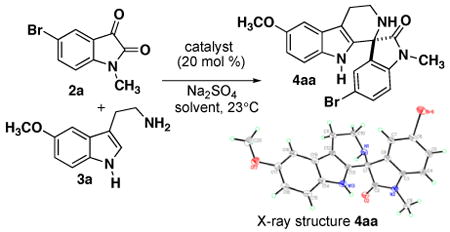
We initiated our investigations for the synthesis of spiroindolones by screening various catalysts for activation of chelating isatin 2a for spirocyclization with tryptamine 3a (Table 1). Initial screening was performed at 20 mol % of acid catalyst in the presence of Na2SO4. Previously in our group, chiral scandium(III) complexes have been shown to catalyze the nucleophilic addition of heteroaromatics to isatins.9 Here we found that scandium(III) chloride exhibits high catalytic activity, affording spirocycle 4aa in 99% yield (entry 1); however, when chiral complexes were investigated, only racemic products were obtained (entries 2-3). Using tin(II) chloride bisoxazoline complex also showed catalytic activity, but no enantioselectivity (entry 4). Because the Pictet-Spengler reaction is known to proceed through an iminium ion intermediate that is capable of forming an iminium-phosphate ion pair, we next investigated BrØnsted acids such as phosphinic acid (7) and chiral BINOL-derived phosphoric acid (S)-8a. These catalysts also promoted spirocycle formation in excellent yield, and (S)-8a afforded a small enantioenrichment for product 4aa (entries 5-6). This initial screening process demonstrates that various Lewis acid and BrØnsted acid catalysts readily promote the spirocyclization with isatins.
Table 1. Catalyst Optimization for Spiroindolone Formation.
 | ||||
|---|---|---|---|---|
| entry | catalyst | solvent | yield (%)[a] |
er (config)[b] |
| 1 | ScCl3 | CH2Cl2 | 99 | -- |
| 2 | ScCl3•5 | CH2Cl2 | 83 | 52:47 (R) |
| 3 | ScCl2(SbF6)•5 | CH2Cl2 | 99 | 54:46 (R) |
| 4 | SnCl2•6 | CH2Cl2 | 86 | 51:49 (R) |
| 5 | 7 | CH2Cl2 | 99 | -- |
| 6 | (S)-8a | CH2Cl2 | 99 | 58:42 (R) |
| 7 | (R)-8b | CH2Cl2 | 99 | 92:8 (S) |
| 8 | (R)-8b | CH3Ph | 91 | 80:20 (S) |
| 9 | (R)-8b | CH2Cl2/DMF[d] | 99 | 56:44 (S) |
| 10 | (R)-8b | DMF | 45 | 65:45 (S) |
| 11 | (R)-8b | EtOAc | 76 | 60:40 (S) |
| 12 | (R)-8c | CH2Cl2 | 76 | 76:24 (S) |
| 13 | (R)-8c | CH3CN | 86 | 85:15 (S) |
| 14 | (R)-8c | THF | 80 | 80:20 (S) |
| 15 | (R)-8c | CHCl3 | 77 | 60:40 (S) |
| 16 | (S)-8d | CH2Cl2 | nd | 73:23 (S) |
| 17 | (S)-8d | CH3Ph | 92 | 82:18 (S) |
| 18 | (S)-8d | CH3Ph[e] | 90 | 83:17 (S) |
| 19 | (S)-8d | CH2Cl2/DMF[c] | 90 | 80:20 (S) |
| 20 | (S)-8d | DMF | 70[d] | 82:18 (S) |
| 21 | (S)-8e | CH2Cl2 | 99 | 68:32 (S) |
| 22 | 9a | CH2Cl2 | 99 | 75:25 (R) |
| 23 | 9a | CH3Ph | 60 | 76:24 (R) |
| 24 | 9b | CH2Cl2 | NR | -- |
| 25 | (R)-8b/MgF2 | CH3Ph | 85 | 77:23 (S) |
Reactions performed with 1.2 equiv of isatin under argon. Reactions run from 48-96h.
