

Abstract
Prostate cancer remains the most commonly diagnosed malignancy and the second leading cause of cancer-related deaths in men in the United States. The current standard of care consists of prostatectomy and radiation therapy, which may often be supplemented with hormonal therapies. Recurrence is common, and many develop metastatic prostate cancer for which chemotherapy is only moderately effective. It is clear that novel therapies are needed for the treatment of the malignant forms of prostate cancer that recur after initial therapies, such as hormone refractory (HRPC) or castration resistant prostate cancer (CRPC). With advances in understanding of the molecular mechanisms of cancer, we have witnessed unprecedented progress in developing new forms of targeted therapy. Several targeted therapeutic agents have been developed and clinically used for the treatment of solid tumors such as breast cancer, non-small cell lung cancer, and renal cancer. Some of these reagents modulate growth factors and/or their receptors, which are abundant in cancer cells. Other reagents target the downstream signal transduction, survival pathways, and angiogenesis pathways that are abnormally activated in transformed cells or metastatic tumors. We will review current developments in this field, focusing specifically on treatments that can be applied to prostate cancers. Finally we will describe aspects of the future direction of the field with respect to discovering biomarkers to aid in identifying responsive prostate cancer patients.
Keywords: Prostate cancer, EGFR, IGF-1R, VEGFR, androgen, hormone resistant
1. Introduction
Currently, prostate cancer is the most commonly diagnosed malignancy and the second leading cause of cancer-related deaths in men in the United States. Based on 2010 estimates, over 200,000 new cases of prostate cancer will be diagnosed and more than 30,000 men will die from this disease annually. The mortality due to prostate cancer has steadily declined for a decade, with a 4% decrease per year between 1999 and 2003, which among several factors, may be attributable to earlier detection and improved treatment of cancer.
Treatment of localized prostate cancer usually includes prostatectomy and radiation therapy, sometimes supplemented with hormonal therapies (either pharmacological anti-androgens and luteinizing hormone-releasing hormone (LH-RH) agonists that prevent testosterone production, or orchiectomy, the surgical removal of testicles) (Fig. 1). Recurrence, usually defined by the observation of surging prostate specific antigen (PSA) levels, occurs in about 15% patients within 5 years after prostatectomy and in about 40% patients within 10 years [1]. Following recurrence, more than 70% of patients can still expect to survive for more than 10 years [2].
Figure 1. Prostate cancer's progression and treatment.
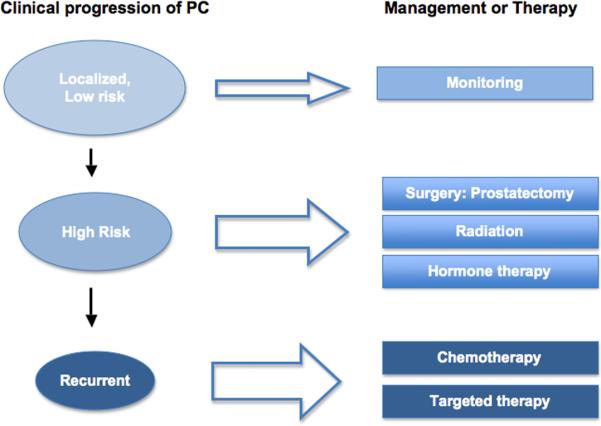
Prostate cancer patients with high risk are recommended to have surgery, radiation, and/or hormone therapies. Although these treatments are efficient in general, some patients have recurrent diseases, at which stage current treatment options, mostly chemotherapy, are limited in terms of clinical outcome. It is hoped that development in targeted therapies can provide more options for the recurrent and metastatic prostate cancers.
In general, localized prostate cancer patients with intermediate or high risk (e.g. higher pathological CAPRA (Cancer of Prostate Risk Assessment) scores have higher mortality rate even after prostatectomy, radiation or hormone therapy [3]. Although only a small percentage of patients who undergo radical prostatectomy are expected to develop metastasis (<3% in 5 years) [4], the mortality rate for metastatic prostate cancer is extremely high. Current treatment options for metastatic prostate cancer are chemotherapies, but metastatic prostate patients can only expect to have a median survival of 12-15 months even with chemotherapies [5]. Thus, novel therapies are on demand for the treatment of the malignant forms of prostate cancer that recur after initial therapies, including hormone refractory (HRPC) or castration resistant prostate cancer (CRPC).
Several targeted therapeutic agents have been developed and clinically used for the treatment of solid tumors such as breast cancer, non-small cell lung cancer, and renal cancer. These reagents modulate growth factors and their receptors that are abundant in cancer cells, or the downstream signal transduction, survival pathways, and angiogenesis pathways that are abnormally activated in transformed cells or metastatic tumors. We review current developments in this field with the focus on treatments that can be applied to prostate cancers (Fig. 2). These targeted therapies in development are for HRPC and CRPC, which are more malignant than localized lesions that can be effectively managed by surgery or radiation therapy.
Figure 2. Molecular targets and designed intervention in targeted therapies for prostate cancer.

In addition to androgen/ androgen receptor (AR) pathways, many growth receptors (EGFR/HER2, IGFR, PDGFR, and VEGFR), and their downstream molecules, have been explored as the targets to block growth signaling of tumor cells or the angiogenesis process that provides nutrients to cancer cells. Antibodies to some prostate cancer specific antigens (e.g. PSMA and the F77 antigen) have also been developed.
2. Inhibitors of Androgens and androgen receptor signaling
Androgens play a major role in the development, growth, and maintenance of the prostate. As with normal prostate development, primary prostate cancers are largely dependent on androgens for growth and survival [6]. Androgens exert their effects via the intracellular androgen receptor (AR), a ligand-dependent transcriptional activator. In fact, androgens and AR represent the very first class of unique targets for therapies tailored for prostate cancers [7]. Despite the standard hormone therapy, the majority of androgen-independent or hormone refractory prostate cancers still express AR [8], and intratumoral androgen levels remain high along with persistent AR activation in CRPC [9]. As a result, novel therapies targeting androgen and AR are needed for prostate cancer patients who suffer relapse after androgen ablation and anti-androgen therapies.
MDV3100 (Fig. 3) is an oral androgen receptor antagonist in development for the treatment of early-stage and advanced prostate cancer [10]. It directly inhibits AR by binding the receptor irreversibly. This interaction impairs AR nuclear translocation, DNA binding, and recruitment of co-activators [11]. . Preclinical studies have demonstrated that MDV3100 binds to the AR receptor with substantially higher affinity compared to Bicalutamide (a clinical AR modulator), resulting in more complete suppression of the androgen receptor pathway [12]. In a Phase I/II study, MDV3100 showed anti-tumor activity in patients with metastatic CRPC [13]. 56% of 140 patients in the trial demonstrated decreases in serum PSA of 50% or more. 61 out of 109 patients had stabilized bone disease after treatment. Currently, MDV3100 is in Phase 3 trials to evaluate activity in advanced prostate cancer patients with or without chemotherapy history [10].
Figure 3.

Structure of small molecule agents in targeted therapies for prostate cancer.
Abiraterone (Fig. 3) inhibits 17-α-hydroxylase/C17-20-lyase (CYP17), an enzyme expressed in testicular, adrenal, and prostate tumor tissues that catalyzes the sequential reactions of the conversion of pregnenolone and progesterone to their 17-α-hydroxy derivatives and the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione [14]. As DHEA and androstenedione are androgens and precursors of testosterone, inhibition of CYP17 activity by abiraterone decreases circulating levels of testosterone. After i.p. injection of 0.5 mmol/kg abiraterone acetate (the pro-drug), the plasma concentration of abiraterone reached more than 1 μM and remained at about 0.4 μM after 24 hours [15]. Potter et al. reported that the inhibitory activity (e.g. IC50) of abiraterone for hydroxylase and lyase activity were both at 3~4 nM [16]. The IC50 is within the range of in vivo abiraterone pharmacological concentration and therefore sufficient to obtain clinical activity.
Activity of abiraterone acetate as a single agent was apparent even in Phase I trials, resulting in significant decreases of serum PSA levels (50% or more) in 55% CRPC patients with or without prior ketoconazole therapy [17], and in 57% of chemotherapy free but hormone therapy resistant prostate cancer patients [18]. Ketoconazole was initially developed as an antimycotic agent but later found to be a nonspecific inhibitor of steroidogenic enzymes. This study has suggested that abiraterone is superior to Ketoconazole in clinically activity. In Phase II trials, when combined with low-dose glucocorticoids such as prednisone, abiraterone acetate caused significant PSA drop (50% or more) in 36% patients with progressive metastatic CRPC who failed docetaxel-based chemotherapy [19], and in 67% of chemotherapy free CRPC patients [20]. In Phase III studies that involved a total of 1195 patients, abiraterone acetate plus prednisone (797 patients), compared to placebo plus prednisone (398 patients), prolonged overall survival among patients with metastatic CRPC who had disease progression after docetaxel-based chemotherapy [21]. The median overall survival was 14.8 months in the abiraterone acetate plus prednisone group vs 10.9 months in the control groups [22]. As a result of the successful phase III trial, the US Food and Drug Administration (FDA) has recently approved abiraterone acetate (Zytiga, Cougar Biotechnology) in combination with prednisone for the treatment of metastatic CRPC in men who have received prior docetaxel chemotherapy.
3. Growth factors and growth factor receptors inhibitors
Multiple growth factors and growth factor receptors have been identified as critical regulatory proteins in signaling networks that are common to many cancer cells. Novel agents currently in clinical treatment are designed to targeted specific protein families such as the epidermal growth factor receptor (EGFR) family and the platelet-derived growth factor receptor (PDGFR) family.
3.1 ErbB inhibitors
The human EGFR family (HER/ErbB) receptors have been recognized as a very important family of receptor tyrosine kinases, which are frequently reported to have significant impacts on the cellular signaling networks within many different solid tumors, including breast cancer, colon cancer, lung cancer and prostate cancer [23]. This family comprises four closely related receptors: EGFR (HER-1/ErbB1), HER-2 (Neu/ErbB2), HER-3 (ErbB3), and HER-4 (ErbB4). These transmembrane glycoproteins contain an extracellular ligand binding domain and an intracellular receptor tyrosine kinase (RTK) domain. It has been reported that EGFR is overexpressed in 18-37% prostate cancers [24-26]. Recently, Neto et al. also reported a significant direct correlation of HER2/neu over-expression with the risk of death and recurrence in prostate cancer [27]. HER2 is also associated with the activation of androgen receptor and androgen-induced PSA expression [28]. These studies indicate that targeted agents for ErbB receptors, including monoclonal antibodies and small molecule tyrosine kinase inhibitors, can potentially provide treatment options for prostate cancer.
3.1.1 Monoclonal Antibodies
Cetuximab (C225/ Erbitux), a monoclonal antibody initially approved by the Food and Drug Administration (FDA) for colorectal cancer in 2004, directly binds to the extracellular domain of EGFR and blocks ligand (EGF) binding. In preclinical research, Karashima et al. reported that Cetuximab had significant anti-tumor effect in a murine prostate cancer model, interfering with cell proliferation/angiogenesis and enhancing apoptosis [29]. Cetuximab is effective for some but not all prostate cancer model systems. For example, Cetuximab caused a significant growth inhibition by inducing cell apoptosis in Du145 cells, but not in PC3 cells [30]. The activity of Cetuximab is associated with its effective inhibition of phosphorylation of EGFR at sites Tyr-845 and Tyr-1173 in Du145 but not in PC3 cells. This result might be related to higher EGFR expression level in Du145 than PC3. A fully humanized EGFR mAb, Panitumumab (Vectibix), demonstrates higher affinity to EGFR than Cetuximab does and shows significant growth inhibition of PC3 cells in vitro [31]. In a phase I clinical study, 3 out of 21 prostate cancer patients showed stable disease after Panitumumab treatment [32].
