

Abstract
A generally applicable, easy-to-use method of focusing a patient's immune system to eradicate or prevent cancer has been elusive. We are attempting to develop a targeted virus to accomplish these aims. We previously created a recombinant replicating Vesicular Stomatitis Virus that preferentially infected Her2/neu expressing breast cancer cells and showed therapeutic efficacy in an implanted Balb/c mouse tumor model. The current work shows that this therapy generated therapeutic anti-tumor CD4 T-cells against multiple tumor antigens. CD4 T-cells transferred directly from cured donor mice could eradicate established tumors in host mice. T-cells were transferred directly from donor mice and were not stimulated ex vivo. Both tumors that expressed Her2/neu and those that did not were cured by transferred T-cells. Analysis of cytokines secreted by anti-tumor memory CD4 T-cells displayed a multifunctional pattern with high levels of IFNγ, IL-4 and IL-17. Anti-tumor memory CD4 T-cells traveled to the mesenteric lymph nodes and were activated there. Treatment with targeted rrVSV is a potent immune adjuvant that generates therapeutic, multifunctional anti-tumor memory CD4 T-cells that recognize multiple tumor antigens. Immunity elicited by viral therapy is independent of host major histocompatibility complex (MHC) or knowledge of tumor antigens. Virus-induced tumor immunity could have great benefit in the prevention and treatment of tumor metastases.
Keywords: anti-tumor memory T-cells, VSV, targeted virus, cancer therapy
Introduction
The immune system can rapidly destroy a large transplanted organ but directing this latent force to eradicate cancer tissue has been very difficult. Cancer immunotherapy began with bacterial vaccinations and now includes passive transfer of antibody or immune cells and tumor vaccination using a variety of strategies.1-5 Despite some successes, a reliable, safe, easy-to-use, reasonably priced, general immunological technique to treat or prevent solid tumor metastases is not available. We are developing a tumor-targeted replicating recombinant VSV to be used both directly for immune-mediated tumor therapy and prophylactically to induce anti-tumor immunity. Prophylactic use would entail administering the virus to the primary tumor at the initial clinical presentation in order to prevent the later development of tumor metastases. The foundation for this strategy is established in part by the data in this paper which shows that treatment with rrVSV generates highly potent anti-tumor memory CD4 T-cells.
VSV is an excellent candidate for development for cancer therapy because it is a safe oncolytic virus that can be genetically engineered.6 Normal tissues are protected from the virus by interferon (IFN) production but most human tumors are insensitive to the effects of IFN and are more susceptible to killing by VSV.7, 8 Viral oncolysis releases multiple tumor antigens in the context of an anti-viral inflammatory response. This inflammatory response stimulates an immune response instead of tolerance to the tumor antigens. The VSV genome can be altered to attenuate the virus or to have it express immune modifiers. Therapeutic effects in animal models have been observed using wild type VSV (wt VSV), VSV modified with cytokines and VSV in combination with chemotherapy.9-12 A theoretical safety concern is whether the virus will be tolerated by cancer patients who are immunologically compromised. Several approaches to address this issue are being developed such as creating an rVSV expressing IFNβ,8 creating a VSV M-protein mutant that induced 20 to 50 times more IFNα than wt VSV or administering IFN systemically during VSV infection.7 We improved the safety profile by creating a recombinant replicating VSV (rrVSV) with an altered surface glycoprotein (gp) that targeted preferentially to breast cancer cells highly expressing the Her2/neu receptor, erbb2. The viral genome was also modified by the inclusion of genes expressing mouse GM-CSF and green fluorescent protein.13 We showed that this rrVSV selectively infected, replicated and killed cells expressing erbb2.13, 14 We then showed that therapy with rrVSV expressing GM-CSF combined with anti-CTLA4 monoclonal antibody (MAb) could eliminate established macroscopic tumor implants. Successful therapy required both CD4 and CD8 T-cell responses in the treated animals. Surviving animals were resistant to tumor re-challenge suggesting a memory immune response.
We now sought to further characterize this anti-tumor memory immune response. We found that CD4 T-cells were responsible for the immune memory response, were highly potent and could support a strategy of dealing with the problem of cancer metastases by immunoprevention. Anti-tumor memory T-cells were obtained from donor animals whose established tumors were cured by therapy with rrVSV and anti-CTLA4 MAb. Cytokine production by these cells was studied using the Milliplex cytokine kit as well as EliSpot and intracellular IFN cytometry. Therapeutic efficacy was studied by transfer to host animals with implanted peritoneal tumors. We found that the memory CD4 T-cells were powerful and able to cure established tumors in host animals 40-50% of the time. T-cells were transferred directly from donor to host mice. No ex vivo stimulation was required. As expected, we found that rrVSV treatment generated memory T-cells to multiple tumor antigens. Unexpectedly, CD4 T-cells alone mediated the memory anti-tumor effect. Transferring CD8 T-cells, B-cells or antibody from cured animals in addition to CD4 T-cells did not improve outcome. Also unexpectedly, the memory anti-tumor CD4 T-cells were not apparently restricted to Th1, Th2 or Th17 type but appeared multifunctional expressing a diverse array of cytokines including IFNγ, IL-4 and IL-17. Cytokine secretion of specific anti-tumor memory T-cells was most effectively studied by in vivo tumor challenge and analysis of lymph node T-cells.
