

Abstract
After many years of debate, there is now general agreement that B cells can participate in the immune response to cardiac transplants. Acute antibody-mediated rejection (AMR) is the best defined manifestation of B cell responses, but diagnostic and mechanistic questions still surround AMR. Many complement dependent mechanisms of antibody-mediated injury have been elucidated. C5 has become a therapeutic target that may not just truncate complement activation, but also may tip the balance away from inflammation by altering macrophage function. More complement independent effects have been identified. These may escape diagnosis and progress to chronic graft injury.
The function of B cell infiltrates in cardiac transplants is even more enigmatic. Nodular endocardial infiltrates that contain B cells and plasma cells have been described in protocol biopsies of cardiac transplants for decades, but an understanding of their significance is still evolving based on more critical morphological and molecular evaluation of these infiltrates. A range of infiltrates containing B cells has also been described in the epicardial fat in transplants with advanced chronic rejection. B cells have been observed in endocardial and epicardial tertiary lymphoid nodules, but their impact on antigen presentation or antibody production remains to be determined. Experimental models in small and large animals suggest that B cells could be essential for the formation of lymphoid nodules through cytokine production. Similarly, the role of proinflammatory adipokines in the formation or function of epicardial lymphoid nodules has not been studied.
These clinical observations provide critical questions to be addressed in experimental models.
Keywords: Antibody mediated rejection, Complement activation, Endocardial lymphoid nodules, Tertiary lymphoid organogenesis, Perivascular adipose tissue
1. Introduction
The potential effects of antibodies and B cell infiltrates on cardiac transplants have been the source of controversy for decades. Antibody-mediated rejection (AMR) was not accepted in the standardized grading system of the International Society for Heart and Lung Transplantation until 2004 (1). Although many questions are not resolved, antibodies are now widely considered to cause injury and even rejection of some heart transplants (2, 3). Diagnosis of AMR is based on a triad of serological, histological and functional findings. The most generally recognized findings include donor specific antibody in the circulation, deposits of complement split products (C4d and/or C3d) in the capillaries of the biopsy and signs of cardiac dysfunction. Based on these criteria, AMR is diagnosed in about 1–10% of biopsies (2–4). The debate now concerns whether AMR is more pervasive than is currently diagnosed. Arguments and mechanisms have been advanced for antibodies causing or at least contributing to rejection in the absence of one or more of the recognized criteria for AMR. For example, complement independent mechanisms of graft injury have been invoked in cases of graft dysfunction associated with circulating donor specific antibodies in the absence of C4d or C3d deposits (5). Advances in knowledge about the effector mechanisms of antibodies are providing new insights to improve diagnosis and treatment of AMR. Therefore, one focus of this review will be effector mechanisms elicited by antibodies in transplants.
Similarly, nodular endocardial infiltrates containing B cells and plasma cells have been described in protocol biopsies of cardiac transplants since 1981 (6), but an understanding of their significance is still evolving based on more critical morphological and molecular evaluations of these infiltrates. A range of infiltrates containing B cells has also been described in the epicardial fat in transplants with advanced chronic rejection (7, 8). The potential importance of these endocardial and epicardial infiltrates will be a second focus of this review.
The final focus of this review will be on experimental approaches to address evolving clinical questions about B cells in cardiac transplants.
2. New Insights into Antibody Mediated Rejection (AMR)
Cardiac transplants are closely monitored by protocol biopsies of the endocardium. The frequent biopsies provide an opportunity for assessing the occurrence of B cells and antibodies in symptomatic and asymptomatic cardiac transplants. However, diagnosis of AMR has been challenging because of the functional properties of antibodies. Although antibodies need to bind target antigens to initiate rejection, the antibodies only need to bind transiently in order to initiate a wide variety of inflammatory functions. The transient binding of antibodies makes them an elusive marker for AMR, and this was the basis for much of the controversy over early reports of AMR. However, the effects initiated by antibodies are more reliably assessed and more relevant to rejection. The most direct effects result from IgG or IgM antibodies cross-linking antigens on tissues. In addition, antibodies can activate the complement system and leukocytes. Whether one or more of these mechanisms is activated depends on many variables including the isotype, concentration, avidity and specificity of the antibodies. With increasing sophistication of serological tests, more data are available about these variables for circulating antibodies. However, it is not clear whether antibodies in the circulation accurately represent antibodies that are bound to the graft.