Wagener et al. showed that Cetuximab increased the treatment effects of radiation for prostate cancer in vitro and in vivo [33]. In a phase Ib/IIa trial, Cetuximab in combination with doxorubicin were administered weekly for 6 times in patients with metastatic CRPC. The median survival of patients under the combination therapy was longer compared to historical control groups, although minimal PSA declines were observed [34].
Trastuzumab (Herceptin) is a recombinant humanized monoclonal antibody that targets HER-2, another member of the ErbB family thought to play a role in regulating cell proliferation and differentiation of cancer cells. Trastuzumab is the first FDA approved therapeutic drug targeting ErbB receptors to treat metastatic breast and stomach cancer. As HER2 overexpression has been observed in prostate cancer patients, ranging from 25% of untreated primary tumors to 78% of castrate metastatic tumors, it is believed Trastuzumab could be effective to treat prostate cancer [35]. An early study indicated that Trastuzumab might have activity on androgen-dependent but not androgen-independent tumor, and combination of Trastuzumab with Paclitaxel leads to inhibition of both type of tumors in xenografted animal models [36].
Docetaxel may induce expression of HER2 in a human prostate cancer model, especially in hormone independent tumor cells [37], suggesting a mechanism for acquired resistance to chemotherapies. Thus, HER2 targeted therapies may provide an approach to overcome the chemo- resistance. However, it has to be investigated if the docetaxel effect is only limited to prostate cancer. Nevertheless, it seems plausible that a docetaxel /trastuzumab combination may become an effective therapeutic approach for hormone refractory prostate cancer. In fact, a phase I study has already demonstrated the safety of administration of docetaxel, estramustine, and trastuzumab in patients with metastatic androgen-independent prostate cancer [38].
3.1.2 Small tyrosine kinase inhibitors (TKI)
A class of tyrosine kinase inhibitors have been developed to dampen the activity of ErbB receptors [39]. Clinical development of small molecule inhibitors of ErbB receptors, including gifitinib (IRESSA/ZD1839), erlotinib, and lapatinib (Fig. 3), has also been explored in the treatment of patients with prostate cancer. Bonaccorsi et al. reported that gefitinib, which affects EGF-stimulated activation of PI3K/Akt pathway, suppressed invasion and proliferation of androgen-independent prostate cancer cell lines PC3 and DU145 [40]. Vicentini and coworkers showed that gefitinib caused cell cycle arrest and initiation of apoptosis in both androgen-sensitive cells (ND1, LNCaP and ALVA-31) and androgen-independent cells [41]. Effects of gefitinib on LNCaP, PC3 and DU145 cell lines was investigated by Sgambato [42], who found that the anti-proliferative effect of gefitinib was mainly cytostatic and associated with a block in the G0/G1 phase of the cell cycle. In DU145 cells, this block was associated with an increase in expression of the CDK inhibitor p27kip1 that was not evident in the LNCaP and PC3 cells.
Little evidence exists suggesting that gefitinib as a single-agent has activity in CRPC. Although phase I studies reported improved pain control, an open-label, single-arm phase II trial of gefitinib in patients with advanced CRPC showed that single-agent gefitinib had no clinical efficacy assessed by PSA response rate [43]. This outcome is in agreement with results from two other phase II trials with HRPC patients, one involving 40 patients who had not received prior chemotherapy and the other involving 58 patients in the US [44, 45]. Both trials showed no response to gefitinib treatments by monitoring PSA levels. Another EGFR TKI, Erlotinib, shows moderate activity in CRPC, leading to reduction of PSA levels in some patients [46] or delay of PSA rise in others [47]. Lapatinib, an inhibitor for both EGFR and HER2, also showed single agent activity in CRPC [48]. Unexpectedly, lapatinib showed no activity in early stage prostate cancer patients [49, 50].
The conflicting results of TKIs in prostate cancer might be traced to the lack of sensitive forms of kinases in the patient population. In non-small cell lung cancer (NSCLC), the response rate to EGFR tyrosine kinase inhibitors was significantly increased when treatment was administered to patients with tumors containing somatic EGFR mutations [51]. It is worth noting that in all the prostate cancer trials as discussed above, patients were not examined for inhibitor sensitive EGFR mutations. One study examined prostate cancer patients who were unresponsive to gefitinib based therapy and revealed no such sensitizing mutations in any patients [52]. In addition, in vitro studies indicate that a low EGFR/HER2 ratio and PTEN absence are also main factors responsible for resistance to erlotinib and gefitinib [53]. Despite the lack of effectiveness of gefitinib as a monotherapy in HRPC, combination of gefitinib with radio- or chemo-therapies have demonstrated some positive results. Joensuu et al. reported that the combination of gefitinib and radiation has promising activity against nonmetastatic prostate cancer in a phase I/II trial [54].
Although the EGFR/HER2 signaling pathways are attractive targets for prostate cancer therapy, there remain several issues that need to be resolved before prostate cancer patients can fully benefit from inhibitors for these targets. The first question relates to the ErbB receptor expression in prostate cancer. It has become clear that tumors need to over-express EGFR or HER2 receptors to be effectively targeted by these inhibitors. However, clinical tests for HER2 and especially EGFR (including identification of drug sensitive mutants) need to be developed to efficiently identify patients for therapies. Secondly, recent understanding of the effect of downstream molecules, such as the k-ras mutation, on the resistance to EGFR targeted therapies has to be incorporated into the screening of prostate patients. This will help to identify patients that are less likely to be benefited from the treatment. Meanwhile, simultaneously targeting ErbB receptors and downstream molecules, such as Src [55], might provide an option for patients who are resistant to the antibody-based therapies.
3.2 Inhibitors for Insulin-like growth factor (IGF) receptors
The insulin-like-growth factor (IGF)- IGF receptor (IGF-R) signaling pathway plays a crucial role in the cell growth and development of both normal and tumor cells [56]. From an evolutionary standpoint, this highly conserved pathway arose to regulate cellular proliferation in response to nutrient availability. The IGF signaling pathway consists of two ligands (IGF-I and IGF-II), two cell surface receptors (IGF-IR and IGF-IIR), six specific high affinity binding proteins (IGFBP-1 to IGFBP-6), and several other IGFBP-interacting molecules that regulate and propagate IGF activity in various tissues [57]. Epidemiological studies indicate that circulating IGF-I levels are positively correlated with increased risk of prostate cancer [58]. Cardillo et al. analyzed 43 paraffin-embeded prostate cancer samples and found kinetic changing of IGF system as prostate tissue progressed from a normal to malignant state, suggesting that differential expression of IGF may be associated with the malignancy of tumor phenotype [59].
Given the substantial evidence that the IGF pathway plays a role in prostate cancer, interference with this pathway appears to be a potential approach for targeted therapies. Cixutumumab (IMC-A12), which is currently in Phase II for prostate cancer [60], is a monoclonal antibody directed against IGF-1R. The antibody selectively binds to the membrane-bound IGF-I receptor, thereby down-regulating the PI3K/AKT survival pathway. In human tumor xenograft models, blocking of IGF-IR by IMC-A12 resulted in rapid and profound growth inhibition of cancers of the breast, lung, colon and pancreas [61]. Wu et al. found that IMC-A12 was effective in both androgen-dependent (AD) and androgen-independent (AI) variant human prostate cancer xenografts [62]. Interestingly, IMC-A12 treatment induces both G1 arrest and apoptosis in the AD tumors, whereas G2-M arrest was the predominant cell cycle effect seen in AI tumors. These studies indicate that IGF-IR may play distinct roles in the growth and maintenance of AD and AI prostate cancers [56].
As IGF-IR signaling is known to mediate resistance to cytotoxic chemotherapy and radiation, targeted disruption of the IGF-IR axis combined with conventional cancer treatments may be more effective in the inhibition of tumor growth. This idea is supported by the finding that IMC-A12 enhances the therapeutic effect of docetaxel on advanced prostate cancer in animal models [62].
Alternatively, IGF signaling can be intercepted by using kinase inhibitors targeting IGFRs. Unfortunately, the tyrosine kinase domain of IGF-IR and the Insulin Receptor (IR) are highly conserved, with a homology of 84% [63]. This poses a major challenge to identifying IGF-IR specific therapeutic inhibitors, as cross-activity against IR would carry a great risk of diabetogenesis. INSM-18 (nordihydroguaiaretic acid, NDGA, Fig. 3) is a TKI that selectively inhibits IGF-1R and currently in phase II trial for prostate cancer [64]. In an animal model of prostate cancer, INSM-18 demonstrated anti-tumor activity but had no effect on blood glucose levels, indicating that it has minimal off-target activity for IR. Interestingly, INSM-18 can also inhibit HER2, another kinase that plays a role in prostate cancer. INSM-18 has an IC50 of 31 μM for the inhibition of IGF-1 induced autophosphorylation of IGF-IR, and its activity to inhibit ligand-independent tyrosine phosphorylation of HER2 is determined to be 15 μM [65]. This characteristic of INSM-18 renders it more appealing as a therapy for prostate cancer.
3.3 Platelet-derived growth factor inhibitors
Platelet-derived growth factors (PDGFs) contain four members of different polypeptides: PDGF-A, PDGF-B, PDGF-C and PDGF-D [66]. PDGFs can form homodimers or heterodimers via disulfide bonds, five of which have so far been described: PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD. These factors exert their cellular effects through receptors PDGFR-α and PDGFR-β. PDGFR-α can be activated by PDGF-AA, PDGF-AB, PDGF-BB and PDGF-CC, whilst PDGFR-β is only bound and activated by PDGF-BB and PDGF-DD. There is an increasing body of evidence implicating PDGFs in the development of solid tumors. Govindarajan demonstrated that SV7tert cells overexpressing PDGF-BB induce tumor formation when transplanted into nude mice [67], documenting that constitutive PDGF signaling can act as a critical factor in the malignant transformation of human cells. In prostate cancer, overexpression of PDGFRα has been detected in epithelial and stromal cells of prostate adenocarcinomas as well as in bone marrow of metastatic androgen–independent disease, indicating a role for this receptor in both primary and progression of prostate cancer [68, 69].
Expression of PDGFR in prostate tumor-associated endothelial cells was much higher than prostate tumor cells [68]. Tumor cells appear to interact with host factors in the microenvironment to induce growth and the expansion of vasculature. PDGFR inhibitors can, therefore, be considered as a class of antivascular therapies that destroy tumor cells by blocking the required oxygen and nutrients for survival. We will discuss anti-angiogenesis inhibitors in a later section.
A study by Dolloff et al. found that PC3-ML cells, which possess high bone-metastatic potential, express significantly higher levels of PDGFR compared to malignant but nonmetastatic PC3-N cells [70]. When cells were exposed to PDGF, the Akt pathway was activated more profoundly in PC3-ML cells. In contrast, there was no difference in EGFR expression and signaling between these two types of cells, suggesting a determinant role for PDGF, but not EGF, in regulating survival of bone-metastatic prostate cells. The study also postulates that the expression of PDGFR can identify cells within the primitive tumor with the highest propensity to metastasize to the skeleton.