Isolated yield.
Enantioselectivity determined by HPLC analysis using chiral stationary phase.
4:1 DCM/DMF.
40°C.
Reaction performed at -15°C.
We proceeded to compare a series of optically-pure BINOL-derived phosphoric acid catalysts and conditions to optimize the enantioselectivity.10 Overall, high yields were observed with all phosphoric acid catalysts (8a-e), but anthracenyl catalyst (R)-8b in DCM was identified as the optimal catalyst, giving the highest enantioselectivity for the model reaction with 5-bromo-N-methylisatin (entry 7). In the process of optimization, the effect of solvent and temperature were examined for the phosphoric acid catalyzed reactions in order to improve the enantioselectivity. A variety of solvents were investigated and the choice of solvent proved to be important for the rate and enantioselectivity (entries 7-21). The use of DCM afforded high 98:2 enantioselectivity with catalyst (R)-8b while reduced enantioselectivity was observed with other solvents tested. With catalysts (S)-8c and (S)-8d, the use of several different solvents (e.g. DCM, toluene, and DMF) provided consistent, albeit moderate, enantioselectivity. Investigating lower reaction temperatures with (S)-8d did not improve the enantioselectivity (entry 18). The structure and absolute configuration of spiroindolone 4aa was confirmed to be the (S)-enantiomer by X-ray crystallographic analysis.11 It is particularly notable that the same (S)-enantiomer of spiroindolone was obtained for catalysts (R)-8b-c and (S)-8d-e, despite having opposite configurations of axial chirality.12 Thus, the substituents on the binaphthyl system (e.g. anthracenyl vs. 2,4,6-tri-1-Pr-phenyl) direct the stereoinduction for the spirocyclization. Several instances have been reported previously where the 3,3′-substitution on the chiral phosphoric acid catalyst has been shown to reverse the sense of enantioselection.10,13
Based on results reported for previous thiourea-catalyzed enantioselective Pictet-Spengler reactions with aldehydes,6 the catalytic activity and enantioselectivity for two thiourea catalysts (9) were also investigated for isatins. Although high catalytic activity was observed with thiourea 9a, only modest enantioselectivity for the (R)-spiroindolone was obtained in both DCM and toluene (entries 22, 23). Thiourea 9b showed no catalytic activity in this reaction (entry 24), which indicates that the amide group of the thiourea catalyst must play an important role for the mechanism of this reaction. We also investigated the potential for dual catalysis using Lewis acid enhanced BrØnsted acidity in order to enhance the reaction rate,14 but this led to a significant decrease in the enantioselectivity compared to the use of only phosphoric acid (R)-8b (entry 25).
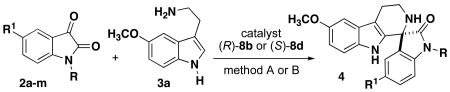
Using the optimized conditions with catalyst (R)-8b, the scope of the isatin electrophile was examined with 5-methoxy tryptamine 3a (Table 2). We also investigated the (S)-TRIP catalyst 8d, previously reported by Bencivenni and coworkers,8 as a second method for comparison between catalysts (S)-8d and (R)-8b to understand the effect of catalyst with respect to substrate scope and the observed enantioinversion. A variety of N-alkylated and NH isatins are successful in this reaction with moderate to high enantioselectivity for both catalyst systems. Based on the optimization of conditions with the 5-bromo-N-methylisatin, method A conditions with catalyst (R)-8b give improved enantioselectivity for halide-substituted isatins relative to catalyst (S)-8d. The DMF conditions of method B are particularly well-suited for NH isatins, which we attribute to solubility. Generally, the formation of the imine intermediate was rarely observed and not isolated for this reaction. One exception is based on investigations of the N-acetyl isatin, which afforded only the imine product with trace amount of spirocyclization (entry 10). The N-propargyl spiroindolone can also be accessed in good yield and enantioselectivity (entry 8), providing an alkyne functional handle for further diversification.