Increasingly detailed phenotypic and molecular studies of biopsies are providing greater insights about different effects of antibodies relative to activation of complement, leukocytes and endothelial cells. We will discuss each of these actions of antibodies in the following sections.
2.1 Complement activation
Activation of complement is the most extensively studied and diagnostically best characterized function of antibodies. In humans, IgG1 and 3 and to a lesser degree IgG2 can activate complement through C1 and Mannose Binding Lectin (MBL). The relative binding of C1 and MBL depends upon the structure of the carbohydrate side chains on the Fc of IgG antibodies (9). The glycosylation of antibodies is not invariable. Variations in the carbohydrate structure of IgG autoantibodies have been recognized for many years. Autoantibodies that lack the terminal sialic acid and galactose (referred to as G0 antibodies) expose terminal N-acetylglucosamine (GlcNAc) residues. Mannose binding lectin binds avidly to GlcNAc. As a result, G0 autoantibodies activate complement through the lectin pathway in both humans and mice (9, 10). The dynamics of glycosylation of antibodies requires further investigation, but it has been reported to be influenced by pregnancy and treatment with infliximab and possibly mycophenolate mofetil (11).
Detection of C1 or MBL has not been a sensitive or reliable indicator of AMR in biopsies. This is consistent with the facts that C1 and MBL are bound in lower concentrations than antibody (one C1 or MBL binds to 2 or more IgG molecules) and they are transiently bound to tissues through antibodies.
Both C1 and MBL enzymatically cleave numerous molecules of C4, and the C4 split product C4b can covalently bind to tissues. The biological importance of C4b is two fold. First, C4b is one of the ligands for complement receptor 1 (CR1; CD 35), which is expressed on circulating leukocytes. Second, C4b complexes with the split product of C2 to form the classical convertase that cleaves C3. C4b is important diagnostically because during regulation of the complement cascade, C4b is cleaved to an inactive split product C4d that remains covalently linked to the tissue. Moreover, the cleavage process reveals a cryptic epitope on C4d. As a result, monoclonal and polyclonal reagents to this cryptic epitope produce sensitive and specific immunohistological stains for complement activation (Fig 1). However, the weak inflammatory properties of C4b and biological inactivity of C4d mean that positive stains for C4d do not necessarily correlate with rejection. In fact, C4 can moderate immune responses as evidenced by the fact that deficiencies in C4 are associated with increased autoimmune diseases. In transplantation, C4d deposits in the absence of accompanying inflammatory infiltrates are not associated with graft dysfunction, but with a state that has been termed accommodation. Accommodation has been most frequently observed in ABO blood group incompatible renal transplants. These transplants are feasible with living donors because they require extensive scheduled treatments of the recipient to decrease antibody titers before transplantation. ABO incompatible heart transplants are not intentionally performed with the notable exception of neonatal recipients. West and colleagues have successfully transplanted ABO incompatible hearts to infants before the age when they develop “natural” antibodies to A and B presumably in response to bacterial colonization of their gut. These transplants induce tolerance to the donor blood group (12). The donor antigens remain expressed on the graft endothelium, but recipient B cells do not produce antibodies to donor or recipient blood group antigens.
Fig. 1.

Endomyocardial biopsy with pathological evidence of AMR. Evidence of edema separating myocytes on routine hematoxylin and eosin stain. Large numbers of interstitial and intracapillary macrophages stained for CD68. Strong linear stains of capillaries for C4d and C3d.
