

Abstract
Parasitic diseases, such as African sleeping sickness, have a significant impact on the health and well-being in the poorest regions of the world. Pragmatic drug discovery efforts are needed to find new therapeutic agents. In this report we describe target repurposing efforts focused on trypanosomal phosphodiesterases. We outline the synthesis and biological evaluation of analogs of sildenafil (1), a human PDE5 inhibitor, for activities against trypanosomal PDEB1 (TbrPDEB1). We find that, while low potency analogs can be prepared, this chemical class is a sub-optimal starting point for further development of TbrPDE inhibitors.
Keywords: Neglected disease, Trypanosoma brucei, Target repurposing, Phosphodiesterase inhibitors, TbrPDEB1, PDE5, sildenafil
Tropical diseases such as human African trypanosomiasis (HAT), leishmaniasis, and Chagas’ disease are often referred to as “neglected” diseases, as the low level of funding of drug discovery research is disproportionate to the total disease burden. For example, collectively these three diseases endanger over 73 million people globally1 and create a burden of 4.5 million disability-adjusted life-years.2,3 Yet, few compounds have advanced to clinical studies for these diseases.4 The primary reason for this disparity is the high cost of drug discovery and unlikelihood that these research costs will be recouped by a research organization via drug sales to highly impoverished populations.
As a result, cost-effective methods for identification of lead matter and expediting compound optimization efforts are acutely needed. We apply the method of “target repurposing”, wherein essential parasite enzymes are matched with proven, druggable human homologs.4 By doing so, the research outputs of large and expensive programs of historical drug discovery projects can be used to initiate and drive optimization projects against parasitic targets.
Phosphodiesterases (PDEs) are a family of enzymes that maintain the balance of cAMP and cGMP in the cell, opposed to adenylate and guanylate cyclase, respectively. Humans possess eleven PDEs, several of which have been fruitfully pursued for drug discovery. The most well-known of these is PDE5, an enzyme that is inhibited by erectile dysfunction drugs such as Viagra™ (sildenafil, 1), Cialis™ (tadalafil, 2), and Levitra™ (vardenafil, 3), Figure 1. Other PDEs are of demonstrated relevance to inflammatory conditions and CNS indications, such as schizophrenia.5–7 Trypanosoma brucei, the causative agent of HAT, expresses five cAMP-specific PDEs,8 two of which (TbrPDEB1 and TbrPDEB2) have together been shown by both RNAi9,10 and small molecules10,11 to be essential for parasite proliferation. While initial benchmarking and early optimization experiments have uncovered a susceptibility of TbrPDEB1 and B2 to human PDE4 inhibitors,11 the two trypanosomal enzymes are also 31% identical to PDE5 within their respective catalytic domains, and previous reports cited low potency of 1 and 2 against the trypanosomal enzymes; thus, we describe here early structure-activity relationships (SAR) of (1), and report such work for 2 in a subsequent article.12
Figure 1.

Human PDE5 inhibitors marketed for erectile dysfunction.
We initially tested 1 for its inhibitory activity against the catalytic domain of TbrPDEB1, and observed low potency (51% at 100 µM). However, we deemed this chemotype worth further exploration given the extensive knowledge of SAR and structural biology around it. We performed activity scoping experiments by making strategic changes to the structure of 1, to explore the impact of variation of the N-methyl group and propyl chain on the pyrazolopyrimidinone core (Region A), and of the specific functionality on Region B. We were particularly interested in exploring the western portion of 1, as this region of the molecule was proposed to project into a parasite-specific pocket (the P-pocket). This pocket was first observed in the Leishmania major phosphodiesterase LmjPDEB113 and is predicted to also exist in TbrPDEB1,11 but, importantly, is absent from all human PDEs.
Compounds that explore Regions A and B were synthesized using the routes outlined in Schemes 1 and 2. The known aminopyrazole 4a14 or 4b15 was acylated with the appropriate benzoic acid and cyclized under basic conditions to give 5–10. Pyrazole N-arylation was achieved using copper catalysis16 to prepare 11–13. Alkylation of 7 with bromoacetamide provided 14.
Scheme 1.

Synthesis of 5–14. Reagents and Conditions. (a) PyBroP, TEA, DCE, 120 °C, MW, 10 min; (b) NH4OH, dioxane, rt; (c) NaOEt/EtOH, 120 °C, MW, 10 min; (d) R1-I, CuI, trans-cyclohexane-1,2-diamine, Cs2CO3,DMF, 110 °C; (e) NaH, 2-bromoacetamide, 0 °C to rt. Refer to Table 1 for the identity of R-groups.
Scheme 2.