Imatinib (Fig. 3) was initially found to block the kinase activity of the Bcr-Abl fusion oncoprotein that is involved in chronic myeloid leukemia. Later Imatinib was found to be a potent inhibitor of PDGFR kinase as well [71]. Imatinib showed profound activity for prostate cancer in animal models. Bone lesions of xenografted mice carrying PC-3MM2-MDR tumors responded to systemic administration of imatinib and paclitaxel (but not to paclitaxel alone), raising the possibility that imatinib could have sensitized the tumor cells to paclitaxel [72]. Meanwhile, after the co-treatment, extensive apoptosis was observed in both tumor cells and tumor associated endothelial cells [73]. A phase I study of 28 androgen-independent patients further confirmed the activity of imatinib in combination with docetaxel [74]. When imatinib was administered alone, only 2 out 28 patients showed PSA decline (both < 50%). After the patients were treated by the combination of imatinib and docetaxel, decline in PSA levels were seen in 59% of patients. However, in a followup trial with metastatic CRPC patients, no difference in progression-free survival was observed for the Imatinib plus docetaxel intervention [75]. Additional investigation is required to determine the activity of imatinib for prostate cancer. The unexpected outcomes of the clinical trial might be explained by the preferential activity of imatinib against the osteoclastic response, which is dominant in the PC3-MM2 orthotopic model, over the osteoblastic response, which is much more commonly seen in human prostate cancer bone metastases [75].
SU101 (ARAVA, leflunomide, Fig. 3) is a small organic molecule that selectively inhibits PDGFRα and PDGFRβ in vitro [76]. SU101 was first developed as a dihydroorotate dehydrogenase inhibitor to prevent pyrimidine synthesis. Clinically SU101 exhibits significant anti-inflammatory effects and is currently used as a disease-modulating agent in the treatment of rheumatoid arthritis. In 1997, Shawver and co-workers reported that SU101 mediated inhibition of mitogenesis induced by PDGF, but not by EGF {Shawver, 1997 #38}. In a phase II trial with SU101 in hormone refractory prostate cancer, PDGFR expression was detected in the majority of patients (80% of metastases and 88% of primary prostate cancers). For patients treated with SU101 as a single agent, 3 out of 39 demonstrated PSA reduction of more than 50%, one out of 19 patients had partial response for measurable disease, and 9 out of 35 people reported significant improvement of pain [77]. SU101 has recently completed a phase II/III trial with mitixantrone and prednisone for stage IV prostate cancers.
Tandutinib (MLN518, Fig. 3), previously known as CT53518, is a small-molecule inhibitor of the type III receptor tyrosine kinases, including the Fms-like tyrosine kinase 3 receptor (FLT3), platelet-derived growth factor receptor (PDGFR), and c-Kit receptor tyrosine kinase. Matthew et. al. conducted a phase II trial studying Tandutinib in treating patients with progressive prostate cancer and bone metastases. Unfortunately, worsened disease progression was observed in patients treated with Tandutinib, although PDGFR inhibition was observed [78]. It is suspected that PDGF may play multiple roles prostate cancer, including the regulation of physiologic equilibrium (homeostasis) of bone metastases associated with advanced prostate cancer. Meanwhile, Tandutinib needs to be improved to have more selective activity targeting only PDGFR before it can be further tested in prostate cancer.
4. Anti-angiogensis targeting therapy
Angiogenesis, the process of new blood vessel formation, is a crucial step in the propagation of malignant tumor growth and metastasis. Tumor growth is highly dependent on diffusion of nutrients and wastes, so a steady blood supply is critical to tumor development [79]. Among the multiple pro-angiogenic factors that promote the process of vessel formation, vascular endothelial growth factor (VEGF) is one of the most important. Clinical trials have demonstrated the efficacy of anti-VEGF therapy as a treatment for many types of cancers. The VEGF family of polypeptide growth factors, of which at least 7 members have been described, activate VEGFR receptor tyrosine kinases resulting in multiple downstream effects [80]. VEGF-A has been shown to associate with key events in tumor angiogenesis. VEGF-A binds two major receptor VEGF tyrosine receptor kinases, namely VEGFR-1 that is expressed mainly in vascular endothelial cells, and VEGFR-2, which is important in cell trafficking [81, 82]. The third VEGF receptor, VEGFR3, is primarily associated with lymphangiogenesis [83].
Angiogenesis plays a critical role in prostate cancer and is associated with higher Gleason grade, metastasis, and worse clinical outcomes. Weidner and colleagues demonstrated that microvessel density (MVD) was significantly higher in prostate cancer samples for those patients with metastatic disease when compared with those without metastatic disease [84]. A study by Borre et. al. in 221 prostate cancer patients followed for a median of 15 years revealed that MVD of tumor samples at diagnosis was statistically significantly correlated with stage, grade, and disease survival [85]. Furthermore, serum levels of the ligand VEGF were found to be significantly higher in those prostate cancer patients with metastatic disease [86]. Plasma VEGF levels have also been shown to be an independent prognostic factor in men with metastatic prostate cancer [87, 88]. Based on these findings, angiogenesis has been targeted as a strategy to treat prostate cancer.
Bevacizumab (Avastin) is a humanized murine monoclonal antibody against the VEGF receptor and has been shown to have activity in multiple cancer cell lines. Bevacizumab is currently FDA-approved for the treatment of several malignancies including colorectal carcinoma, NSCLC, recurrent glioblastoma and, most recently, metastatic renal cell carcinoma. Initial preclinical studies showed that VEGF inhibition by Bevacizumab prevented further tumor growth of the prostate cancer cell line DU145 implanted in nude mice [89]. Unfortunately, use of Bevacizumab as a single agent in prostate cancer was disappointing.
In a phase II trial, in which 15 patients were treated with 10 mg/kg Bevacizumab every 14 days for six infusions, objective responses or meaningful PSA level declines were not observed [90]. However, in 2010, two phaseII clinical trials involving the use of bevacizumab in combination with standard chemotherapy had positive results. 60 patients with progressive metastatic CRPC were enrolled and received intravenous docetaxel and bevacizumab plus oral thalidomide and prednisone. 90% of the patients had a PSA decline of greater than 50%. Progression free survival was estimated to be 18.2 months, with median overall survival reported as 28.2 months [91].
In another phase II study, 79 patients were enrolled, and 77 of those patients were eligible and assessable. Therapy was based on 21-day cycles. Patients received oral estramustine phosphate 3 times daily for 5 days, decadron twice daily for 3 days and intravenous docetaxel (70mg/m2) and bevacizumab (15 mg/kg). A 50% PSA decline was observed in 58 patients (75%). Twenty-three of 39 patients (59%) with measurable disease had a partial response [92]. More recently, a randomized phase III trial has been conducted comparing the combined use of docetaxel, prednisone, and bevacizumab with docetaxel. This study randomized 1050 chemotherapy-naïve metastatic CRPC patients into each treatment group using overall survival as a primary endpoint. Despite an improvement in progression-free survival, disease response and PSA decline, adding bevacizumab to docetaxel and prednisone did not improve overall survival in patients with metastatic CRPC, and was associated with greater morbidity and mortality [93].
Inhibition of tyrosine kinase activity of VEGF receptors is another approach to inhibit angiogenesis. Two VEGFR tyrosine kinase inhibitors, Sorafenib and Sunitinib (Fig. 3), have been studied for the treatment of prostate cancer. Sorafenib was initially developed to inhibit Raf-1/ BRAF but was subsequently discovered to inhibit some receptor tyrosine kinase receptors, including human VEGFR-2, with IC50 in the nanomolar range [94]. In prostate cancer cells, Sorafenib has been shown to block the MAPK pathway and induce apoptosis and autophagy [95]. It is expected that Sorafenib, with anti- angiogenesis capability, can have more profound in vivo activity. Interestingly, in a transgenic mouse adenocarcinoma prostate model, Sorafenib, mainly through its anti-angiogenesis effect, reduced progression of high grade prostate intraepithelial neoplasia to adenocarcinoma [96]. In clinical studies, the activity of Sorafenib was initially unclear when PSA was used as the biomarker to measure response. In one open-label phase II study using Sorafenib as the single agent therapy, two patients appeared to have worsening PSA scores after treatment but both had dramatic reduction of bone metastatic lesions as determined by bone scan [97]. In another phase II trial with CRPC, the PSA response rate was only 3.6% during the trial, but 10 out of 16 Sorafenib treated patients had PSA declines of 7%-52% after the trial is finished [98]. It is suspected that Sorafenib treatment might cause PSA release. Thus, response to treatment should be evaluated independent of PSA. A further study concluded that Sorafenib has moderate activity as a second-line treatment for metastatic CRPC, with one partial response, 10 stable disease and median survival of 18.3 months for a total of 46 patients [99]. In comparison, treatment with cytotoxic agents in CRPC after docetaxel led to median overall survival in the range of 9.8 months to 17 months.
Sunitinib is another multi-specific tyrosine kinase inhibitor that targets VEGFR1-3, PDGFRα/β, CSF-1R, and Flt-3 [100]. It is FDA-approved for gastrointestinal stromal tumor (as a second line treatment after imatinib), metastatic renal cell carcinoma, and recently pancreatic neuroendocrine tumors. In vivo studies showed additive anti-tumor effect of Sunitinib in combination with docetaxel and/or anti-EGFR antibody Cetuximab in the prostate cancer PC3 and DU145 xenografts[101, 102].
In a phaseIIclinical study, 17 patients with no prior chemotherapy and 17 patients with docetaxel-resistant prostate cancer were treated with sunitinib[103]. One patient in each group showed PSA reduction of more than 50% from the base line. About half of patients in each group also reported stable PSA levels at week 12. In addition, improvement of radiological imaging was seen in some people even without PSA decline. The activity of sunitinib was also confirmed in other phase II studies in CRPC [104, 105]. In a very recent phase III trial of sunitinib in combination with prednisone as compared to prednisone alone in metastatic CRPC, treated patients had significant improvement of progression-free survival, and higher overall response rate were documented. Overall survival was also longer for sunitinib treated patients but the difference was not significant (13.1 vs 11.7 months) [106]. Because of the lack of significant improvement of overall survival in the treatment regimen, Pfizer has discontinued the trial.
5. Other molecular targets for prostate cancer therapy
Prostate-specific membrane antigen (PSMA)
PSMA is a type II transmembrane glycoprotein predominantly expressed in the prostate epithelium. Significantly increased expression of PSMA is observed in prostate cancer, particularly in poorly differentiated, metastatic, and hormone refractory carcinomas [107]. The first PSMA mAb, 7E11-C5.3, recognizes intracellular domain of PSMA. As a result, 7E11-C5.3 (or the antibody conjugate Capromab pendetide or CYT-365) only binds to late stage tumors or soft tissue metastases when cell membrane becomes permeable due to tumor necrosis [108].
An antibody named J591 was developed to target the external domain of PSMA {Chang, 1999 #285}. Radiolabeled J591 has been shown to accurately target bone and soft tissue metastatic prostate cancer sites and may be useful as therapeutic and diagnostic imaging agents {Morris, 2005 #286}. This antibody is currently under evaluation for prostate and non-prostate radio-immunotherapies. MLN2704 (Millennium Pharmaceuticals, Inc., Cambridge, MA) is the humanized J591 conjugated with the microtubule- depolymerizing drug maytansinoid 1 (DM1, Immunogen, Cambridge, MA)[109]. Phase I/II trials using MLN2704 have revealed acceptable safety of the compound and the non-immunogenicity of the antibody. In addition, two patients showed sustained PSA decline of more than 50% and six patients had stable PSA levels for up to 86 days [110]. In other trials, huJ591 was radio-labeled with 111indium (for imaging), 90yttrium (for therapy), or 177lutetium (for both imaging and therapy) [111]. The labeled antibody was shown to target bone and/or soft tissue lesions. Kuroda et al. reported the conjugation of saporin toxin with huJ591 and demonstrated anti-tumor activity in animal models [112].