Materials and Methods
Cells, antibodies, chemicals and animals
D2F2/E2 cells, a mouse mammary tumor line that has been stably transfected with a vector expressing the human Her2/neu gene and its parent cell line, D2F2 were a generous gift from Dr. Wei-Zen Wei, Karmanos Cancer Institute, Wayne State University, Detroit, MI. Anti-CTLA4 (9H10) 15 ascites was prepared from a hybridoma generously supplied by Dr. James P. Allison, Memorial Sloan Kettering Cancer Center, New York, NY or obtained commercially (BioXcell Fermentation/Purification Services #BE0131, West Lebanon, NH). Cytoxan (Cyclophosphamide, #NDC 0015-0502-42, Bristol-Myers Squibb Co., Princeton, NJ) was freshly diluted in sterile water to a stock concentration of 20 mg/ml. Stock solution of 125 μl was freshly diluted in 375 μl of PBS and administered IP. All animal studies were conducted using female BALB/c mice, 8 to 20 weeks of age, weighing 20-25g, obtained from Taconic (Hudson, NY). These animal studies were approved by the institutional Animal Research and Care Committee.
rrVSV
rrVSV targeted to cells expressing Her2/neu was created from vector components as previously described.13 In brief, vectors expressing the VSV genome (XN2) and the individual VSV genes P, L, N and G (pBS-P, L, N and G respectively) on a T7 promoter were a very generous gift of Dr. John K. Rose, Yale University School of Medicine. Vectors expressing Sindbis glycoprotein (gp) and Sindbis gp modified between amino acids 71 and 74 to express two IgG binding domains (Sindbis-ZZ) were generously supplied by Dr. Irvin S. Y. Chen, University of California, Los Angeles Medical School. A vector expressing a single chain antibody (SCA) based on the 4D5 anti-erbb2 antibody was a generous gift by Genentech Inc. As previously described, we used PCR to create a chimeric Sindbis gp which consisted of the first 71 amino acids of the Sindbis E2 gp followed in order by a poly-glycine linker, SCA to erbb2, CH1 linker, the remainder of the E2 gp and the entire E1 Sindbis gp. 16 The gene for the native VSV-G gp was removed from the VSV genome (XN2) and replaced with a gene coding for the chimeric Sindbis gp. In addition, genes coding for enhanced green fluorescent protein (EGFP) and mouse GM-CSF were added to the VSV genome producing a genome of 14,838 bases. Replicating recombinant VSV was created using standard techniques13, 17 that expressed only the chimeric Sindbis gp on its surface and also expressed EGFP and GM-CSF. This rrVSV was then adapted to grow well on D2F2/E2 cells by serial passage in vitro on this cell line.18 rrVSV for animal trials was made by infecting D2F2/E2 cells at a multiplicity of infection (MOI) =0.001 in 168 cm2 tissue culture flask (Corning/CoStar, Corning, NY) and harvesting supernatant 48h later. Titers of rrVSV in the supernatant were typically 1-3 × 108/ml on D2F2/E2 cells assayed by counting green cells as previously described.13
Cell collection
Animals were sacrificed prior to cell harvesting. Spleens were harvested, minced and ground through a 70 μM nylon cell strainer (#352350, BD Falcon, Franklin Lakes, NJ). RBC were lysed by incubating the cell suspension in 0.16M tris-buffered NH4CL for 5 minutes. Bone marrow cells were aspirated from both femurs and tibias, passed through the cell strainer and underwent RBC lysis. Mesenteric lymph node cells were collected by careful dissection of the lymph nodes in the peritoneum and grounding through the cell strainer. RBC lysis was performed when necessary. Peritoneal washings were performed by injecting 10 ml of sterile PBS into the peritoneum through a 16 gauge needle which was left in place. Five minutes later all the fluid that could be aspirated easily into the syringe was collected. Usually 9.0 ml was collected. Mononuclear cells were collected, when necessary, by centrifugation over lymphocyte separation media (#25-072-CV, Mediatech, Inc., Manassas, VA). All cells were washed twice with PBS and re-suspended in PBS.
T and B cell isolation
Total T-cells, B-cells, CD4 T-cells and CD8 T-cells were isolated by positive selection using the autoMACS™ separator and the appropriate antibody microbeads according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA): CD90 (Thy1.2, #130-049-101), CD19 (#130-052-201), CD4 (L3T4, #130-049-201), CD8a (Ly-2, #130-049-401).
Flow cytometric intracellular cytokine analysis
Intracellular staining for mouse IFNγ was performed using the Cytofix/Cytoperm Plus Kit with GolgiPlug (#555028, Becton Dickinson, Mountainview, CA) as recommended by the manufacturer. 5 × 106 spleen cells per well were plated in 1 ml of DMEM containing 10% FCS in Linbro 24 well tissue culture plates (#76-033-05, ICN Biomedicals, Aurora, Ohio) and incubated overnight at 37°C with 5% CO2. Following incubation with brefeldin A (final concentration at 10 μg/ml) for 4 hrs, T-cells were isolated by positive selection as above. 1 × 106 T-cells were suspended in ice-cold PBS/0.1% BSA/0.2% Azide and stained with phycoerythrin (PE)-conjugated antibodies to either CD4 or CD8 or isotype control antibody (BD #12-0042-82, 12-0081-82 and 12-4321-82). The cells were fixed and permeabilized by incubation with 250 ul cytofix/cytoperm solution for 20 min on ice and stained with APC-conjugated antibody to mouse IFNγ or isotype control antibody(BD #12-7311-81 and 17-4714-71). Immunofluorescence was quantified using a FACStarPlus cytometer (Becton Dickinson, Mountainview, CA).
EliSpot
Elispot analyses for IFNγ producing T-cells were performed using the mouse IFN-γ ELISpot PLUS kit with white precoated plates, HRP (#3321-4HPW-4, MABTECH). Following in vivo challenge, 2 × 105 T-cells from spleen, lymph nodes and peritoneal fluid were incubated in 0.1 ml media in separate wells without further stimulation for 48 hours at 37°C in a 5% CO2 incubator. Exposure of the spots was performed according to manufacturer's recommendations and the spots were visualized and quantified with an Elispot microscope reader (Carl Zeiss MicroImaging Inc., Thronwood, NY).