Table 2. Scope of Isatins for Phosphoric acid-catalyzed Spirocyclization.
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | R1 | product | method | yield (%)[a] | er[b] |
| 1 | CH3 | Br | 4aa | A | 99 | 91:9 |
| B | 70 | 82:18 | ||||
| 2 | CH3 | Cl | 4ba | A | 99 | 92:8 |
| B | 90 | 88:12 | ||||
| 3 | CH3 | H | 4ca | A | 99 | 88:12 |
| B | 99 | 95:5 | ||||
| 4 | CH3 | OCH3 | 4da | A | 99 | 58:42 |
| B | 79 | 70:30 | ||||
| 5 | Bn | H | 4ea | A | 99 | 82:18 |
| B | 95 | 97:3 | ||||
| 6 | PMB | H | 4fa | A | 72 | 87:13 |
| B | 87 | 93:7 | ||||
| 7 | PMB | Br | 4ga | A | 86 | 87:13 |
| B | 81 | 75:25 | ||||
| 8 | CH2CCH | F | 4ha | A | 60 | 92:8 |
| B | 86 | 92:8 | ||||
| 9 | Ph | H | 4ia | A | 99 | 85:15 |
| B | 44 | 97:3 | ||||
| 10 | Ac | H | 4ja | A | 87[c] | -- |
| B | 53[c] | -- | ||||
| 11 | H | H | 4ka | A | 66 | 94:6 |
| B | 99 | 94:6 | ||||
| 12 | H | Cl | 4la | A | 99 | 84:16 |
| B | 88 | 80:20 | ||||
| 13 | H | OCH3 | 4ma | A | 84 | 97:3 |
| B | 92 | 96:4 | ||||
Method A: 20 mol % catalyst (R)-8b in CH2Cl2 at 23°C for 72-96h.
Method B: 10 mol % catalyst (S)-8d in DMF at 40°C for 24-48h.
Isolated yield.
Enantioselectivity determined by chiral phase HPLC.
Yield is for imine.
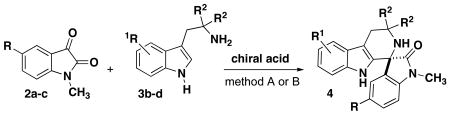
The scope of the tryptamine was also examined for spirocyclization, providing insight into the limitations of this methodology (Table 3). While the 5-methoxy tryptamine (Table 2) and unsubstituted tryptamine both proceed with high yield and moderate to high enantioselectivity, the enantioselectivity of the spirocyclization deteriorates with other substitution patterns. The position of the methoxy group on tryptamine also effects the level of enantioslectivity, where the 6-methoxytryptamine proceeds with poor enantioselectivity (60:40 er) relative to the 5-methoxy tryptamine (92:8 er) (Table 3 entries 1-2 vs Table 2, entry 2). Although the addition of unsubstituted tryptamines to isatin have been previously investigated by Bencivenni and coworkers,8 we found that halide substitution at the 5-position of N-methyl isatin leads to an errosion of selectivity in comparison with the unsubstitiuted methyl isatin (entries 3-6). The geminally-disubstituted tryptamine 3d was also investigated because this substrate has been utilized previously to promote cyclization with aldehydes by exploiting the Thorpe-Ingold effect.7a While tryptamine 3d was surprisingly unreactive with catalyst (R)-8b and (S)-8d, using catalyst (S)-8e afforded high yields of product, albeit with minimal enantioselectivity (entries 7-9). This result indicates that the steric effects of the geminal disubstitution may inhibit formation of the iminium-phosphate ion complex necessary to induce asymmetry, although product formation is still observed with a more acidic catalyst.15
Table 3. Tryptamine Investigation for Phosphoric Acid-catalyzed Spirocyclization.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | product | R | R1 | R2 | method | chiral acid | yield %[a] | er[b] |
| 1 | 4bb | Cl | 6-OCH3 | H | A | 8b | 99 | 60:40 |
| 2 | 4bb | Cl | 6-OCH3 | H | B | 8d | 39 | 80:20 |
| 3 | 4bc | Cl | H | H | A | 8b | 93 | 67:33 |
| 4 | 4bc | Cl | H | H | B | 8d | 89 | 65:35 |
| 5 | 4cb | H | H | H | A | 8b | 35 | 95:5 |
| 6 | 4cb | H | H | H | B | 8d | 71 | 95:5 |
| 7 | 4bd | Cl | H | CO2CH3 | A | 8b | no rxn | -- |
| 8 | 4ad | Br | H | CO2CH3 | B | 8d | 21 | 52:48 |
| 9 | 4ad | Br | H | CO2CH3 | A | 8e | 92 | 59:41 |
Method A: 20 mol % catalyst in CH2Cl2 at 23°C for 72-96h.