Heat shock protein (Hsp)
Hsp is a family of chaperones that assist in the posttranslational folding of proteins. They play roles in a variety of cellular process including proliferation, differentiation, and survival by maintaining the stability of regulatory proteins [113]. One such Hsp is Hsp90. Inhibition of Hsp90 function has been shown to cause degradation of client proteins via the ubiquitinproteasome pathway, resulting in the depletion of multiple cancer-related proteins, including HER2. 17-(allylamino)-17-demethoxygeldanamycin (17-AAG, Fig. 3), an Hsp90 inhibitor derived from geldanamycin, binds to the ATP-binding pocket in the N-terminal domain of Hsp90 and blocks the binding of the adenine nucleotides to Hsp90. Interestingly, 17-AAG has much higher affinity to Hsp90 from tumor than that from normal cells[114]. However, in a phase II clinical study 17-AGG did not show any activity in metastatic androgen independent prostate cancer patients with regard to PSA response [115]. F4, a novobiocin analogue designed to inhibit the C-terminal portion of Hsp90, demonstrated improved cytotoxicity in prostate cancer cell lines as compared to 17-AAG [116]. Recently, a class of small molecule Hsp90 inhibitor gamitrinibs showed preclinical activity and favorable safety in the PC3 xenograft model for drug-resistant and bone metastatic prostate cancer [117]. Gamitrinibs are ATPase antagonists but only accumulate in the mitochondria of tumor cells, and thus has much less toxicity to normal cells [118].
mTOR inhibitor
The phosphoinositide-3 kinase pathway (PI3K) is another critical signal transduction pathway that is involved in cancer development. This pathway may be activated by growth factors such as EGF, and it leads to the activation of a number of important downstream cellular signaling components. One such component is the mammalian target of rapamycin (mTOR) protein. Activation of mTOR results in sequential activation of downstream molecules, which ultimately leads to cell division. Current mTOR Inhibitors in clinical trials are mainly rapamycin and temsirolimus (CCI-779) (Fig. 3). The response of prostate cancer cell lines to a combination of rapamycin and the nonsteroidal androgen receptor (AR) antagonist bicalutamide has been documented [119]. In a clinical study involving HRPC patients, two patients treated with Rapamycin experienced a response with PSA reduction, and 4 patients were reported to have stable disease after the treatment [120]. Wu et. al. studied the effects of Temsirolimus on hormone resistant PC3 cells, which are PTEN-negative and have higher Akt and mTOR activity, and DU145 cells, which are PTEN positive. Temsirolimus inhibited the growth of xenografts derived from both cell lines but better activity was observed with PC3 tumors [121].
Histone Deacetylase (HDAC) inhibitors
Epigenetic modifications play a key role in the patho-physiology of cancer. Histone deacetylases (HDACs), whose substrates are not limited to histone, are involved in cancer progression. HDACs are part of a transcriptional co-repressor complex that influences various tumor suppressor genes. Because of the significant roles played by HDACs in various human cancers, HDAC inhibitors are emerging as a new class of chemotherapeutic agents. HDAC inhibitors have been shown to induce growth arrest, differentiation, and/or apoptosis of prostate cancer cells. Up to now, more than 100 clinical trials are ongoing with HDAC inhibitors (HDACi) either as monotherapy or in various combination therapies. In one report on a phase II clinical trial with the HDAC inhibitor romidepsin (Fig. 3), two of 35 HRPC patients achieved a confirmed radiological response along with a PSA decline of >50% [122]. However, 11 people experienced toxicity and were removed from the trial, which raises safety concerns. HDAC inhibitors have to be improved for specificity, and combination therapies should be considered.
6. New targets and Biomarkers for prostate cancer therapy
One of the validated and widely accepted prostate cancer biomarkers is PSA. Although the measurement of PSA is clinically practiced to provide a way to predict response to therapy, this test fails for some patients. While PSA is elevated in the presence of prostate cancer, it is also present in small amounts in the serum of healthy males. The effectiveness is thus limited with a positive predictive value of 28-35% [123]. A recent study also revealed that the change of the PSA levels (PSA velocity) was a poor biomarker for prostate cancer diagnosis and unable to predict the grade (e.g. Gleason score) of the disease [124]. For some experimental therapies as we mentioned above, reduction of PSA levels are not observed in patients showing radiological responses. In some cases, the PSA levels can even rise in patients showing response to therapies by other measurements [97].
A great effort in the field of prostate cancer has been made to discover novel prostate markers. This includes extensive effort at the proteomic scale [125] and the expansion of biomarker candidates from protein to DNA/RNA [126] and circulating tumor cells [127]. Reviews of prostate biomarkers and recent developments are presented in other articles [128, 129].
In many cases, the molecular target for a targeted therapy can be used as a biomarker for prostate cancer. Whether or not these biomarkers are specific to prostate, their existence in the tumor provides guidance to targeted therapies to obtain higher response in a sub-category of prostate cancer. In fact, the unique linkage between the target and the therapy differentiates targeted therapies from the conventional cytotoxic chemtherapeutic therapies for cancers. In addition to targeted proteins such as the PSMA we mentioned above, several molecules have recently emerged to be potential “targeted” biomarkers that can be used to guide therapies.
The F77 antigen
Recently, F77, a monoclonal antibody developed by immunizing mice with prostate cancer cells PC3 , was shown to recognize small molecular species with glycolipid properties [130]. The F77 antigen, termed prostate cancer lipid antigen (PCLA), is concentrated in the lipid raft microdomains that serve as platforms in the assembly of associating protein complexes. Due to its glycolipid properties, PCLA holds significant value as a cancer biomarker. It is expressed on both androgen dependent and androgen independent prostate cancer, making it a target for early diagnosis and treatment of advanced diseases. Tissue microarray studies show that F77 stained 112 of 166 primary and 29 of 34 metastatic prostate cancer specimens [130]. By eliciting ADCC/ACD activity towards the cancer cells, the F77 antibody was useful as a therapeutic reagent to limit the growth of prostate cancer growth in xenografted mice [130]. Currently the antibody is subjected to humanization. The humanized antibody will be tested if F77 provides a novel treatment for human prostate cancer.
Serine peptidase inhibitor Kazal type 1 (SPINK1)
SPINK1 is a biomarker specific to a subset of aggressive prostate cancer that does not carry genetic rearrangement of the ETS (E26 transformation specific) transcription factors into the TMPRSS2 promoter region. In 2008, Tomlins et al. demonstrated that high levels of SPINK1 in this subset were correlated with a greater rate of cancer recurrence [131]. Recent in vivo data suggests that SPINK1 promotes prostate tumor growth through EGFR [132]. SPINK1 has structural similarities with EGF and binds to EGFR, inducing the dimerization and sustained phosphorylation of the receptor. Inhibiting SPINK1 attenuates key downstream mediators of the EGFR pathway, including MEK, ERK, and AKT. These results support the potential of EGFR inhibitors to treat SPINK1 positive prostate cancer. In fact, the anti-EGFR mAb Cetuximab specifically decreased SPINK1+ tumor growth in mice [132].
Survivin
Survivin is a member of the Inhibitor of Apoptosis (IAP) family that functions to inhibit caspase activation, leading to the prevention of apoptosis [133]. As this protein is highly expressed during fetal development and in many human tumors, but also absent in normal terminally differentiated tissues, it is useful as both a cancer diagnostic marker and therapeutic target [134]. Survivin is overexpressed in multiple types of adenocarcinomas, including prostate [135]. Zhang et al. demonstrated Survivin's contribution to the development of hormonal therapy resistance in prostate cancer cells by studying its expression in LNCaP, an androgen-dependent cell line [136]. It was shown that inhibiting Survivin could enhance the therapeutic effect of Flutamide, an anti-androgen agent. However, it was reported that in locally advanced prostate cancer cytoplasmic overexpression of Survivin predicted local progression but nuclear expression was associated with improved survival [137]. Recent studies have revealed that acetylation promotes the translocation of Survivin into the nucleus, where it functions as a repressor of STAT3 oncogenic activity [138]. These data indicate that Survivin may have multiple roles in the cell.
Several small-molecule inhibitors and natural compounds that suppress Survivin expression have been developed and proven effective in suppressing prostate cancer tumor growth and enhancing docetaxel- induced apoptosis. One such agent is YM155 (Fig. 3), a small molecule suppressant of Survivin expression, promoting apoptosis in vitro and in a xenograft mouse model in vivo [139]. YM155 has completed various phase I and II trials for its safety and efficacy in several types of cancers [140, 141]. As a single-agent, YM155 induced decline in PSA concentrations in two out of nine advanced prostate cancer patients [142].
Sun et al. have identified Sanguinarine (Fig. 3), a benzophenanthridine alkaloid primarily derived from the bloodroot plant, as a novel inhibitor of Survivin [143]. Sanguinarine selectively kills prostate cancer cells over “normal” prostate epithelial cells. By promoting Survivin degradation via the ubiquitin-proteasome system, Sanguinarine was found to induce apoptosis and inhibit in vivo tumor formation.
Recently, an allosteric modulator S12 (Fig. 3) was developed to bind to a cavity near the dimerization domain of Survivin [144]. S12 was able to change spindle formation and halt mitosis, leading to cell death. It showed anti-tumor activity in several animal models [144].
7. Summary
The rapid expansion of targeted therapies in the past decade has provided new strategies for the treatment of prostate cancer, especially the most malignant hormone independent and castration resistant forms. However, the most advanced therapies in this category (Table 1), except androgen inhibitors, are still under clinical trials. Further studies will determine the clinical use of those agents that show promises in early stage trials. Extensive studies will also be needed to optimize some lead compounds for better specificity and higher efficacy. Although some of the initial excitements, including the endothelin A receptor antagonist, zibotentan, failed to be materialized, the extensive research activities in this field have generated insights for the development of future therapies that could be more efficient to control this disease. In addition to these antibody or small molecule based approaches, dendritic cell (DC) based therapies have also been explored [145], leading to the FDA approved Provenge (sipuleucel-T). The hope is that, with targeted therapies, prostate cancer will be divided into sub- categories and treated in a more targeted manner in the future.
Table 1.