Cytokine and Chemokine analyses
Cytokine secretion was quantified using the Milliplex map mouse cytokine/chemokine kit (Millipore Co., Billerica, MA) which simultaneously measured the following cytokines: G-CSF, GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, IP-10, KC, MCP-1, MIG, MIP-1α, RANTES, TNF-α. CD4 and CD8 T-cells were fractionated as above and 1 × 106 cells suspended in 200 μl DMEM with 10% FCS were plated at 37°C in a 5% CO2 incubator in individual wells of a 96 well round bottom tray for 24 hours(Corning Inc., Corning, NY). A Luminex xMAP reader (Bio-Rad, Hercules, CA) was used to measure cytokine concentration in 25 μl aliquots of supernatant from each well using standard samples supplied by the manufacturer and assayed according to the manufacturers instructions
In vitro stimulation studies were performed by first incubating 1 × 105 DC with 1 × 104 tumor cells for 6 hrs in 200 μl media at 37°C in a 5% CO2 incubator. Tumor cells were either live, killed and disrupted by 3 rounds of freeze-thaw or killed by exposure to mitomycin (25 μg/ml). Then, 5 × 103 DC were incubated with 5 × 104 CD4 or CD8 T-cells in 250 μl media in individual wells of a 96 well round bottom tray for 24 hours. Cytokine concentration in supernatant was measured as above.
Treatment trials
Female BALB/c mice were implanted intraperitoneally (IP) with 2 × 106 D2F2/E2 cells in 500 μl PBS. All viral and antibody treatments were administered IP. Adoptive cell transfer was usually administered IP except for 2 animals that received IV treatment as noted in the text and figure. Animals were assessed three times per week for ascites, abdominal nodules and signs of poor health such as low activity, poor grooming, rough coat, hunched posture and dehydration and sacrificed if they developed ascites, nodules or any of these signs. The animals were considered cured if they survived for 100 days after tumor.
Statistics
The log rank statistic was used to compare survival among the treatment groups. The Mann-Whitney one-tailed test was used to compare cytokine secretion in the CD4 T-cells from various treatment groups. This non-parametric test was used because some values in the Milliplex analyses were too low to be accurately measured and were arbitrarily assigned a value lower than the lowest recorded value. PRISM software was used to analyze the data (GraphPad Software, Inc., La Jolla, CA).
Results
We had previously used re-challenge experiments to demonstrate that successful viral therapy produced anti-tumor immunity. A Balb/c mouse mammary cancer cell line, D2F2, had been stably transfected by others with a vector expressing the human Her2/neu receptor creating the cell line, D2F2/E2.19 D2F2/E2 cells were implanted in the peritoneum and mice were treated 3 days later with rrVSV targeted to Her2/neu and anti-CTLA4 antibody. Animals surviving for 100 days were considered cured and then re-challenged with intraperitoneal tumor. In a small series of animals, we showed that most animals were able to resist re-challenge with first the Her2/neu expressing D2F2/E2 cells and then the non-Her2/neu expressing parental cell line, D2F2.20 We now confirmed this finding in a larger series of animals and used transfer experiments to identify the effective memory cells.
Re-challenge cured animals
Fifteen long term survivors of D2F2/E2 implantation (including 5 previously reported) who had been treated with rrVSV and aCTLA4 MAb were re-challenged with D2F2/E2 cells. These mice did not receive any therapy. Fourteen survived >100 days indicating the presence of immunity to this tumor cell line. One died 34 days after challenge. It was important to determine whether survival was determined by immunity only to the foreign Her2/neu receptor protein which had been introduced into the D2F2/E2 cells or whether the immunity extended to the parent D2F2 cells. Twelve of these animals were then challenged with D2F2 cells. Nine never developed tumor thereby demonstrating that rrVSV therapy had resulted in immunity to the fully syngeneic D2F2 cells. Three animals died 31-40 days after challenge indicating partial immunity because the median time to death in naïve animals following tumor challenge has been 16 to 20 days.16 Five long term survivors were challenged directly with D2F2 cells and all survived indicating that immunity to D2F2 antigens developed after initial viral therapy and did not require a first re-challenge with D2F2/E2.
Transfer T-cells from cured animals
A series of transfer experiments was performed to prove that viral immunotherapy was generating anti-tumor memory T-cells. In agreement with previous work, we found that transferred cells from cured donor animals were only effective when host animals were pre-treated with a single dose of cyclophosphamide (CPM) at 100-125 mg/kg.21 Pre-treatment with CPM is apparently required to make room for the transferred cells, to stimulate production of host cytokines such as IL-7 or IL-17 or to suppress inhibitory T-cell subsets.21-23
The first experiment used spleen cells from cured donor animals to prevent tumor development. Spleen cells, 4-6 × 107 cells, were administered intravenously (IV, 2 animals) or intraperitoneally (IP, 2 animals) one day prior to intraperitoneal tumor challenge (Fig. 1). None of the animals given donor spleen cells ever developed tumors whereas the animals given CPM alone promptly developed tumors and were sacrificed (median survival=24.5 days). Thus, as reported by others, spleen cells from cured donor animals were able prevent tumor establishment.24
Fig. 1.

Survival curves of D2F2/E2 implants treated with adoptive transfer of spleen cells from cured animals. Spleen cells, 4-6 × 107 cells, were administered intravenously (IV, 2 animals) or intraperitoneally (IP, 2 animals) one day prior to intraperitoneal tumor challenge. All treated animals survived. Controls received CPM alone and all died with a median survival of 24.5 days (log rank statistic p=0.018).
Next, we attempted a more stringent model which asked whether transferred cells could eradicate established tumors in host animals. Animals were implanted with tumor cells IP and treated 3 days later with cells transferred from cured mice. Spleen cells (mean: 2.7 × 107 cells) which contain about 1/3 T-cells eradicated these established macroscopic tumor in about 50% of cases (Fig. 2). Antibody did not appear to play a significant role because adding serum from cured mice to the spleen cells did not improve survival. In addition, bone marrow cells, which include memory B-cells, administered at about the same number as spleen cells (mean: 2.4 × 107 cells) were ineffective. Finally, lymph node cells which contain about 2/3 T-cells were also ineffective but the dose of transferred cells was 10-fold lower (mean: 2.4 × 106 cells). In addition, it is possible that more anti-tumor memory T-cells resided in the spleen than the lymph nodes in resting cured donor mice.