Method B: 10 mol % catalyst in DMF at 40°C for 24-48h.
Isolated yield.
Enantioselectivity determined by chiral phase HPLC.
The proposed mechanism for the spiroindolone synthesis is initiated by formation of an iminium-phosphate ion complex with the chiral phosphoric acid catalyst (Scheme 1).5 Intramolecular attack of the nucleophilic indole from the 2-position leads to spirocyclization and formation of the new chiral carbon center. The resulting indolenium ion undergoes an elimination to restore aromaticity, generating spiroindolone 4 and regenerating the catalyst. We have noted that using different 3,3′-substitution on the binaphthyl system with the opposite axial chiral configuration (e.g. (S)-8d and (R)-8b) affords the spiroindolone with the same absolute configuration. Therefore, the same axial configuration of catalyst can afford an inversion in the sense of enantioselection for the spirocyclization. This inversion is attributed to the steric interactions that exist upon formation of the iminium-phosphate ion complex with the 9-anthracenyl catalyst (R)-8b, thereby reversing the approach of the indole to the iminium ion.
Scheme 1. Catalytic Cycle for Spirocyclization.

In conclusion, we have developed a catalytic asymmetric method for accessing a variety of functionalized spiroindolones from isatins, including halide substitution patterns found in the anti-malaria lead compound NITD609. A comparison of the 9-anthracenyl catalyst (R)-8b and the (S)-TRIP catalyst (8d) provides insight into the substrate scope as well as the effect of different solvent and catalyst combinations that are needed in order to obtain optimal yields and enantioselectivities. The (S)-spiroindolone product is obtained for both the (S)-TRIP and (R)-anthracenyl catalysts, which indicates that the substitution on the binaphthyl system directs the sense of enantioselection. Transition state studies to investigate this observed inversion are currently in progress and will be reported in due course.
Supplementary Material
Figure 2. Structures of Chiral Ligands and Catalysts Investigated.

Acknowledgments
This research is supported by funds from the University of California, Davis, the donors of the American Chemical Society Petroleum Research Fund, and NIH/NIGMS (P41-GM0089153). We thank the National Science Foundation (Grant 0840444) for the Dual source diffractometer. A.K.F. acknowledges 3M Corporation for a Nontenured Faculty Award, J.J.B. acknowledges support from the NSF in the form of a graduate research fellowship, and A.S. thanks Bristol-Myers Squibb for an undergraduate research fellowship. Special thanks to Dr. James Fettinger for solving X-ray structures.