Therapeutic agents and their targets in prostate cancer.
| Drugs | Targets | Class of Agent/Mechanism | Stage of Development | Ref |
|---|---|---|---|---|
| MDV3100 | AR | SMI/AR antagonist | Phase III | [10] |
| Abiraterone | CYP17 | SMI/androgen inhibitor | FDA approved in 2011 | [22] |
| Panitumumab | EGFR | mAb | Phase I | [32] |
| Cetuximab | EGFR | mAb/ ligand blocking | Phase Ia/IIb (with doxo) | [34] |
| Trastuzumab | HER2 | mAb | Phase I (with DCT) | [38] |
| Gifinitib | EGFR | SMI/ Tyrosine kinase inhibitor | Phase I/II (with radiation) | [54] |
| Erolotinib | EGFR | SMI/ Tyrosine kinase inhibitor | Phase II | [46, 47] |
| Lapatinib | EGFR/HER2 | SMI/ Tyrosine kinase inhibitor | Phase II | [48] |
| Cixutumumab | IGF-1R | mAb | Phase II | [60] |
| INSM-18 | IGF-1R/HER2 | SMI/ Tyrosine kinase inhibitor | Phase II | [64] |
| Imatinib | PDGFR | SMI/ Tyrosine kinase inhibitor | Phase I and II | [74, 75] |
| SU101 | PDGFR | SMI/ Tyrosine kinase inhibitor | Phase II | [77] |
| Tandutinib | Type III RTK (PDGFR) | SMI/ Tyrosine kinase inhibitor | Failed in phase II | [78] |
| Bevacizumab | VEGF receptor | mAb | Phase III (with DCT; PFS observed but no OS benefit) | [146] |
| Sorafenib | VEGFR2 (Raf-1/BRAF) | SMI/ Tyrosine kinase | Phase II | [97, 98] |
| Sunitinib | Type III RTK (PDGFR, VEGFR) | SMI/ Tyrosine kinase inhibitor | Phase III (discontinued, no significant OS benefit) | [106] |
| Thalidomide | Angiogenesis | Phase II (with bevacizumab, DCT) | [91] | |
| huJ591 | PSMA | mAb | Phase I/II (lDM1 or radio-labeled ) | [110, 111] |
| 17-AAG | Hsp90 | SMI/blocking ATP binding | Phase II (failed) | [115] |
| F4 | Hsp90 | SMI/allosteric modulator | Preclinical | [116] |
| Rapamycin | SMI/mTOR inhibitor | Phase I | [120] | |
| F77 | PCLA | mAb | Preclinical | [130] |
| YM155 | Survivin | SMI/expression repressor | Phase II | [142] |
| Sanguinarine | Survivin | Natural product/ promoting Survivin degradation | Preclinical | [143] |
| S12 | Survivin | SMI/allosteric modulator | Preclinical | [144] |
| EGFR inhibitors | SPINK1 | Preclinical | [132] |
mAb: monoclonal antibody; SMI: small molecule inhibitor; TKI: Tyrosine kinase inhibitor; doxo: doxorubicin; DCT: docetaxel; RTK: receptor tyrosine kinase; PFS: progression free survival; OS: overall survival;
Acknowledgement
This work was supported by grants from the National Institutes of Health (1R01CA157766-01 (A.T.) and R01 CA055306 (M.I.G.)) and the Abramson Family Cancer Research Institute (to M.I.G.). We thank Dr. Mark I. Greene for his comments on this manuscript.
Abbreviations
- AR
androgen receptor
- CAPRA
Cancer of Prostate Risk Assessment
- CRPC
castration resistant prostate cancer
- CYP17
17-α-hydroxylase/C17-20-lyase
- DC
dendritic cell
- DHEA
dehydroepiandrosterone
- EGF
epidermal growth factor
- EGFR
EGF receptor
- FDA
Food and Drug Administration
- HDAC
Histone deacetylases
- HER
human EGFR family (HER/ErbB) receptors
- HRPC
hormone refractory prostate cancer
- HSP
heat shock protein
- IAP
Inhibitor of Apoptosis
- IGF
insulin-like-growth factor
- IGF-R
IGF receptor
- IR
Insulin Receptor
- LH-RH
luteinizing hormone
- LH-RH
luteinizing hormone-releasing hormone
- NSCLC
non-small cell lung cancer
- PDGF
platelet-derived growth factor
- PDGFR
PDGF receptor
- PSA
prostate specific antigen
- PSMA
Prostate-specific membrane antigen
- RTK
receptor tyrosine kinase
- SPINK1
Serine peptidase inhibitor Kazal type 1
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Han M, Partin AW, Zahurak M, Piantadosi S, Epstein JI, Walsh PC. Biochemical (prostate specific antigen) recurrence probability following radical prostatectomy for clinically localized prostate cancer. J Urol. 2003;169:517–523. doi: 10.1097/01.ju.0000045749.90353.c7. [DOI] [PubMed] [Google Scholar]
- 2.Freedland SJ, Humphreys EB, Mangold LA, Eisenberger M, Dorey FJ, Walsh PC, Partin AW. Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. Jama. 2005;294:433–439. doi: 10.1001/jama.294.4.433. [DOI] [PubMed] [Google Scholar]
- 3.Cooperberg MR, Vickers AJ, Broering JM, Carroll PR. Comparative risk-adjusted mortality outcomes after primary surgery, radiotherapy, or androgen-deprivation therapy for localized prostate cancer. Cancer. 2010;116:5226–5234. doi: 10.1002/cncr.25456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephenson AJ, Kattan MW, Eastham JA, Dotan ZA, Bianco FJ, Jr., Lilja H, Scardino PT. Defining biochemical recurrence of prostate cancer after radical prostatectomy: a proposal for a standardized definition. J Clin Oncol. 2006;24:3973–3978. doi: 10.1200/JCO.2005.04.0756. [DOI] [PubMed] [Google Scholar]
- 5.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, Roessner M, Gupta S, Sartor AO. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 6.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 7.Santos AF, Huang H, Tindall DJ. The androgen receptor: a potential target for therapy of prostate cancer. Steroids. 2004;69:79–85. doi: 10.1016/j.steroids.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Agoulnik IU, Weigel NL. Androgen receptor action in hormone-dependent and recurrent prostate cancer. J Cell Biochem. 2006;99:362–372. doi: 10.1002/jcb.20811. [DOI] [PubMed] [Google Scholar]
- 9.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 10.Wu Y, Rosenberg JE, Taplin ME. Novel agents and new therapeutics in castration-resistant prostate cancer. Curr Opin Oncol. 2011;23:290–296. doi: 10.1097/CCO.0b013e3283449400. [DOI] [PubMed] [Google Scholar]
- 11.Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, Wongvipat J. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC) J Med Chem. 2010;53:2779–2796. doi: 10.1021/jm901488g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haidar S, Ehmer PB, Barassin S, Batzl-Hartmann C, Hartmann RW. Effects of novel 17alpha-hydroxylase/C17, 20-lyase (P450 17, CYP 17) inhibitors on androgen biosynthesis in vitro and in vivo. J Steroid Biochem Mol Biol. 2003;84:555–562. doi: 10.1016/s0960-0760(03)00070-0. [DOI] [PubMed] [Google Scholar]
- 15.Barrie SE, Potter GA, Goddard PM, Haynes BP, Dowsett M, Jarman M. Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17-20 lyase) J Steroid Biochem Mol Biol. 1994;50:267–273. doi: 10.1016/0960-0760(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 16.Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. 1995;38:2463–2471. doi: 10.1021/jm00013a022. [DOI] [PubMed] [Google Scholar]
- 17.Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, Martins V, Lee G, Kheoh T, Kim J, Molina A, Small EJ. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28:1481–1488. doi: 10.1200/JCO.2009.24.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, de Bono JS. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 19.Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, Smith MR, Taplin ME, Bubley GJ, Kheoh T, Haqq C, Molina A, Anand A, Koscuiszka M, Larson SM, Schwartz LH, Fleisher M, Scher HI. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol. 2010;28:1496–1501. doi: 10.1200/JCO.2009.25.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryan CJ, Shah S, Efstathiou E, Smith MR, Taplin ME, Bubley GJ, Logothetis CJ, Kheoh T, Kilian C, Haqq CM, Molina A, Small EJ. Phase II study of abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer displaying bone flare discordant with serologic response. Clin Cancer Res. 2011;17:4854–4861. doi: 10.1158/1078-0432.CCR-11-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pal SK, Sartor O. Phase III data for abiraterone in an evolving landscape for castration-resistant prostate cancer. Maturitas. 2011;68:103–105. doi: 10.1016/j.maturitas.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr., Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blackledge G. Growth factor receptor tyrosine kinase inhibitors; clinical development and potential for prostate cancer therapy. J Urol. 2003;170:S77–83. doi: 10.1097/01.ju.0000095022.80033.d3. discussion S83. [DOI] [PubMed] [Google Scholar]
- 24.de Muga S, Hernandez S, Agell L, Salido M, Juanpere N, Lorenzo M, Lorente JA, Serrano S, Lloreta J. Molecular alterations of EGFR and PTEN in prostate cancer: association with high-grade and advanced-stage carcinomas. Mod Pathol. 2010;23:703–712. doi: 10.1038/modpathol.2010.45. [DOI] [PubMed] [Google Scholar]
- 25.Schlomm T, Kirstein P, Iwers L, Daniel B, Steuber T, Walz J, Chun FH, Haese A, Kollermann J, Graefen M, Huland H, Sauter G, Simon R, Erbersdobler A. Clinical significance of epidermal growth factor receptor protein overexpression and gene copy number gains in prostate cancer. Clin Cancer Res. 2007;13:6579–6584. doi: 10.1158/1078-0432.CCR-07-1257. [DOI] [PubMed] [Google Scholar]
- 26.Shuch B, Mikhail M, Satagopan J, Lee P, Yee H, Chang C, Cordon-Cardo C, Taneja SS, Osman I. Racial disparity of epidermal growth factor receptor expression in prostate cancer. J Clin Oncol. 2004;22:4725–4729. doi: 10.1200/JCO.2004.06.134. [DOI] [PubMed] [Google Scholar]
- 27.Neto AS, Tobias-Machado M, Wroclawski ML, Fonseca FL, Teixeira GK, Amarante RD, Wroclawski ER, Del Giglio A. Her-2/neu expression in prostate adenocarcinoma: a systematic review and meta-analysis. J Urol. 2010;184:842–850. doi: 10.1016/j.juro.2010.04.077. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Majumder S, McCall W, Sartor CI, Mohler JL, Gregory CW, Earp HS, Whang YE. Inhibition of HER-2/neu kinase impairs androgen receptor recruitment to the androgen responsive enhancer. Cancer Res. 2005;65:3404–3409. doi: 10.1158/0008-5472.CAN-04-4292. [DOI] [PubMed] [Google Scholar]
- 29.Karashima T, Sweeney P, Slaton JW, Kim SJ, Kedar D, Izawa JI, Fan Z, Pettaway C, Hicklin DJ, Shuin T, Dinney CP. Inhibition of angiogenesis by the antiepidermal growth factor receptor antibody ImClone C225 in androgen-independent prostate cancer growing orthotopically in nude mice. Clin Cancer Res. 2002;8:1253–1264. [PubMed] [Google Scholar]
- 30.Dhupkar P, Dowling M, Cengel K, Chen B. Effects of anti-EGFR antibody cetuximab on androgen-independent prostate cancer cells. Anticancer Res. 2010;30:1905–1910. [PubMed] [Google Scholar]
- 31.Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol. 2001;38:17–23. doi: 10.1016/s1040-8428(00)00134-7. [DOI] [PubMed] [Google Scholar]