Fig. 2.

Survival curves of D2F2/E2 implants treated with adoptive transfer of various cells from cured mice. Animals were implanted with tumor cells IP and treated 3 days later IP with transferred spleen cells ((mean: 2.7 × 107 cells), bone marrow cells (mean: 2.4 × 107 cells) lymph node cells (mean: 2.4 × 106 cells) or CPM only (n=5 for each group). 100 ul of serum from cured mice was added to one group that received spleen cells (n=4). (Compared to CPM control, survival was significantly improved in the group treated with spleen cells, log rank statistic p=0.013 and the group treated with spleen cells and serum, log rank statistic p=0.0047).
In order to determine whether memory T-cells alone were responsible for cure or a combination of memory lymphocytes was required, we compared therapy using spleen T-cells with therapy using a combination of T and B-cells (Fig. 3). We found no difference between the groups indicating that the anti-tumor effect was carried by the T-cells alone.
Fig. 3.

Survival curves of D2F2/E2 implants treated with adoptive transfer of T and B cells from cured mice. Animals were implanted with tumor cells IP and treated 3 days later IP with transferred T-cells (mean: 2.8 × 107 cells) with and without B cells (mean: 5.6 × 107 cells). (n=8 pairs). There was no statistical difference in outcome between the two groups (log rank statistic p=0.57).
We then asked whether the memory anti-tumor effect was mediated by CD4 T-cells, CD8 T-cells or both. We found that transfer of memory CD4 T-cells produced cure in 37.5% of cases (Fig. 4). Adding CD8 T-cells to the CD4 T-cells produced no significant additional benefit and CD8 T-cells alone were not more effective than CPM alone. The larger number of transferred CD4 than CD8 T-cells reflected the increased number of CD4 T-cells harvested from individual mouse spleen.
Fig. 4.

Survival curves of D2F2/E2 implants treated with adoptive transfer of CD4 and CD8 T-cells from cured mice. Animals were implanted with tumor cells IP and treated 3 days later IP with transferred CD4 T-cells ((mean: 2.0 × 107 cells), CD8 T-cells (mean: 3.9 × 106 cells) or both. (n=8 sets). Control animals received CPM only (n=5). Treatment with CD4 T-cells (log rank statistic p=0.012) and CD4 plus CD8 T-cells (log rank statistic p=0.0005) were statistically superior to control but not different from each other (log rank statistic p=0.58). Treatment with CD8 T-cells was not statistically different from control (log rank statistic p=0.18).
In further extensions of these transfer experiments, we found that T-cells from cured mice were able to eradicate not only 3 day old but also 5 day old established tumor in all five of a set of 5 host animals. More importantly we proved that anti-tumor memory T-cells recognized antigens distinct from Her2/neu. T-cells from cured mice were administered to host animals bearing 3 day established tumor of the parent cell line, D2F2. Tumors were eradicated in 50% of animals (n=8).
Long lasting immunity was transferred from donor to host animals as demonstrated by re-challenge experiments (Table 1). Sixteen host animals cured by transfer of T-cells from donor animals were re-challenged with D2F2/E2 cells. Fourteen survived and 2 died at 22 and 38 days after challenge. Twelve of these animals were then re-challenged with D2F2 cells. Eight survived and 4 died at 24, 29, 29 and 63 days after challenge. It is most likely that transferred CD4 T-cells survived in the hosts and were responsible for this permanent immunity but we did not pursue proof of this point
Table 1. Re-challenge with either erbb2 expressing D2F2/E2 or parent tumor D2F2 in animals cured by T-cell transfer.
| Cells | Total | Survival | |
|---|---|---|---|
| First re-challenge | D2F2/E2 | 16 | 14 (88%) |
| Second re-challenge* | D2F2 | 12 | 8 (67%) |
All of these animals survived a first re-challenge with D2F2/E2
Characterize the anti-tumor memory T-cells
We sought to determine whether viral immunotherapy generated predominantly a single functional CD4 T-cells response such as Th1, Th2 or Th17 or a multifunctional response. In addition, we wanted to clearly demonstrate that the memory CD4 T-cells responded to native antigens from the parent D2F2 cells and not just the foreign Her2/neu receptor protein which had been introduced into the D2F2/E2 cells. Cured animals, >100 days after tumor implant and viral therapy, were therefore re-challenged with either D2F2/E2 cells or D2F2 cells. Three days later, CD4 and CD8 T-cells were harvested from spleen, mesenteric lymph nodes and peritoneal lavage and allowed to secrete cytokines overnight without further stimulation. Supernatants were harvested and assayed for a wide variety of cytokines and chemokines. A standard control group consisted of naïve animals that were challenged with D2F2/E2 cells and sacrificed 3 days later. However, we required a more stringent control group because the experimental groups received virus and anti-CTLA4 in addition to tumor and we wanted to be sure that we were assaying anti-tumor and not anti-viral T-cells. The crucial control group, therefore, consisted of animals that did not receive tumor but were treated with virus and anti-CTLA4 antibody. Sixty to 100 days later, they were challenged with D2F2/E2 cells just like the experimental group and T-cells were harvested 3 days after challenge. The T-cells were placed in wells without any stimulation and cytokine concentration was measured after 24 hours of secretion. The primary question was whether the pattern of cytokine secretion in the experimental groups was different than the virus only control, indicating a specific memory anti-tumor response. A consistent anti-tumor memory response was found in lymph node CD4 T-cells. Lymph node CD4 T-cells from the D2F2/E2 challenged experimental animals had significant elevations of the following cytokines compared to D2F2/E2 challenged virus controls (p≤0.02): IFNγ, IL-4, IL-5, IL-17, IL-2, IL-3, IL-10, GM-CSF, IP-10, MIG, MIP-1α, and RANTES (Table 2). These CD4 T-cells were multifunctional, secreting cytokines characteristic of Th1, Th2 and Th17 T-cells.25 In almost all cases the cytokine response was mildly higher when the animals were challenged with D2F2/E2 than the parent cell D2F2. However, in all cases the response in animals challenged with D2F2 was higher than in the control groups. The T-cell cytokine response in naïve animals challenged with D2F2/E2 was very low indicating that the high responses found in the experimental animals were generated by the viral therapy and that memory T-cells responded to native antigens found in D2F2 cells as well as the Her2/neu antigen expressed in D2F2/E2 cells.