Whether accommodation occurs in cardiac transplants remains to be determined, but Tan and colleagues have reported a cohort of adult recipients of ABO compatible cardiac transplants, whose endocardial biopsies have strong linear deposits of C4d in the absence of apparent graft dysfunction. Only a third of these patients had detectable circulating donor specific antibodies, and therefore, may represent a clinical situation in which low levels of antibodies activate complement at a gradual pace or in limited amounts that allow C4 to be continually inactivated before significant amounts of C3 are cleaved. In fact, many of these biopsies had detectable decay accelerating factor (DAF; CD55) and protectin (CD59), two complement regulatory molecules that have been shown to be upregulated in accommodation. DAF accelerates the disassociation of C3 convertase and leaves C4b attached to tissue. It is not known what mechanisms control the expression of complement regulators in transplants, but TNFα and IFNγ increase DAF expression by endothelial cells in vitro (13). Mediators associated with chronic inflammation, such as basic FGF and VEGF also upregulate the expression of DAF.
Although structurally similar to C4, the split products of C3 have more inflammatory functions. Similar to C4b, C3b can bind covalently to tissues, but C3b represents a focal point of the complement system because C3b can initiate the alternative pathway of complement, which serves as an amplification loop. This can result in tenfold more C3b than C4b bound to the tissues, and greatly increases the density of ligands for CR1, which binds both C3b and C4b. Moreover, C3a, the small split product of C3, is a chemoattractant for neutrophils and macrophages and serves to bring these cells towards the tissue bound ligands. This is one mechanism to account for the association of neutrophils and macrophages with AMR.
The immunological balance is further shifted towards inflammation by the participation of C3b in the formation of C5 convertases with both classical and alternative complement components. These C5 convertases cleave C5 into C5a and C5b. C5b initiates the formation of the membrane attack complex (MAC; C5b-C9). This is an inefficient process and is countered by various regulators on allografts, including CD59. As a result, endothelial lysis is not widespread in AMR. However, sublytic amounts of MAC can activate endothelial cells as has been documented extensively using cell cultures and purified C5b through C9 (14–16). The immediate effect of endothelial cell activation is retraction of the plasma membrane from the underlying substrate (17), which would result in edema (Fig 1). Edema has been proposed as a secondary diagnostic indicator of AMR (3). At the same time, endothelial cells exocytose preformed von Willebrand factor (vWf) and P-selectin from their Weibel-Palade storage granules (14), which can promote adhesion of platelets and monocytes to the endothelium and stimulate their production of inflammatory mediators (18). In addition to releasing preformed mediators, endothelial cells activated by MAC in synergy with TNFα or IL1α synthesize IL-8, monocyte chemoattractant protein 1 (MCP-1; CCL2), P-selectin, E-selectin and ICAM-1 (16). These chemokines and adhesion molecules participate in the recruitment and activation of leukocytes in the transplant. Through these mechanisms, MAC could contribute to the accumulation of macrophages in capillaries that is observed in acute AMR (Fig 1). Of relevance to chronic rejection, MAC also induces endothelial cells to produce growth factors (platelet-derived growth factor and basic fibroblast growth factor; PDGF and FGF) and to proliferate (15).
C5a has pivotal impact in AMR (Fig 2). Not only is C5a an extremely potent chemoattractant for neutrophils and macrophages, but it also causes macrophages to upregulate the stimulatory FcgRIII and down regulate the inhibitory FcgRIIB for IgG (19). These receptors make macrophages more responsive to antibodies and complement split products. In addition, C5a increases vascular leakage through C5a receptors on mast cells and endothelial cells. These are among the reasons that treatment with antibodies to C5 or C5a receptor antagonists are effective in inhibiting AMR (20). Decreasing C5a is also likely to moderate antigen release by endothelial cells and antigen presentation by macrophages and dendritic cells.
Fig. 2.