- 32.Weiner LM, Belldegrun AS, Crawford J, Tolcher AW, Lockbaum P, Arends RH, Navale L, Amado RG, Schwab G, Figlin RA. Dose and schedule study of panitumumab monotherapy in patients with advanced solid malignancies. Clin Cancer Res. 2008;14:502–508. doi: 10.1158/1078-0432.CCR-07-1509. [DOI] [PubMed] [Google Scholar]
- 33.Wagener M, Zhang X, Villarreal HG, Levy L, Allen P, Shentu S, Fang B, Krishnan S, Chang JY, Cheung MR. Effect of combining anti-epidermal growth factor receptor antibody C225 and radiation on DU145 prostate cancer. Oncol Rep. 2008;19:1071–1077. [PubMed] [Google Scholar]
- 34.Slovin SF, Kelly WK, Wilton A, Kattan M, Myskowski P, Mendelsohn J, Scher HI. Anti-epidermal growth factor receptor monoclonal antibody cetuximab plus Doxorubicin in the treatment of metastatic castration-resistant prostate cancer. Clin Genitourin Cancer. 2009;7:E77–82. doi: 10.3816/CGC.2009.n.028. [DOI] [PubMed] [Google Scholar]
- 35.Signoretti S, Montironi R, Manola J, Altimari A, Tam C, Bubley G, Balk S, Thomas G, Kaplan I, Hlatky L, Hahnfeldt P, Kantoff P, Loda M. Her-2-neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst. 2000;92:1918–1925. doi: 10.1093/jnci/92.23.1918. [DOI] [PubMed] [Google Scholar]
- 36.Agus DB, Scher HI, Higgins B, Fox WD, Heller G, Fazzari M, Cordon-Cardo C, Golde DW. Response of prostate cancer to anti-Her-2/neu antibody in androgen-dependent and -independent human xenograft models. Cancer Res. 1999;59:4761–4764. [PubMed] [Google Scholar]
- 37.Legrier ME, Oudard S, Judde JG, Guyader C, de Pinieux G, Boye K, de Cremoux P, Dutrillaux B, Poupon MF. Potentiation of antitumour activity of docetaxel by combination with trastuzumab in a human prostate cancer xenograft model and underlying mechanisms. Br J Cancer. 2007;96:269–276. doi: 10.1038/sj.bjc.6603553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Small EJ, Bok R, Reese DM, Sudilovsky D, Frohlich M. Docetaxel, estramustine, plus trastuzumab in patients with metastatic androgen-independent prostate cancer. Semin Oncol. 2001;28:71–76. doi: 10.1016/s0093-7754(01)90159-9. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonaccorsi L, Marchiani S, Muratori M, Forti G, Baldi E. Gefitinib (‘IRESSA’, ZD1839) inhibits EGF-induced invasion in prostate cancer cells by suppressing PI3 K/AKT activation. J Cancer Res Clin Oncol. 2004;130:604–614. doi: 10.1007/s00432-004-0581-8. [DOI] [PubMed] [Google Scholar]
- 41.Vicentini C, Festuccia C, Gravina GL, Angelucci A, Marronaro A, Bologna M. Prostate cancer cell proliferation is strongly reduced by the epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in vitro on human cell lines and primary cultures. J Cancer Res Clin Oncol. 2003;129:165–174. doi: 10.1007/s00432-003-0420-3. [DOI] [PubMed] [Google Scholar]
- 42.Sgambato A, Camerini A, Faraglia B, Ardito R, Bianchino G, Spada D, Boninsegna A, Valentini V, Cittadini A. Targeted inhibition of the epidermal growth factor receptor-tyrosine kinase by ZD1839 (‘Iressa’) induces cell-cycle arrest and inhibits proliferation in prostate cancer cells. J Cell Physiol. 2004;201:97–105. doi: 10.1002/jcp.20045. [DOI] [PubMed] [Google Scholar]
- 43.Pezaro C, Rosenthal MA, Gurney H, Davis ID, Underhill C, Boyer MJ, Kotasek D, Solomon B, Toner GC. An open-label, single-arm phase two trial of gefitinib in patients with advanced or metastatic castration-resistant prostate cancer. Am J Clin Oncol. 2009;32:338–341. doi: 10.1097/COC.0b013e31818b946b. [DOI] [PubMed] [Google Scholar]
- 44.Canil CM, Moore MJ, Winquist E, Baetz T, Pollak M, Chi KN, Berry S, Ernst DS, Douglas L, Brundage M, Fisher B, McKenna A, Seymour L. Randomized phase II study of two doses of gefitinib in hormone-refractory prostate cancer: a trial of the National Cancer Institute of Canada-Clinical Trials Group. J Clin Oncol. 2005;23:455–460. doi: 10.1200/JCO.2005.02.129. [DOI] [PubMed] [Google Scholar]
- 45.Small EJ, Fontana J, Tannir N, DiPaola RS, Wilding G, Rubin M, Iacona RB, Kabbinavar FF. A phase II trial of gefitinib in patients with non-metastatic hormone-refractory prostate cancer. BJU Int. 2007;100:765–769. doi: 10.1111/j.1464-410X.2007.07121.x. [DOI] [PubMed] [Google Scholar]
- 46.Nabhan C, Lestingi TM, Galvez A, Tolzien K, Kelby SK, Tsarwhas D, Newman S, Bitran JD. Erlotinib has moderate single-agent activity in chemotherapy-naive castration-resistant prostate cancer: final results of a phase II trial. Urology. 2009;74:665–671. doi: 10.1016/j.urology.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 47.Gravis G, Bladou F, Salem N, Goncalves A, Esterni B, Walz J, Bagattini S, Marcy M, Brunelle S, Viens P. Results from a monocentric phase II trial of erlotinib in patients with metastatic prostate cancer. Ann Oncol. 2008;19:1624–1628. doi: 10.1093/annonc/mdn174. [DOI] [PubMed] [Google Scholar]
- 48.Whang YE, Armstrong AJ, Rathmell WK, Godley PA, Kim WY, Pruthi RS, Wallen EM, Crane JM, Moore DT, Grigson G, Morris K, Watkins CP, George DJ. A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urol Oncol. 2011 doi: 10.1016/j.urolonc.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 49.Sridhar SS, Hotte SJ, Chin JL, Hudes GR, Gregg R, Trachtenberg J, Wang L, Tran-Thanh D, Pham NA, Tsao MS, Hedley D, Dancey JE, Moore MJ. A multicenter phase II clinical trial of lapatinib (GW572016) in hormonally untreated advanced prostate cancer. Am J Clin Oncol. 2010;33:609–613. doi: 10.1097/COC.0b013e3181beac33. [DOI] [PubMed] [Google Scholar]
- 50.Liu G, Chen YH, Kolesar J, Huang W, Dipaola R, Pins M, Carducci M, Stein M, Bubley GJ, Wilding G. Eastern Cooperative Oncology Group Phase II Trial of lapatinib in men with biochemically relapsed, androgen dependent prostate cancer(,) Urol Oncol. 2011 doi: 10.1016/j.urolonc.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 52.Curigliano G, Pelosi G, De Pas T, Renne G, De Cobelli O, Manzotti M, Spitaleri G, de Braud F. Absence of epidermal growth factor receptor gene mutations in patients with hormone refractory prostate cancer not responding to gefitinib. Prostate. 2007;67:603–604. doi: 10.1002/pros.20530. [DOI] [PubMed] [Google Scholar]
- 53.Festuccia C, Gravina GL, Biordi L, D'Ascenzo S, Dolo V, Ficorella C, Ricevuto E, Tombolini V. Effects of EGFR tyrosine kinase inhibitor erlotinib in prostate cancer cells in vitro. Prostate. 2009;69:1529–1537. doi: 10.1002/pros.20995. [DOI] [PubMed] [Google Scholar]
- 54.Joensuu G, Joensuu T, Nokisalmi P, Reddy C, Isola J, Ruutu M, Kouri M, Kupelian PA, Collan J, Pesonen S, Hemminki A. A phase I/II trial of gefitinib given concurrently with radiotherapy in patients with nonmetastatic prostate cancer. Int J Radiat Oncol Biol Phys. 2010;78:42–49. doi: 10.1016/j.ijrobp.2009.07.1731. [DOI] [PubMed] [Google Scholar]
- 55.Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, Sahin AA, Esteva FJ, Hortobagyi GN, Yu D. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461–469. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rowinsky EK, Youssoufian H, Tonra JR, Solomon P, Burtrum D, Ludwig DL. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res. 2007;13:5549s–5555s. doi: 10.1158/1078-0432.CCR-07-1109. [DOI] [PubMed] [Google Scholar]
- 57.Monti S, Proietti-Pannunzi L, Sciarra A, Lolli F, Falasca P, Poggi M, Celi FS, Toscano V. The IGF axis in prostate cancer. Curr Pharm Des. 2007;13:719–727. doi: 10.2174/138161207780249128. [DOI] [PubMed] [Google Scholar]
- 58.Kojima S, Inahara M, Suzuki H, Ichikawa T, Furuya Y. Implications of insulin-like growth factor-I for prostate cancer therapies. Int J Urol. 2009;16:161–167. doi: 10.1111/j.1442-2042.2008.02224.x. [DOI] [PubMed] [Google Scholar]
- 59.Cardillo MR, Monti S, Di Silverio F, Gentile V, Sciarra F, Toscano V. Insulin-like growth factor (IGF)-I, IGF-II and IGF type I receptor (IGFR-I) expression in prostatic cancer. Anticancer Res. 2003;23:3825–3835. [PubMed] [Google Scholar]
- 60.Higano CS, Alumkal JJ, Ryan CJ, Yu EY, Beer TM, Fox FE, Dontabhaktuni A, Youssoufian H, Schwartz JD. A phase II study of cixutumumab (IMC-A12), a monoclonal antibody (MAb) against the insulin-like growth factor 1 receptor (IGF-IR), monotherapy in metastatic castration-resistant prostate cancer (mCRPC) 2010 Genitourinary Cancers Symposium. 2010 Abstract # 189. [Google Scholar]
- 61.Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, Bassi R, Abdullah R, Hooper AT, Koo H, Jimenez X, Johnson D, Apblett R, Kussie P, Bohlen P, Witte L, Hicklin DJ, Ludwig DL. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res. 2003;63:8912–8921. [PubMed] [Google Scholar]
- 62.Wu JD, Haugk K, Coleman I, Woodke L, Vessella R, Nelson P, Montgomery RB, Ludwig DL, Plymate SR. Combined in vivo effect of A12, a type 1 insulin-like growth factor receptor antibody, and docetaxel against prostate cancer tumors. Clin Cancer Res. 2006;12:6153–6160. doi: 10.1158/1078-0432.CCR-06-0443. [DOI] [PubMed] [Google Scholar]
- 63.Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E, et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. Embo J. 1986;5:2503–2512. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Ji QS, Mulvihill M, Pachter JA. Inhibition of the IGF-I receptor for treatment of cancer. Kinase inhibitors and monoclonal antibodies as alternative approaches. Recent Results Cancer Res. 2007;172:59–76. doi: 10.1007/978-3-540-31209-3_5. [DOI] [PubMed] [Google Scholar]
- 65.Youngren JF, Gable K, Penaranda C, Maddux BA, Zavodovskaya M, Lobo M, Campbell M, Kerner J, Goldfine ID. Nordihydroguaiaretic acid (NDGA) inhibits the IGF-1 and c-erbB2/HER2/neu receptors and suppresses growth in breast cancer cells. Breast Cancer Res Treat. 2005;94:37–46. doi: 10.1007/s10549-005-6939-z. [DOI] [PubMed] [Google Scholar]
- 66.Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 67.Govindarajan B, Shah A, Cohen C, Arnold RS, Schechner J, Chung J, Mercurio AM, Alani R, Ryu B, Fan CY, Cuezva JM, Martinez M, Arbiser JL. Malignant transformation of human cells by constitutive expression of platelet-derived growth factor-BB. J Biol Chem. 2005;280:13936–13943. doi: 10.1074/jbc.M500411200. [DOI] [PubMed] [Google Scholar]
- 68.Fudge K, Wang CY, Stearns ME. Immunohistochemistry analysis of platelet-derived growth factor A and B chains and platelet-derived growth factor alpha and beta receptor expression in benign prostatic hyperplasias and Gleason-graded human prostate adenocarcinomas. Mod Pathol. 1994;7:549–554. [PubMed] [Google Scholar]