Table 2.
Cytokine production by CD4 T-cells harvested from lymph nodes in animals receiving various treatment.
A. Implanted with D2F2/E2. Treated with rrVSV and anti-CTLA4 MAb. Challenged >100 days later with D2F2/E2 (erbb2 expressing).
B. Implanted with D2F2/E2. Treated with rrVSV and anti-CTLA4 MAb. Challenged >100 days later with D2F2 (non-erbb2 expressing).
C. No tumor implanted. Treated with rrVSV and anti-CTLA4 MAb. Challenged >60-100 days later with D2F2/E2 (erbb2 expressing).
D. No tumor. No treatment. Challenged with D2F2/E2 (erbb2 expressing). Values are concentration of cytokine in supernatant, pg/ml. Means of 4 experiments.
| A | B | C | D | |
|---|---|---|---|---|
|
| ||||
| IFNγ | 500 | 302 | 68 | 2 |
|
| ||||
| IL-4 | 165 | 73 | 18 | 7 |
|
| ||||
| IL-5 | 557 | 385 | 173 | 1 |
|
| ||||
| IL-17 | 8 | 9 | 0 | 0 |
|
| ||||
| IL-2 | 236 | 203 | 69 | 5 |
|
| ||||
| IL-10 | 122 | 55 | 17 | 2 |
|
| ||||
| GM-CSF | 633 | 354 | 100 | 2 |
|
| ||||
| IL-3 | 475 | 347 | 95 | 5 |
|
| ||||
| MIP-1α | 154 | 99 | 12 | 6 |
|
| ||||
| Cured tumor | yes | yes | no | no |
| Virus | yes | yes | yes | no |
| anti-CTLA4 | yes | yes | yes | no |
| Re-challenge | D2F2/E2 | D2F2 | D2F2/E2 | D2F2/E2 |
Unlike CD4 T-cells, CD8 T-cells from lymph nodes did not show a clear pattern of cytokine secretion that was different in the experimental from the control group. CD4 and CD8 T-cells isolated from spleen and peritoneal cells following in vivo tumor challenge also did not show a pattern of cytokine secretion that was consistently different in the experimental from the control groups. Finally, cytokine concentrations in blood and peritoneal fluid following in vivo tumor challenge did not show distinct patterns in the experimental group.
Analysis by EliSpot supported the cytokine secretion data. IFNγ secreting CD4 T-cells were assayed from mesenteric lymph nodes following challenge with D2F2/E2 or D2F2. The most positive cells were seen in the experimental group challenged with D2F2/E2 but this group and the group challenged with D2F2 had more positive cells than the control groups challenged with D2F2/E2 (Fig. 5).
Fig. 5.

Number of IFNγ secreting CD4 T-cells harvested from lymph nodes in animals receiving various treatments. Means of 3 experiments with standard error bars.
A. Implanted with D2F2/E2. Treated with rrVSV and anti-CTLA4 MAb. Challenged >100 days later with D2F2/E2 (erbb2 expressing).
B. Implanted with D2F2/E2. Treated with rrVSV and anti-CTLA4 MAb. Challenged >100 days later with D2F2 (non-erbb2 expressing).
C. No tumor implanted. Treated with rrVSV and anti-CTLA4 MAb. Challenged >60-100 days later with D2F2/E2 (erbb2 expressing).
D. No tumor. No treatment. Challenged with D2F2/E2 (erbb2 expressing).
These experiments which assayed T-cells after in vivo stimulation were more successful in demonstrating a memory T-cell response to tumor antigens than experiments utilizing in vitro stimulation. In a single experiment, we harvested T-cells from spleens of cured animals, challenged them with dendritic cells (DC) loaded with tumor antigens and assayed cytokine response using the Milliplex cytokine kit. Tumor antigens were obtained from freeze/thawed tumor cells, mitomycin treated tumor cells and live tumor cells. Results in the experimental animals were not clearly different than the control animal who received virus and anti-CTLA4 MAb but no tumor. The in vitro conditions were not able to adequately simulate antigen presentation as it occurs in the lymph nodes of live animals challenged with tumor.
Attempts to identify and quantify the anti-tumor T-cells response at earlier times following therapy were clouded by a very strong anti-viral T-cell response, as noted by others.26 In one set of 3 independent paired experiments, an experimental group implanted with D2F2/E2 and treated with virus and anti-CTLA4 was compared with a control group receiving virus and anti-CTLA4 but no tumor. All mice were challenged with D2F2/E2 30 days after treatment. Three days later mesenteric lymph nodes were harvested and EliSpot analyses performed on T-cells. The mean number of activated IFNγ secreting CD4 T-cells was 168 per 105 lymph node cells in the experimental group (range 104-216) and 112 in the control group (range 61-169). This difference was not statistically different. In another set of 3 independent experiments, experimental groups implanted with D2F2/E2 and treated with virus and anti-CTLA4 were compared with control groups receiving tumor and treated with virus alone, groups receiving tumor alone or groups receiving virus plus anti-CTLA4 but no tumor. Spleens were harvested 4 days after viral therapy and intracellular flow cytometric analyses performed on T-cells. In all experiments, the experimental group had about 3% of CD4 T-cells expressing IFNγ (Fig. 6) but values in the no tumor control group receiving virus and anti-CTLA4 varied from 1.6%-3.6% and the differences were not statistically significant.
Fig. 6.