Synthesis of 20–22. Reagents and Conditions.(a) CDI, A,70 °C, EtOAc, o/n; (b) PyBroP, A, Et3N, DCE, MW 120 °C 20 min; (c) SOCl2; (d) NH3, iPrOH; (e) NaOEt, EtOH, MW 120 °C 10 min.
The preparation of Strategy B analogs of compound 1 is illustrated in Scheme 2. The appropriate aminopyrazole 15,17 16,18 or 1719 is acylated with A using either CDI or PyBroP; these reaction conditions surprisingly resulted in the partial-to-complete hydrolysis of the primary amide (of 14 and 15) or ester (of 16). Thus, the resulting carboxylic acid 18 was converted to the primary amide 19 via treatment with thionyl chloride, followed by ammonia in isopropanol. Cyclization to the desired products was effected under basic conditions.
Following synthesis, the analogs were tested in biochemical assays11 at a single concentration. Notably, with one exception (7), none of the analogs that varied the pyrazole N1 substituent (H, Me, 3-pyridyl, or acetamide) nor the C3 position (H, Me, Pr, Ph) showed improved potency over 1.
The removal of the N-methylpiperazinyl sulfonamide head group resulted in compounds with significant loss in solubility, and as such biochemical screening data was not possible with some analogs (Table 1, entries 8, 12–14).
Table 1.
Results of biochemical screening of analogs of 1.
| Entry | Compd | R1 | R2 | R3 | R4 | TbrPDEB1 (%inh)a |
|---|---|---|---|---|---|---|
| 1 | 1 | CH3 | Pr | OEt | 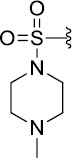 |
51.5 |
| 2 | 7 | H | Ph | OEt | 70.9 | |
| 3 | 13 | 3-Pyr | Pr | OEt | 32.4 | |
| 4 | 22 | CH3 | Ph | OEt | 17.5 | |
| 5 | 10 | H | Pr | OEt | 16.0 | |
| 6 | 20 | CH3 | H | OEt | 13.6 | |
| 7 | 8 | CH3 | Pr | H | 8.4 | |
| 8 | 21 | CH3 | CH3 | OEt | NDb | |
| 9 | 6 | H | Ph | OEt | H | 22.9 |
| 10 | 9 | CH3 | Pr | OEt | H | 21.8 |
| 11 | 14 | CH2CONH2 | Ph | OEt | H | 7 |
| 12 | 11 | Ph | Ph | OEt | H | NDb |
| 13 | 12 | 3-Pyr | Ph | OEt | H | NDb |
| 14 | 5 | H | Pr | OEt | H | NDb |
Standard assay conditions: 100µM, 10% DMSO.
Compound showed lack of solubility, which precluded testing.
A broad exploration of heterocyclic substitutions in Region B was undertaken by application of parallel synthesis (Scheme 3). This was achieved by condensing the commercially available pyrazole amino amide 23 with various monocyclic heteroaromatic carboxylic acids that were available in pre-weighed quantities from a commercial vendor (ASDI, Inc). Following this amidation reaction, cyclization was achieved by treatment with sodium ethoxide in ethanol.
Scheme 3.

Synthesis of 24a–d. Reagents and Conditions. (a) R5CO2H, imidazole-HCl, CDI, then 23, 50 °C, o/n;. b) EtOH, NaOEt, 120 °C, 2 h. Refer to Table 2 for the identity of R-groups.
A series of 2-alkoxy-3-pyridyl variations were prepared as shown in Scheme 4. The acylated aminopyrazole 25 was cyclized under basic conditions, using an appropriate alcoholic solvent, which yielded the 2-alkoxypyridyl derivatives 26.
Scheme 4.

Synthesis of 26a–c. Reagents and Conditions. a) 2-chloronicotinic acid, PyBroP, Et3N; (b) R-OH, KOtBu.
Table 2 summarizes the biochemical results for analogs prepared using this parallel methodology that display >25% inhibition at 100 µM (characterization and biochemical data for all compounds are listed in the Supporting Information). In brief, heterocycles show improved potency at TbrPDEB1, specifically, 2-methoxy-3-pyridyl and 3-methyl-2-pyridyl show the best potency (entries 1–2); larger 2-alkoxy groups (entries 3 and 4) lead to reduced activity. Unfortunately, dose-response experiments with 26c were precluded by poor solubility at higher concentrations.
Table 2.
Analogs of sildenafil (Strategy A).
| # | |||
|---|---|---|---|
| Compound | R5 | TbrPDEB1 (%inh)a |
|
| 1 | 26c | 2-methoxy-3-pyridyl | 77.2 |
| 2 | 24a | 3-methyl-2-pyridyl | 72.1 |
| 3 | 26a | 2-isopropoxy-3-pyridyl | 53.8 |
| 4 | 26b | 2-ethoxy-3-pyridyl | 41.5 |
| 5 | 24b | 2-methyl-5-pyrazinyl | 30.0 |
| 6 | 24c | 2,4-dimethyloxazol-5-yl | 29.0 |
| 7 | 24d | thiazol-4-yl | 25.4 |
Standard assay conditions: 100µM, 10% DMSO
While we appreciate that the potencies of the compounds shown in Tables 1 and 2 are modest, it is clear that 7, 26c and 24a represent qualitative potency improvement over 1. Nonetheless, the current exploration of the various regions of the sildenafil template is not suggestive of promising structure-activity relationship trends.
In conclusion, with the broad interest in repurposing PDE inhibitors for neglected diseases11,20,21 and the extensive precedent in medicinal chemistry for human PDE5 inhibitors, we note our finding that these initial sildenafil analogs do not represent as strong starting points for further optimization for TbrPDEB inhibition as the PDE4 chemotypes previously reported.11 This information may be deemed important for reprioritizing the focus of these efforts.
Supplementary Material
Figure 2.