Diagram of increasingly proinflammatory effects of complement activation by antibody. Leukocytes with Fc receptors (e.g., neutrophils and macrophages) can be stimulated by antibodies bound to antigen. The strength of Fc receptor interactions with IgG is dependent on the carbohydrate side chains (yellow hexagons) on the antibodies. The carbohydrate side chains on IgG also interact with the first components of the classical and lectin complement pathways. At least 2 closely spaced IgG antibodies are required to initiate binding of one C1 or MBL molecule. C1 or MBL can enzymatically cleave many molecules of C4, which produce C4b that can bind covalently to cell membranes. C4b anchors the classical convertase (a complex of C4b and C2a) that cleaves large numbers of C3 producing C3b, which can bind covalently to tissues, and C3a, which is chemoattractant for neutrophils and macrophages. C3b joins C4bC2a in the formation of classical C5 convertase, which cleaves C5 into C5a and C5b. C5b initiates the formation of the membrane attack complex (MAC; C5b-C9). Both the C3 and C5 convertases are the target of several regulators of complement activation (RCA). As a result, endothelial lysis is not widespread in AMR, but sublytic amounts of MAC can stimulate endothelial cells to produce cytokines, express adhesion molecules and shed microparticles containing the offending MAC. C5a is pivotal inflammatory mediator because it is an extremely potent chemoattractant for neutrophils and macrophages, and it also causes macrophages to upregulate the stimulatory FcγRIII and down regulate the inhibitory FcγRIIB for IgG. These receptors make macrophages more responsive to antibodies and complement split products.
Several corollaries emerge from the view that the complement cascade progresses from components with anti-inflammatory functions (C1 and C4 in particular) to terminal components (beginning with C5) with extremely inflammatory effects (as depicted in Fig 2). Of most immediate relevance to transplantation is the application of inhibitors to C5, which are currently being tested clinically. For example, Eculizumab, a monoclonal antibody to C5, has been used with the general rationale of blocking terminal complement activation. To most investigators this is envisioned primarily as blocking possible lysis of endothelial cells. While this may be a part of the mechanism, there are at least two other important mechanisms relative to antigenic stimulation. One is that Eculizumab also prevents formation of sublytic amounts of MAC that stimulate endothelial cells to produce cytokines, to express adhesion molecules and to shed microparticles containing the offending MAC. These microparticles presumably also contain donor antigens, and thus increase the antigen delivered to recipient lymphoid tissues. In addition, Eculizumab decreases the levels of C5a and as a result decreases chemotaxis and activation of neutrophils and macrophages. Through these two mechanisms, Eculizumab could have benefits beyond merely stopping AMR. Actually, one could argue that by leaving proximal complement activation intact and truncating terminal complement activation, the conditions approximate accommodation, in which C4d deposition occurs but chemotaxis and activation of leukocytes does not. Much more experience with complement inhibitors is required to predict their effects on longterm outcomes. As discussed in the next 2 sections, complement independent actions of antibodies on leukocytes and endothelial cells could contribute to chronic rejection. In fact, endomyocardial biopsies from patients with late rejections that are attributed to antibodies contain macrophage infiltrates more frequently than C4d or C3d deposits (21).
2.2 Leukocyte activation
Accumulation of neutrophils or macrophages in capillaries is a key characteristic of AMR (22). As discussed in the previous section, complement activation can greatly augment the influx of neutrophils and macrophages, but in the absence of complement, these cells can be stimulated through their Fc receptors by antibodies.
Macrophages express the high affinity FcγRI (CD64) that is capable of binding monomeric IgG as well as the low affinity FcγRIIA (CD32), FcγRIII (CD16) and FcγRIV that are engaged by clusters of IgG on antigen. These FcgR signal through immunoreceptor tyrosine-based activation motifs (ITAMs). Activation of macrophages through FcgR upregulates many proinflammatory functions, including the production MCP-1 and IL-8, and promotes survival of macrophages at sites of inflammation. Experimental models have demonstrated that macrophage infiltrates decline within days after antibodies and complement deposits are terminated (23). FcγRIII is expressed by NK cells as well as macrophages and neutrophils. This receptor signals through ITAM to initiate antibody-dependent cell-mediated cytotoxicity (ADCC) by NK cells. In experimental models, passive transfers of non-complement activating antibodies have been demonstrated to be associated with infiltrates of NK cells (24).