- 69.Chott A, Sun Z, Morganstern D, Pan J, Li T, Susani M, Mosberger I, Upton MP, Bubley GJ, Balk SP. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of the type 1 insulin-like growth factor receptor. Am J Pathol. 1999;155:1271–1279. doi: 10.1016/S0002-9440(10)65229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dolloff NG, Shulby SS, Nelson AV, Stearns ME, Johannes GJ, Thomas JD, Meucci O, Fatatis A. Bone-metastatic potential of human prostate cancer cells correlates with Akt/PKB activation by alpha platelet-derived growth factor receptor. Oncogene. 2005;24:6848–6854. doi: 10.1038/sj.onc.1208815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.George D. Targeting PDGF receptors in cancer--rationales and proof of concept clinical trials. Adv Exp Med Biol. 2003;532:141–151. doi: 10.1007/978-1-4615-0081-0_12. [DOI] [PubMed] [Google Scholar]
- 72.Uehara H, Kim SJ, Karashima T, Shepherd DL, Fan D, Tsan R, Killion JJ, Logothetis C, Mathew P, Fidler IJ. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases. J Natl Cancer Inst. 2003;95:458–470. doi: 10.1093/jnci/95.6.458. [DOI] [PubMed] [Google Scholar]
- 73.Kim SJ, Uehara H, Yazici S, Busby JE, Nakamura T, He J, Maya M, Logothetis C, Mathew P, Wang X, Do KA, Fan D, Fidler IJ. Targeting platelet-derived growth factor receptor on endothelial cells of multidrug-resistant prostate cancer. J Natl Cancer Inst. 2006;98:783–793. doi: 10.1093/jnci/djj211. [DOI] [PubMed] [Google Scholar]
- 74.Mathew P, Thall PF, Jones D, Perez C, Bucana C, Troncoso P, Kim SJ, Fidler IJ, Logothetis C. Platelet-derived growth factor receptor inhibitor imatinib mesylate and docetaxel: a modular phase I trial in androgen-independent prostate cancer. J Clin Oncol. 2004;22:3323–3329. doi: 10.1200/JCO.2004.10.116. [DOI] [PubMed] [Google Scholar]
- 75.Mathew P, Thall PF, Bucana CD, Oh WK, Morris MJ, Jones DM, Johnson MM, Wen S, Pagliaro LC, Tannir NM, Tu SM, Meluch AA, Smith L, Cohen L, Kim SJ, Troncoso P, Fidler IJ, Logothetis CJ. Platelet-derived growth factor receptor inhibition and chemotherapy for castration-resistant prostate cancer with bone metastases. Clin Cancer Res. 2007;13:5816–5824. doi: 10.1158/1078-0432.CCR-07-1269. [DOI] [PubMed] [Google Scholar]
- 76.Shawver LK, Schwartz DP, Mann E, Chen H, Tsai J, Chu L, Taylorson L, Longhi M, Meredith S, Germain L, Jacobs JS, Tang C, Ullrich A, Berens ME, Hersh E, McMahon G, Hirth KP, Powell TJ. Inhibition of platelet-derived growth factor-mediated signal transduction and tumor growth by N-[4-(trifluoromethyl)-phenyl]5-methylisoxazole-4-carboxamide. Clin Cancer Res. 1997;3:1167–1177. [PubMed] [Google Scholar]
- 77.Ko YJ, Small EJ, Kabbinavar F, Chachoua A, Taneja S, Reese D, DePaoli A, Hannah A, Balk SP, Bubley GJ. A multi-institutional phase ii study of SU101, a platelet-derived growth factor receptor inhibitor, for patients with hormone-refractory prostate cancer. Clin Cancer Res. 2001;7:800–805. [PubMed] [Google Scholar]
- 78.Mathew P, Tannir N, Tu SM, Wen S, Guo CC, Marcott V, Bekele BN, Pagliaro L. Accelerated disease progression in prostate cancer and bone metastases with platelet-derived growth factor receptor inhibition: observations with tandutinib. Cancer Chemother Pharmacol. 2011 doi: 10.1007/s00280-011-1567-2. [DOI] [PubMed] [Google Scholar]
- 79.Hwang C, Heath EI. Angiogenesis inhibitors in the treatment of prostate cancer. J Hematol Oncol. 2010;3:26. doi: 10.1186/1756-8722-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Epstein RJ. VEGF signaling inhibitors: more pro-apoptotic than anti-angiogenic. Cancer Metastasis Rev. 2007;26:443–452. doi: 10.1007/s10555-007-9071-1. [DOI] [PubMed] [Google Scholar]
- 81.Zhao Q, Egashira K, Hiasa K, Ishibashi M, Inoue S, Ohtani K, Tan C, Shibuya M, Takeshita A, Sunagawa K. Essential role of vascular endothelial growth factor and Flt-1 signals in neointimal formation after periadventitial injury. Arterioscler Thromb Vasc Biol. 2004;24:2284–2289. doi: 10.1161/01.ATV.0000147161.42956.80. [DOI] [PubMed] [Google Scholar]
- 82.Zhong H, Bowen JP. Molecular design and clinical development of VEGFR kinase inhibitors. Curr Top Med Chem. 2007;7:1379–1393. doi: 10.2174/156802607781696855. [DOI] [PubMed] [Google Scholar]
- 83.Paavonen K, Puolakkainen P, Jussila L, Jahkola T, Alitalo K. Vascular endothelial growth factor receptor-3 in lymphangiogenesis in wound healing. Am J Pathol. 2000;156:1499–1504. doi: 10.1016/S0002-9440(10)65021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143:401–409. [PMC free article] [PubMed] [Google Scholar]
- 85.Borre M, Offersen BV, Nerstrom B, Overgaard J. Microvessel density predicts survival in prostate cancer patients subjected to watchful waiting. Br J Cancer. 1998;78:940–944. doi: 10.1038/bjc.1998.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duque JL, Loughlin KR, Adam RM, Kantoff PW, Zurakowski D, Freeman MR. Plasma levels of vascular endothelial growth factor are increased in patients with metastatic prostate cancer. Urology. 1999;54:523–527. doi: 10.1016/s0090-4295(99)00167-3. [DOI] [PubMed] [Google Scholar]
- 87.George DJ, Halabi S, Shepard TF, Vogelzang NJ, Hayes DF, Small EJ, Kantoff PW. Prognostic significance of plasma vascular endothelial growth factor levels in patients with hormone-refractory prostate cancer treated on Cancer and Leukemia Group B 9480. Clin Cancer Res. 2001;7:1932–1936. [PubMed] [Google Scholar]
- 88.El-Gohary YM, Silverman JF, Olson PR, Liu YL, Cohen JK, Miller R, Saad RS. Endoglin (CD105) and vascular endothelial growth factor as prognostic markers in prostatic adenocarcinoma. Am J Clin Pathol. 2007;127:572–579. doi: 10.1309/X6NXYE57DLUE2NQ8. [DOI] [PubMed] [Google Scholar]
- 89.Borgstrom P, Bourdon MA, Hillan KJ, Sriramarao P, Ferrara N. Neutralizing anti-vascular endothelial growth factor antibody completely inhibits angiogenesis and growth of human prostate carcinoma micro tumors in vivo. Prostate. 1998;35:1–10. doi: 10.1002/(sici)1097-0045(19980401)35:1<1::aid-pros1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 90.Reese David M., Fratesi P, Corry M, Novotny W, Holmgren E, Small Eric J. A Phase II Trial of Humanized Anti-Vascular Endothelial Growth Factor Antibody for the Treatment of Androgen-Independent Prostate Cancer. The Prostate Journal. 2001;3:65–70. [Google Scholar]
- 91.Ning YM, Gulley JL, Arlen PM, Woo S, Steinberg SM, Wright JJ, Parnes HL, Trepel JB, Lee MJ, Kim YS, Sun H, Madan RA, Latham L, Jones E, Chen CC, Figg WD, Dahut WL. Phase II trial of bevacizumab, thalidomide, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:2070–2076. doi: 10.1200/JCO.2009.25.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Picus J, Halabi S, Kelly WK, Vogelzang NJ, Whang YE, Kaplan EB, Stadler WM, Small EJ. A phase 2 study of estramustine, docetaxel, and bevacizumab in men with castrate-resistant prostate cancer: results from Cancer and Leukemia Group B Study 90006. Cancer. 2011;117:526–533. doi: 10.1002/cncr.25421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pouessel D, Culine S. [Actualities in prostate cancer in ASCO annual meeting 2010] Bull Cancer. 2010;97:1563–1572. doi: 10.1684/bdc.2010.1222. [DOI] [PubMed] [Google Scholar]
- 94.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 95.Ullen A, Farnebo M, Thyrell L, Mahmoudi S, Kharaziha P, Lennartsson L, Grander D, Panaretakis T, Nilsson S. Sorafenib induces apoptosis and autophagy in prostate cancer cells in vitro. Int J Oncol. 2010;37:15–20. doi: 10.3892/ijo_00000648. [DOI] [PubMed] [Google Scholar]
- 96.Bono AV, Pannellini T, Liberatore M, Montironi R, Cunico SC, Cheng L, Sasso F, Musiani P, Iezzi M. Sorafenib's inhibition of prostate cancer growth in transgenic adenocarcinoma mouse prostate mice and its differential effects on endothelial and pericyte growth during tumor angiogenesis. Anal Quant Cytol Histol. 2010;32:136–145. [PubMed] [Google Scholar]
- 97.Dahut WL, Scripture C, Posadas E, Jain L, Gulley JL, Arlen PM, Wright JJ, Yu Y, Cao L, Steinberg SM, Aragon-Ching JB, Venitz J, Jones E, Chen CC, Figg WD. A phase II clinical trial of sorafenib in androgen-independent prostate cancer. Clin Cancer Res. 2008;14:209–214. doi: 10.1158/1078-0432.CCR-07-1355. [DOI] [PubMed] [Google Scholar]
- 98.Chi KN, Ellard SL, Hotte SJ, Czaykowski P, Moore M, Ruether JD, Schell AJ, Taylor S, Hansen C, Gauthier I, Walsh W, Seymour L. A phase II study of sorafenib in patients with chemo-naive castration-resistant prostate cancer. Ann Oncol. 2008;19:746–751. doi: 10.1093/annonc/mdm554. [DOI] [PubMed] [Google Scholar]
- 99.Aragon-Ching JB, Jain L, Gulley JL, Arlen PM, Wright JJ, Steinberg SM, Draper D, Venitz J, Jones E, Chen CC, Figg WD, Dahut WL. Final analysis of a phase II trial using sorafenib for metastatic castration-resistant prostate cancer. BJU Int. 2009;103:1636–1640. doi: 10.1111/j.1464-410X.2008.08327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roskoski R., Jr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356:323–328. doi: 10.1016/j.bbrc.2007.02.156. [DOI] [PubMed] [Google Scholar]
- 101.Guerin O, Formento P, Lo Nigro C, Hofman P, Fischel JL, Etienne-Grimaldi MC, Merlano M, Ferrero JM, Milano G. Supra-additive antitumor effect of sunitinib malate (SU11248, Sutent) combined with docetaxel. A new therapeutic perspective in hormone refractory prostate cancer. J Cancer Res Clin Oncol. 2008;134:51–57. doi: 10.1007/s00432-007-0247-4. [DOI] [PubMed] [Google Scholar]
- 102.Cumashi A, Tinari N, Rossi C, Lattanzio R, Natoli C, Piantelli M, Iacobelli S. Sunitinib malate (SU-11248) alone or in combination with low-dose docetaxel inhibits the growth of DU-145 prostate cancer xenografts. Cancer Lett. 2008;270:229–233. doi: 10.1016/j.canlet.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 103.Dror Michaelson M, Regan MM, Oh WK, Kaufman DS, Olivier K, Michaelson SZ, Spicer B, Gurski C, Kantoff PW, Smith MR. Phase II study of sunitinib in men with advanced prostate cancer. Ann Oncol. 2009;20:913–920. doi: 10.1093/annonc/mdp111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sonpavde G, Periman PO, Bernold D, Weckstein D, Fleming MT, Galsky MD, Berry WR, Zhan F, Boehm KA, Asmar L, Hutson TE. Sunitinib malate for metastatic castration-resistant prostate cancer following docetaxel-based chemotherapy. Ann Oncol. 2010;21:319–324. doi: 10.1093/annonc/mdp323. [DOI] [PubMed] [Google Scholar]