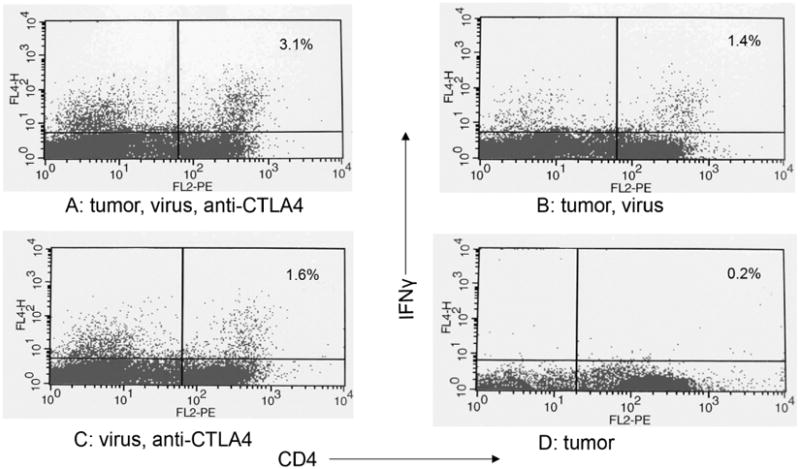
Mononuclear cells were collected from spleens and incubated overnight to let macrophages adhere tightly to the flask. Following incubation with brefeldin A for 4 hrs, T-cells were isolated by positive selection using the autoMACS™ separator and CD90 (Thy1.2) microbeads (Miltenyi Biotec, Auburn, CA). T-cells were stained with phycoerythrin (PE)-conjugated antibodies (FL2) to CD4. The cells were then fixed and permeabilized and stained with APC-conjugated antibody to mouse IFNγ (FL4). Immunofluorescence was quantified using a FACStarPlus (Becton Dickinson, Mountainview, CA). One experiment of 3.
A. Animal implanted with tumor and treated with rrVSV and anti-CTLA4
B. Animal implanted with tumor and treated with rrVSV
C. Animal was not implanted with tumor but was treated with rrVSV and anti-CTLA4
D. Animal was implanted with tumor but received no treatment.
Discussion
Cancer therapy has been attempted using viruses and using immunotherapy. Passive immunotherapy using antibodies or ex vivo activated amplified CD8 T-cells has had some success against specific cancers.4, 27 Active immunization using a variety of techniques has had limited impact.3, 28, 29 Virus therapy alone has also been unsuccessful. Recently, viral therapy with immunotherapy has shown some clinical benefit.26 Our goal is to combine virus and immune therapy by developing a safe, easy-to-use targeted virus that consistently evokes anti-tumor immunity that eradicates or prevents tumor metastases. Our previous work has shown that a targeted rrVSV expressing GM-CSF combined with anti-CTLA4 MAb can eliminate established small macroscopic tumor implants through an immunologic mechanism. This paper proves conclusively that targeted rrVSV generates long-lived therapeutic anti-tumor memory CD4 T-cells that recognize multiple tumor antigens. These CD4 T-cells are capable of orchestrating a curative anti-tumor response that eradicates small established tumor implants.
A major advantage of using an oncolytic virus to induce active immunotherapy is that multiple tumor antigens are released in situ in the context of an anti-viral inflammatory response producing the ultimate personalized therapy. Each individual in the genetically heterogeneous human population will generate immunity to the antigen or antigens that best fit their unique MHC profile. This therapy requires no knowledge of specific tumor antigens and is independent of the patient's MHC. The development of potent immune responses against multiple antigens was clearly shown in this model. As expected, the most potent immune response was generated against the foreign Her2/neu receptor protein as shown by cure rates in transfer experiments and analysis of cytokine secretion following in vivo tumor challenge. However, curative responses in transfer experiments and stimulation of tumor-specific CD4 T-cells as shown by cytokine secretion were also found with challenge by D2F2 cells which did not express Her2/neu. We recognize that it is important to confirm these findings in a fully syngeneic model and are currently performing studies in a transgenic mouse which expresses the human Her2/neu receptor under the murine mammary tumor virus promoter.30 We will also attempt to treat spontaneous tumors which develop in 76% of animals in this model system.
An unexpected finding in these studies was the anti-tumor potency of memory CD4 T-cells. Transfer of these cells alone was able to cure established tumors in host animals without apparent additional benefit by also transferring CD8 T-cells or B-cells. We were able to harvest and transfer more CD4 T-cells than CD8 T-cells from each donor animal and it is possible that transferring a larger number of CD8 T-cells would have shown greater effect. A contributing role for memory CD8 T-cells can therefore not be excluded. Importantly, host animals had endogenous CD8 T-cells and B-cells which may have contributed to the therapeutic response but the only memory cells required from the donor mice were CD4 T-cells. Most clinical programs of cancer immunotherapy attempt to generate or activate anti-tumor CD8 T-cells.31, 32 These T-cells can directly attack tumor cells by attaching to their surface class I antigens. In contrast, CD4 T-cells can not directly recognize tumor cells because tumor cells do not generally express class II antigens on their surface. Instead, CD4 T-cells detect tumor cells indirectly via the antigens that they shed and are picked up and presented by dendritic cells and macrophages. Previous work has shown that transferred CD4 T-cells can eliminate tumors in host mice but these studies have either used T-cells from transgenic mice that express a monoclonal CD4 T-cell population or activated the cells ex vivo or used additional therapy in conjunction with the transferred cells.33-40 A single patient has been treated successfully with autologous CD4 T-cells activated ex vivo but other patients treated with the same protocol did not have a successful outcome.25, 41 The anti-tumor memory T-cells generated by therapy with rrVSV came from animals with a normally diverse immune system, were not stimulated ex vivo and did not require any adjunctive therapy to cure established tumors at least 5 days old and to cure tumors composed of fully syngeneic tumor cells. The location and mechanism of activation of transferred CD4 T-cells has previously been questioned.33 This paper demonstrates that anti-tumor memory CD4 T-cells traveled to the mesenteric lymph nodes and were activated there, presumably by DCs and macrophages that migrated with tumor antigens from the peritoneal implants of tumor.24, 33, 42 We did not explore the mechanisms by which CD4 T-cells lead to tumor elimination but previous work in other model systems has shown dependence on macrophages, PMN and NK cells.24, 39 These findings support the development of adoptive transfer of memory CD4 T-cells for cancer therapy despite technical challenges such as expanding the number of cells without inhibiting their activity and dealing with the Class II heterogeneity in the human population.25
Another unexpected finding was the apparent multifunctional nature of the anti-tumor memory CD4 T-cell response. The usual emphasis for tumor immunotherapy is to try to stimulate a Th1 response and thereby activate a cellular response which can directly kill tumor cells.43 Recently there is interest in activating a Th17 response which is thought to be involved with autoimmunity and can be directed against the autologous tumor cells.34, 44, 45 Th2 responses are generally thought to counteract antitumor immunity28 though this has not been true in every model system.36 This study found that anti-tumor memory CD4 T-cells stimulated by tumor antigens in the mesenteric lymph nodes secreted a heterogeneous group of cytokines characteristic of Th1, Th2 and Th17 cells. The key to generating a sustained immune response that eradicates all cancer cells may be a balance among the CD4 T-cell subsets.46 We recognize that further work is required to prove that each subset is not only present but necessary to achieve tumor cure and to characterize the balance required among the subsets, quantitatively, temporally and topographically.