Footnotes
Supporting Information Available: Spectral data for all new compounds and X-ray crystal structure for 4aa and 4ha. This material is available free of charge via the Internet at www.elsevier.com.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Badillo JJ, Hanhan NV, Franz AK. Curr Opin Drug Discovery Devel. 2010;13:758–776. [PubMed] [Google Scholar]; (b) Vintonyak 5, Warburg K, Kruse H, Grimme S, Hübel K, Rauh D, Waldmann H. Angew Chem, Int Ed. 2010;49:5902–5905. doi: 10.1002/anie.201002138. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Brennan MK. Synthesis. 2009:3003–3025. [Google Scholar]; (d) Galliford CV, Scheidt KA. Angew Chem, Int Ed. 2007;46:8748–8758. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]; (e) Yong SR, Ung AT, Pyne SG, Skelton BW, White AH. Tetrahedron. 2007;63:5579–5586. [Google Scholar]
- 2.(a) Chen XH, Wei Q, Luo SW, Xiao H, Gong LZ. J Am Chem Soc. 2009;131:13819–13825. doi: 10.1021/ja905302f. [DOI] [PubMed] [Google Scholar]; (b) Jiang 10, Cao Y, Wang Y, Liu L, Shen F, Wang R. J Am Chem Soc. 2010;132:15328–15333. doi: 10.1021/ja106349m. [DOI] [PubMed] [Google Scholar]; (c) Wang J, Crane EA, Scheidt KA. Org Lett. 2011;13:3086–3089. doi: 10.1021/ol200987c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tan B, Candeias NR, Barbas CF. J Am Chem Soc. 2011;133:4672–4675. doi: 10.1021/ja110147w. [DOI] [PubMed] [Google Scholar]; (e) Badillo JJ, Arevalo GE, Fettinger JC, Franz AK. Org Lett. 2011;13:418–421. doi: 10.1021/ol1027305. [DOI] [PubMed] [Google Scholar]; (f) Chen WB, Wu ZJ, Hu J, Cun LF, Zhang XM, Yuan WC. Org Lett. 2011;13:2472–2475. doi: 10.1021/ol200724q. [DOI] [PubMed] [Google Scholar]; (g) Tan B, Hernandez-Torres G, Barbas CF. J Am Chem Soc. 2011;133:12354–12357. doi: 10.1021/ja203812h. [DOI] [PubMed] [Google Scholar]; (h) Tan B, Candeias NR, Barbas CF. Nat Chem. 2011;3:473–477. doi: 10.1038/nchem.1039. [DOI] [PubMed] [Google Scholar]
- 3.Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 4.(a) Yeung BKS, Zou B, Rottmann M, Lakshminarayana SB, Ang SH, Leong SY, Tan J, Wong J, Keller-Maerki S, Fischli C, Goh A, Schmitt EK, Krastel P, Francotte E, Kuhen K, Plouffe D, Henson K, Wagner T, Winzeler EA, Petersen F, Brun R, Dartois 5, Diagana TT, Keller TH. J Med Chem. 2010;53:5155–5164. doi: 10.1021/jm100410f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois 5, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For recent reviews of Pictet-Spengler reactions, see: Stöckigt J, Antonchick AP, Wu F, Waldmann H. Angew Chem, Int Ed. 2011 doi: 10.1002/anie.201008071. asap.Pulka K. Curr Opin Drug Discovery Devel. 2010;13:669–684.Cox ED, Cook JM. Chem Rev. 1995;95:1797–1842.