- 105.Castellano D, Gonzalez-Larriba JL, Anton-Aparicio LM, Cassinello J, Grande E, Esteban E, Sepulveda J. Experience in the use of sunitinib given as a single agent in metastatic chemoresistant and castration-resistant prostate cancer patients. Expert Opin Pharmacother. 2011 doi: 10.1517/14656566.2011.590132. [DOI] [PubMed] [Google Scholar]
- 106.Ou Y, Michaelson MD, Sengeløv L. Randomized, placebo-controlled, phase III trial of sunitinib in combination with prednisone (SU+P) versus prednisone (P) alone in men with progressive metastatic castration-resistant prostate cancer (mCRPC).. ASCO Annual Meeting; 2011. e. al. Abstract 4515. [DOI] [PubMed] [Google Scholar]
- 107.Gregorakis AK, Holmes EH, Murphy GP. Prostate-specific membrane antigen: current and future utility. Semin Urol Oncol. 1998;16:2–12. [PubMed] [Google Scholar]
- 108.Smith-Jones PM, Vallabhajosula S, Navarro V, Bastidas D, Goldsmith SJ, Bander NH. Radiolabeled monoclonal antibodies specific to the extracellular domain of prostate-specific membrane antigen: preclinical studies in nude mice bearing LNCaP human prostate tumor. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2003;44:610–617. [PubMed] [Google Scholar]
- 109.Henry MD, Wen S, Silva MD, Chandra S, Milton M, Worland PJ. A prostate-specific membrane antigen-targeted monoclonal antibody-chemotherapeutic conjugate designed for the treatment of prostate cancer. Cancer Res. 2004;64:7995–8001. doi: 10.1158/0008-5472.CAN-04-1722. [DOI] [PubMed] [Google Scholar]
- 110.Galsky MD, Eisenberger M, Moore-Cooper S, Kelly WK, Slovin SF, DeLaCruz A, Lee Y, Webb IJ, Scher HI. Phase I trial of the prostate-specific membrane antigen-directed immunoconjugate MLN2704 in patients with progressive metastatic castration-resistant prostate cancer. J Clin Oncol. 2008;26:2147–2154. doi: 10.1200/JCO.2007.15.0532. [DOI] [PubMed] [Google Scholar]
- 111.Bander NH, Trabulsi EJ, Kostakoglu L, Yao D, Vallabhajosula S, Smith-Jones P, Joyce MA, Milowsky M, Nanus DM, Goldsmith SJ. Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J Urol. 2003;170:1717–1721. doi: 10.1097/01.ju.0000091655.77601.0c. [DOI] [PubMed] [Google Scholar]
- 112.Kuroda K, Liu H, Kim S, Guo M, Navarro V, Bander NH. Saporin toxin-conjugated monoclonal antibody targeting prostate-specific membrane antigen has potent anticancer activity. Prostate. 2010;70:1286–1294. doi: 10.1002/pros.21164. [DOI] [PubMed] [Google Scholar]
- 113.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 114.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 115.Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, Harper F, Gaskins M, Pitot HC, Tan W, Ivy SP, Pili R, Carducci MA, Erlichman C, Liu G. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Cancer Res. 2008;14:7940–7946. doi: 10.1158/1078-0432.CCR-08-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Matthews SB, Vielhauer GA, Manthe CA, Chaguturu VK, Szabla K, Matts RL, Donnelly AC, Blagg BS, Holzbeierlein JM. Characterization of a novel novobiocin analogue as a putative C-terminal inhibitor of heat shock protein 90 in prostate cancer cells. Prostate. 2010;70:27–36. doi: 10.1002/pros.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kang BH, Siegelin MD, Plescia J, Raskett CM, Garlick DS, Dohi T, Lian JB, Stein GS, Languino LR, Altieri DC. Preclinical characterization of mitochondria-targeted small molecule hsp90 inhibitors, gamitrinibs, in advanced prostate cancer. Clin Cancer Res. 2010;16:4779–4788. doi: 10.1158/1078-0432.CCR-10-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, Altieri DC. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest. 2009;119:454–464. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Y, Mikhailova M, Bose S, Pan CX, deVere White RW, Ghosh PM. Regulation of androgen receptor transcriptional activity by rapamycin in prostate cancer cell proliferation and survival. Oncogene. 2008;27:7106–7117. doi: 10.1038/onc.2008.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Amato RJ, Jac J, Mohammad T, Saxena S. Pilot study of rapamycin in patients with hormone-refractory prostate cancer. Clin Genitourin Cancer. 2008;6:97–102. doi: 10.3816/CGC.2008.n.015. [DOI] [PubMed] [Google Scholar]
- 121.Wu L, Birle DC, Tannock IF. Effects of the mammalian target of rapamycin inhibitor CCI-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res. 2005;65:2825–2831. doi: 10.1158/0008-5472.CAN-04-3137. [DOI] [PubMed] [Google Scholar]
- 122.Molife LR, Attard G, Fong PC, Karavasilis V, Reid AH, Patterson S, Riggs CE, Jr., Higano C, Stadler WM, McCulloch W, Dearnaley D, Parker C, de Bono JS. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC) Ann Oncol. 2010;21:109–113. doi: 10.1093/annonc/mdp270. [DOI] [PubMed] [Google Scholar]
- 123.Ferrini R, Woolf SH. American College of Preventive Medicine practice policy. Screening for prostate cancer in American men. Am J Prev Med. 1998;15:81–84. doi: 10.1016/s0749-3797(98)00050-6. [DOI] [PubMed] [Google Scholar]
- 124.Bittner N, Merrick GS, Andreini H, Taubenslag W, Allen ZA, Butler WM, Anderson RL, Adamovich E, Wallner KE. Prebiopsy PSA velocity not reliable predictor of prostate cancer diagnosis, Gleason score, tumor location, or cancer volume after TTMB. Urology. 2009;74:171–176. doi: 10.1016/j.urology.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 125.Skvortsov S, Schafer G, Stasyk T, Fuchsberger C, Bonn GK, Bartsch G, Klocker H, Huber LA. Proteomics profiling of microdissected low- and high-grade prostate tumors identifies Lamin A as a discriminatory biomarker. J Proteome Res. 2011;10:259–268. doi: 10.1021/pr100921j. [DOI] [PubMed] [Google Scholar]
- 126.de Kok JB, Verhaegh GW, Roelofs RW, Hessels D, Kiemeney LA, Aalders TW, Swinkels DW, Schalken JA. DD3(PCA3), a very sensitive and specific marker to detect prostate tumors. Cancer Res. 2002;62:2695–2698. [PubMed] [Google Scholar]
- 127.Goodman OB, Jr., Symanowski JT, Loudyi A, Fink LM, Ward DC, Vogelzang NJ. Circulating Tumor Cells as a Predictive Biomarker in Patients With Hormone-sensitive Prostate Cancer. Clin Genitourin Cancer. 2011 doi: 10.1016/j.clgc.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 128.Detchokul S, Frauman AG. Recent developments in prostate cancer biomarker research: therapeutic implications. Br J Clin Pharmacol. 2011;71:157–174. doi: 10.1111/j.1365-2125.2010.03766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Madu CO, Lu Y. Novel diagnostic biomarkers for prostate cancer. J Cancer. 2010;1:150–177. doi: 10.7150/jca.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang G, Zhang H, Wang Q, Lal P, Carroll AM, de la Llera-Moya M, Xu X, Greene MI. Suppression of human prostate tumor growth by a unique prostate-specific monoclonal antibody F77 targeting a glycolipid marker. Proc Natl Acad Sci U S A. 2010;107:732–737. doi: 10.1073/pnas.0911397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tomlins SA, Rhodes DR, Yu J, Varambally S, Mehra R, Perner S, Demichelis F, Helgeson BE, Laxman B, Morris DS, Cao Q, Cao X, Andren O, Fall K, Johnson L, Wei JT, Shah RB, Al-Ahmadie H, Eastham JA, Eggener SE, Fine SW, Hotakainen K, Stenman UH, Tsodikov A, Gerald WL, Lilja H, Reuter VE, Kantoff PW, Scardino PT, Rubin MA, Bjartell AS, Chinnaiyan AM. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell. 2008;13:519–528. doi: 10.1016/j.ccr.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ozaki N, Ohmuraya M, Hirota M, Ida S, Wang J, Takamori H, Higashiyama S, Baba H, Yamamura K. Serine protease inhibitor Kazal type 1 promotes proliferation of pancreatic cancer cells through the epidermal growth factor receptor. Mol Cancer Res. 2009;7:1572–1581. doi: 10.1158/1541-7786.MCR-08-0567. [DOI] [PubMed] [Google Scholar]
- 133.Li F, Ling X. Survivin study: an update of “what is the next wave”? J Cell Physiol. 2006;208:476–486. doi: 10.1002/jcp.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sah NK, Khan Z, Khan GJ, Bisen PS. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006;244:164–171. doi: 10.1016/j.canlet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 135.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 136.Zhang M, Latham DE, Delaney MA, Chakravarti A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene. 2005;24:2474–2482. doi: 10.1038/sj.onc.1208490. [DOI] [PubMed] [Google Scholar]
- 137.Zhang M, Ho A, Hammond EH, Suzuki Y, Bermudez RS, Lee RJ, Pilepich M, Shipley WU, Sandler H, Khor LY, Pollack A, Chakravarti A. Prognostic value of survivin in locally advanced prostate cancer: study based on RTOG 8610. Int J Radiat Oncol Biol Phys. 2009;73:1033–1042. doi: 10.1016/j.ijrobp.2008.06.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang H, Holloway MP, Ma L, Cooper ZA, Riolo M, Samkari A, Elenitoba-Johnson KS, Chin YE, Altura RA. Acetylation directs survivin nuclear localization to repress STAT3 oncogenic activity. J Biol Chem. 2010;285:36129–36137. doi: 10.1074/jbc.M110.152777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, Kita A, Tominaga F, Yamanaka K, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 140.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, van Klaveren RJ, Chaudhary S, Gunther A, Shamsili S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 141.Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, Lawson D, Whitman E, Gonzalez R. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2011;29:161–166. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- 142.Tolcher AW, Mita A, Lewis LD, Garrett CR, Till E, Daud AI, Patnaik A, Papadopoulos K, Takimoto C, Bartels P, Keating A, Antonia S. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol. 2008;26:5198–5203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sun M, Lou W, Chun JY, Cho DS, Nadiminty N, Evans CP, Chen J, Yue J, Zhou Q, Gao AC. Sanguinarine Suppresses Prostate Tumor Growth and Inhibits Survivin Expression. Genes Cancer. 2010;1:283–292. doi: 10.1177/1947601910368849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Berezov A, Cai Z, Freudenberg JA, Zhang H, Cheng X, Thompson T, Murali R, Greene MI, Wang Q. Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene. 2011 doi: 10.1038/onc.2011.377. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fishman M. A changing world for DCvax: a PSMA loaded autologous dendritic cell vaccine for prostate cancer. Expert Opin Biol Ther. 2009;9:1565–1575. doi: 10.1517/14712590903446921. [DOI] [PubMed] [Google Scholar]