Immunotherapy would benefit greatly from markers of anti-tumor immune response. In the clinic, survival as a measure of response can take years to quantify. Valid markers could identify ineffective therapies early and indicate the need for new tactics. In addition, markers that correlated with efficacy could help elucidate the mechanisms of successful immunotherapy. This study points out the difficulties in finding useful markers. As noted by others, viral therapy yields a strong anti-viral immune response which overshadows the anti-tumor response.26 Equally problematic, the anti-tumor response may only be characterizable in tissues and under conditions that do not translate to the clinic. In our model, anti-tumor memory CD4T-cells were clearly present in the mesenteric lymph nodes following in vivo stimulation, as expected.46 However, they were not found in spleen and peritoneal fluid following in vivo stimulation and not found in spleen following in vitro stimulation. Cytokine response in blood and peritoneal fluid following in vivo stimulation also did not yield useful markers. A hopeful finding was that the Milliplex cytokine kit was more sensitive than EliSpot or intracellular flow cytometry at detecting anti-tumor T-cells and we are presently undertaking analysis of temporal patterns in CD4 T-cells following acute therapy to identify valid markers of effector anti-tumor CD4 T-cell response. Interestingly, the cytokine with the greatest amplification in experimental animals compared to viral controls was MIP-1α (CCL3). This may be explained by a recent report showing that MIP-1α is secreted by activated CD4 T-cells which are in contact with DC.47 Assay of this chemokine at the appropriate time and location after viral therapy may provide a marker to the development of anti-tumor immunity.
The goal for rrVSV therapy is to eradicate active, growing Her2/neu positive tumor metastases and to prevent growth of occult, dormant metastatic collections. Local disease is not a clinical problem because it can be surgically extirpated. The current study supports the development of rrVSV as an in vivo vaccine to prevent growth of tumor metastases. The plan would be to administer rrVSV to the primary tumor in order to stimulate a powerful anti-tumor memory T-cell response. Major obstacles hindering other cancer vaccines would be obviated because no knowledge of specific tumor antigens is required and immunity would develop in patients with any set of MHC markers.28 The circulating memory CD4 T-cells would orchestrate a curative anti-tumor response whenever occult metastases started growing and released tumor antigens to regional lymph nodes. Nascent metastases would be eliminated before they developed immunosuppressive properties. A good test case for these concepts would be advanced ovarian cancer. rrVSV would be administered to Her2/neu positive cancers at the time of initial tumor therapy in order to kill cancer cells that had implanted in the peritoneum and more importantly, to generate anti-tumor memory CD4 T-cells which would then prevent future outgrowth of intraperitoneal and distant metastases.
Acknowledgments
The contents of this study are solely the responsibility of the authors and do not necessarily represent the official views of the granting institution. We thank Drs. Wei-Zen Wei, John K. Rose, Irvin S. Y. Chen, James P. Allison and Genentech Inc. who very generously supplied materials as noted in the text. We thank Erich Scheller for technical assistance.
Footnotes
Conflict of Interest: None of the authors have any competing financial interests in relation to the work described.
Contributor Information
Yanhua Gao, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA.
Patricia Whitaker-Dowling, Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, PA.
Judith A. Griffin, Department of Pediatrics, University of Pittsburgh, Pittsburgh, PA.
Ira Bergman, Departments of Pediatrics, Neurology, and Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA.