Strategy for exploration of the sildenafil core.
Acknowledgments
This work was supported by the National Institutes of Health (R01AI082577), Boston University and Northeastern University. We gratefully acknowledge a donation of sildenafil 1 from Pfizer, Inc. and a free academic license to the OpenEye suite of software.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information
Full tabulation of all inhibitors prepared and screened in the context of this project is included in the supporting information, along with spectroscopic characterization of new compounds. Screening data will be made publically available as a public data set in the Collaborative Drug Discovery database (www.collaborativedrug.com).
References
- 1.Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. J. Clin. Invest. 2008;118:1301. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization; [Accessed 01/23/2012]. http://www.who.int/healthinfo/global_burden_disease/2004_report_update/en/index.html. [Google Scholar]
- 3.World Health Organization; [Accessed 01/23/2012]. http://www.who.int/healthinfo/global_burden_disease/metrics_daly/en/ [Google Scholar]
- 4.Pollastri MP, Campbell RK. Future Med. Chem. 2011;3:1307. doi: 10.4155/fmc.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menniti FS, Chappie TA, Humphrey JM, Schmidt CJ. Curr. Opin. Investig. Drugs. 2007;8:54. [PubMed] [Google Scholar]
- 6.Menniti FS, Faraci WS, Schmidt CJ. Nat. Rev. Drug. Discov. 2006;5:660. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 7.Lugnier C. Pharmacol. Therap. 2006;109:366. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Gould MK, de Koning HP. FEMS Microbiol Rev. 2011 doi: 10.1111/j.1574-6976.2010.00262.x. [DOI] [PubMed] [Google Scholar]
- 9.Oberholzer M, Marti G, Baresic M, Kunz S, Hemphill A, Seebeck T. FASEB J. 2007;21:720. doi: 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- 10.Shakur Y, de KHP, Ke H, Kambayashi J, Seebeck T. Handb. Exp. Pharmacol. 2011:487. doi: 10.1007/978-3-642-17969-3_20. [DOI] [PubMed] [Google Scholar]
- 11.Bland ND, Wang C, Tallman C, Gustafson AE, Wang Z, Ashton TD, Ochiana SO, McAllister G, Cotter K, Fang AP, Gechijian L, Garceau N, Gangurde R, Ortenberg R, Ondrechen MJ, Campbell RK, Pollastri MP. J. Med. Chem. 2011;54:8188. doi: 10.1021/jm201148s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ochiana SO, Gustafson A, Bland ND, Wang C, Campbell RK, Pollastri MP. Bioorg. Med. Chem. Lett. 2011 doi: 10.1016/j.bmcl.2012.01.118. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Yan Z, Geng J, Kunz S, Seebeck T, Ke H. Mol. Microbiol. 2007;66:1029. doi: 10.1111/j.1365-2958.2007.05976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baxendale IR, Ley SV. Bioorg. Med. Chem. Lett. 2000;10:1983. doi: 10.1016/s0960-894x(00)00383-8. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, He X, Choi H-S, Yang K, Woodmansee D, Wang Z, Ellis DA, Wu B, He Y, Nguyen TN. WO2006047516A2. [Google Scholar]
- 16.Antilla JC, Baskin JM, Barder TE, Buchwald SL. J. Org. Chem. 2004;69:5578. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]
- 17.DeWald HA, Beeson NW, Hershenson FM, Wise LD, Downs DA, Heffner TG, Coughenour LL, Pugsley TA. J. Med. Chem. 1988;31:454. doi: 10.1021/jm00397a032. [DOI] [PubMed] [Google Scholar]
- 18.Yuan J, Gulianello M, De LS, Brodbeck R, Kieltyka A, Hodgetts KJ. Bioorg. Med. Chem. Lett. 2002;12:2133. doi: 10.1016/s0960-894x(02)00358-x. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher SR, Menet CJM, Dykes GJ, Merayo MN, Schmitt BA. WO2008071650A2. [Google Scholar]
- 20.Seebeck T, Sterk GJ, Ke H. Future Med. Chem. 2011;3:1289. doi: 10.4155/fmc.11.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beghyn TB, Charton J, Leroux F, Laconde G, Bourin A, Cos P, Maes L, Deprez B. J. Med. Chem. 2011;54:3222. doi: 10.1021/jm1014617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.