- 6.(a) Taylor MS, Jacobsen EN. J Am Chem Soc. 2004;126:10558–10559. doi: 10.1021/ja046259p. [DOI] [PubMed] [Google Scholar]; (b) Raheem IT, Thiara PS, Jacobsen EN. Org Lett. 2008;10:1577–1580. doi: 10.1021/ol800256j. [DOI] [PubMed] [Google Scholar]; (b) Klausen RS, Jacobsen EN. Org Lett. 2009;11:887–890. doi: 10.1021/ol802887h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Seayad J, Seayad AM, List B. J Am Chem Soc. 2006;128:1086–1087. doi: 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]; (b) Sewgobind NV, Wanner MJ, Ingemann S, de Gelder R, van Maarseveen JH, Hiemstra H. J Org Chem. 2008;73:6405–6408. doi: 10.1021/jo8010478. [DOI] [PubMed] [Google Scholar]; (c) Bou-Hamdan FR, Leighton JL. Angew Chem, Int Ed. 2009;48:2403–2406. doi: 10.1002/anie.200806110. [DOI] [PubMed] [Google Scholar]; (d) Muratore ME, Holloway CA, Pilling AW, Storer RI, Trevitt G, Dixon DJ. J Am Chem Soc. 2009;131:10796–10797. doi: 10.1021/ja9024885. [DOI] [PubMed] [Google Scholar]; (e) Holloway CA, Muratore ME, Storer RI, Dixon DJ. Org Lett. 2010;12:4720–4723. doi: 10.1021/ol101651t. [DOI] [PubMed] [Google Scholar]; (f) Fang H, Wu 10, Nie L, Dai 10, Chen J, Cao W, Zhao G. Org Lett. 2010;12:5366–5369. doi: 10.1021/ol101922h. [DOI] [PubMed] [Google Scholar]
- 8.While this work was in progress, Bencivenni and coworkers described a related Pictet-Spengler reaction of isatins. In these reports, catalyst (R)-8b was not reported and different substrates and scope were investigated, see: Duce S, Pesciaioli F, Gramigna L, Bernardi L, Mazzanti A, Ricci A, Bartoli G, Bencivenni G. Adv Synth Catal. 2011;353:860–864.
- 9.Hanhan NV, Sahin AH, Chang TW, Fettinger JC, Franz AK. Angew Chem, Int Ed. 2010;49:744–747. doi: 10.1002/anie.200904393. [DOI] [PubMed] [Google Scholar]
- 10.For a recent review of chiral phosphoric acid catalysis, see: Terada M. Synthesis. 2010:1929–1982.
- 11.This X-ray structure confirms the absolute configuration that was previously determined as the (S)-enantiomer by simulation of the electronic circular (ECD) dichroism spectra, see reference 8.
- 12.The absolute configurations for chiral phosphoric acids in this study are based on products purchased from Sigma-Aldrich. (R)-3,3′-Bis(9-anthracenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate is product #695718 and lot# MKBG2357V was used for these studies with an optical rotation of +139.3 (c = 1%, chloroform), as provided by Sigma-Aldrich. Akiyama and co-workers report [α]D26 −24.9 (c 1.00, EtOH), for the (R)-anthracenyl acid, see: Akiyama T, Morita H, Fuchibe K. J Am Chem Soc. 2006;128:13070–13071. doi: 10.1021/ja064676r.
- 13.(a) Terada M, Yokoyama S, Sorimachi K, Uraguchi D. Adv Synth Catal. 2007;349:1863–1867. [Google Scholar]; (b) Wanner MJ, Hauwert P, Schoemaker HE, de Gelder R, van Maarseveen JH, Hiemstra H. Eur J Org Chem. 2008:180–185. [Google Scholar]; (c) Sheng YF, Gu Q, Zhang AJ, You SL. J Org Chem. 2009;74:6899–6901. doi: 10.1021/jo9013029. [DOI] [PubMed] [Google Scholar]; (d) Yu 10, Lu A, Wang Y, Wu G, Song H, Zhou Z, Tang C. Eur J Org Chem. 2011:892–897. doi: 10.1021/jo2002825. [DOI] [PubMed] [Google Scholar]
- 14.(a) Yamamoto H, Futatsugi K. Angew Chem, Int Ed. 2005;44:1924–1942. doi: 10.1002/anie.200460394. [DOI] [PubMed] [Google Scholar]; (b) Lv J, Li 10, Zhong L, Luo S, Cheng JP. Org Lett. 2010;12:1096–1099. doi: 10.1021/ol1000928. [DOI] [PubMed] [Google Scholar]; (c) Rueping M, Koenigs RM, Atodiresei 1. Chem Eur J. 2010;16:9350–9365. doi: 10.1002/chem.201001140. [DOI] [PubMed] [Google Scholar]
- 15.We have also investigated reactions of racemic α- and β-substituted tryptamines to determine if a chiral resolution and stereospecific cyclization would occur. In both cases, moderate yield is observed, and only racemic products were obtained resulting from the diastereoselective nature of the reaction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.