References
- 1.Wei MQ, Mengesha A, Good D, Anne J, Wei MQ, Mengesha A, et al. Bacterial targeted tumour therapy-dawn of a new era. Cancer Lett. 2008;259:16–27. doi: 10.1016/j.canlet.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 2.Finn OJ, Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–2715. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drake CG, Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10:580–593. doi: 10.1038/nri2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Mattos CA, de Mattos CC, Rupprecht CE. Rhabdoviruses. In: Knipe D, Howley P, editors. Fundamental Virology. 4. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 1245–1277. [Google Scholar]
- 7.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 8.Obuchi M, Fernandez M, Barber GN. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J Virol. 2003;77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 11.Porosnicu M, Mian A, Barber GN. The oncolytic effect of recombinant vesicular stomatitis virus is enhanced by expression of the fusion cytosine deaminase/uracil phosphoribosyltransferase suicide gene. Cancer Res. 2003;63:8366–8376. [PubMed] [Google Scholar]
- 12.Ebert O, Shinozaki K, Huang TG, Savontaus MJ, Garcia-Sastre A, Woo SL. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 2003;63:3605–3611. [PubMed] [Google Scholar]
- 13.Bergman I, Whitaker-Dowling P, Gao Y, Griffin JA. Preferential targeting of vesicular stomatitis virus to breast cancer cells. Virology. 2004;330:24–33. doi: 10.1016/j.virol.2004.06.048. [DOI] [PubMed] [Google Scholar]
- 14.Pilon SA, Kelly C, Wei WZ. Broadening of epitope recognition during immune rejection of ErbB-2-positive tumor prevents growth of ErbB-2-negative tumor. J Immunol. 2003;170:1202–1208. doi: 10.4049/jimmunol.170.3.1202. [DOI] [PubMed] [Google Scholar]
- 15.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11:728–734. [PubMed] [Google Scholar]
- 16.Bergman I, Griffin JA, Gao Y, Whitaker-Dowling P, Bergman I, Griffin JA, et al. Treatment of implanted mammary tumors with recombinant vesicular stomatitis virus targeted to Her2/neu. Int J Cancer. 2007;121:425–430. doi: 10.1002/ijc.22680. [DOI] [PubMed] [Google Scholar]
- 17.Lawson ND, Stillman EA, Whitt MA, Rose JK. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci U S A. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Y, Whitaker-Dowling P, Watkins SC, Griffin JA, Bergman I. Rapid adaptation of a recombinant vesicular stomatitis virus to a targeted cell line. J Virol. 2006;80:8603–8612. doi: 10.1128/JVI.00142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei WZ, Shi WP, Galy A, Lichlyter D, Hernandez S, Groner B, et al. Protection against mammary tumor growth by vaccination with full-length, modified human ErbB-2 DNA. Int J Cancer. 1999;81:748–754. doi: 10.1002/(sici)1097-0215(19990531)81:5<748::aid-ijc14>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 20.Gao Y, Whitaker-Dowling P, Griffin JA, Barmada MA, Bergman I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009;16:44–52. doi: 10.1038/cgt.2008.55. [DOI] [PubMed] [Google Scholar]
- 21.Bracci L, Moschella F, Sestili P, LaS V, Valentini M, Canini I, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007;13:644–653. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 22.Taieb J, Chaput N, Schartz N, Roux S, Novault S, Menard C, et al. Chemoimmunotherapy of tumors: cyclophosphamide synergizes with exosome based vaccines. J Immunol. 2006;176:2722–2729. doi: 10.4049/jimmunol.176.5.2722. [DOI] [PubMed] [Google Scholar]
- 23.Viaud S, Flament C, Zoubir M, Pautier P, LeCesne A, Ribrag V, et al. Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res. 2011;71:661–665. doi: 10.1158/0008-5472.CAN-10-1259. [DOI] [PubMed] [Google Scholar]
- 24.Liu Z, Noh HS, Chen J, Kim JH, Falo LD, Jr, You Z, et al. Potent tumor-specific protection ignited by adoptively transferred CD4+ T cells. J Immunol. 2008;181:4363–4370. doi: 10.4049/jimmunol.181.6.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muranski P, Restifo NP, Muranski P, Restifo NP. Adoptive immunotherapy of cancer using CD4(+) T cells. Curr Opin Immunol. 2009;21:200–208. doi: 10.1016/j.coi.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, Ugolini M, et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010;70:4297–4309. doi: 10.1158/0008-5472.CAN-09-3567. [DOI] [PubMed] [Google Scholar]
- 27.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 28.Palucka K, Ueno H, Banchereau J, Palucka K, Ueno H, Banchereau J. Recent developments in cancer vaccines. J Immunol. 2011;186:1325–1331. doi: 10.4049/jimmunol.0902539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, et al. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finkle D, Quan ZR, Asghari V, Kloss J, Ghaboosi N, Mai E, et al. HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin Cancer Res. 2004;10:2499–2511. doi: 10.1158/1078-0432.ccr-03-0448. [DOI] [PubMed] [Google Scholar]
- 31.Morgan RA, Dudley ME, Rosenberg SA, Morgan RA, Dudley ME, Rosenberg SA. Adoptive cell therapy: genetic modification to redirect effector cell specificity. Cancer J. 2010;16:336–341. doi: 10.1097/PPO.0b013e3181eb3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg SA, Dudley ME, Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen PA, Peng L, Plautz GE, Kim JA, Weng DE, Shu S, et al. CD4+ T cells in adoptive immunotherapy and the indirect mechanism of tumor rejection. Crit Rev Immunol. 2000;20:17–56. [PubMed] [Google Scholar]
- 34.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lundin KU, Hofgaard PO, Omholt H, Munthe LA, Corthay A, Bogen B, et al. Therapeutic effect of idiotype-specific CD4+ T cells against B-cell lymphoma in the absence of anti-idiotypic antibodies. Blood. 2003;102:605–612. doi: 10.1182/blood-2002-11-3381. [DOI] [PubMed] [Google Scholar]
- 36.Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med. 1999;190:617–627. doi: 10.1084/jem.190.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kennedy R, Celis E, Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–144. doi: 10.1111/j.1600-065X.2008.00616.x. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Tian S, Falo LD, Jr, Sakaguchi S, You Z, Liu Z, et al. Therapeutic immunity by adoptive tumor-primed CD4(+) T-cell transfer in combination with in vivo GITR ligation. Mol Ther. 2009;17:1274–1281. doi: 10.1038/mt.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109:5346–5354. doi: 10.1182/blood-2006-10-051318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–667. doi: 10.1084/jem.20091921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H, Bi XG, Yuan JY, Xu SL, Guo XL, Xiang J. Combined CD4+ Th1 effect and lymphotactin transgene expression enhance CD8+ Tc1 tumor localization and therapy. Gene Ther. 2005;12:999–1010. doi: 10.1038/sj.gt.3302486. [DOI] [PubMed] [Google Scholar]
- 44.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kryczek I, Wei S, Szeliga W, Vatan L, Zou W, Kryczek I, et al. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114:357–359. doi: 10.1182/blood-2008-09-177360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKinstry KK, Strutt TM, Swain SL, McKinstry KK, Strutt TM, Swain SL. The potential of CD4 T-cell memory. Immunology. 2010;130:1–9. doi: 10.1111/j.1365-2567.2010.03259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castellino F, Huang AY, tan-Bonnet G, Stoll S, Scheinecker C, Germain RN, et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440:890–895. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]