Although CD68 has been used to detect macrophages in endomyocardial biopsies (3), macrophages have not been further delineated in terms of subpopulations or activation markers. It could be of diagnostic importance to differentiate between inflammatory versus wound healing macrophages especially early after transplantation when macrophages infiltrate ischemic tissues. Further delineation of macrophage populations infiltrating grafts in response to donor specific antibodies would certainly add to understanding the contributions of macrophages to graft inflammation.
2.3 Endothelial cell activation
In vitro experiments have revealed that antibodies have direct effects on target tissues such as vascular endothelial cells and smooth muscle cells as well as various epithelial cells (5, 25–27). The most rapid effect of cross-linking HLA class I antigens on endothelial cells is the exocytosis of von Willebrand factor (vWf) and P-selectin from the Weibel-Palade storage granules. The exocytosis of these preformed adhesion molecules occurs within minutes in vitro (26). This immediate effect of alloantibodies on endothelial cells is followed by production of cytokines, upregulation of receptors for growth factors and proliferation of endothelial cells (5, 25, 27).
Understanding the range of endothelial cell responses to alloantibodies may be helpful in transforming subjective pathological criteria that are currently used to diagnose AMR in the absence of detectable complement deposits. For example, various grading systems for AMR include the pathological criterion of endothelial activation based on endothelial cell swelling, which is subject to many artifacts. Recognizing the need for more objective measures of endothelial activation induced by alloantibodies, Reed and colleagues tested immunohistological stains for phosphorylated molecules in the signaling cascade initiated by cross-linking of HLA by antibodies (28). They predicted from their in vitro findings that downstream targets in the Akt signaling pathway would be logical markers. Therefore, they stained endomyocardial biopsies for phosphorylated 6RP. As predicted, this identified activated endothelial cells in biopsies with AMR. Interestingly, it also stained macrophages within the capillaries and leukocytes in the interstitium.
Other markers of endothelial cell activation that have been exploited in animal models of AMR include vWf and P-selectin, both of which identify intracapillary aggregates of platelets. This approach detected platelet aggregates in immunodeficient mice that received passively transferred F(ab′)2 fragments of alloantibodies indicating that complement activation is not required (26). Although this approach has not been tested in endomyocarial biopsies, Meehan et al (29) found that the majority of renal biopsies from patients with AMR contained platelet aggregates in peritubular capillaries. The mechanisms through which platelets augment vascular inflammation are under investigation in animal models as discussed in section 5.
3. New Insights into Nodular Endocardial Infiltrates (Quilty effect)
In 1981, Margaret Billingham observed nodular endocardial infiltrates of lymphocytes in a patient named Quilty (6). Subsequently referred to as the Quilty effect, these nodules were first suspected to be the consequence of Cyclosporin A treatment. Later studies, indicated that Quilty lesions were not related to individual immunosuppressive drugs or to viral infections.
Attempts to link the Quilty effect to acute or chronic rejection in large cohorts of patients from different transplant centers produced conflicting conclusions (6, 30, 31). Consequently, in the 2004 grading system for endocardial biopsies, Quilty lesions were categorized as a frequent (occurring in 10 to 20% of biopsies) finding that was not related to acute rejection (1). Quilty lesions were identified as consisting of a dense infiltrate containing T cells, B cells and plasma cells in a background of fibrosis and prominent vascularity (1). These infiltrates have continued to receive attention because of their variable cellular composition and size. Initially, two types of Quilty effect were recognized: Quilty A infiltrates that were confined to the endocardial surface and Quilty B infiltrates that extended into the underlying myocardium and could cause adjacent myocyte injury mimicking acute cellular rejection (Fig 3). This distinction was discarded in the 2004 grading system because it lacked general clinical value. A further subset of Quilty lesions has been found deep within the endocardium or adjacent to coronary vessels (32).
Fig. 3.
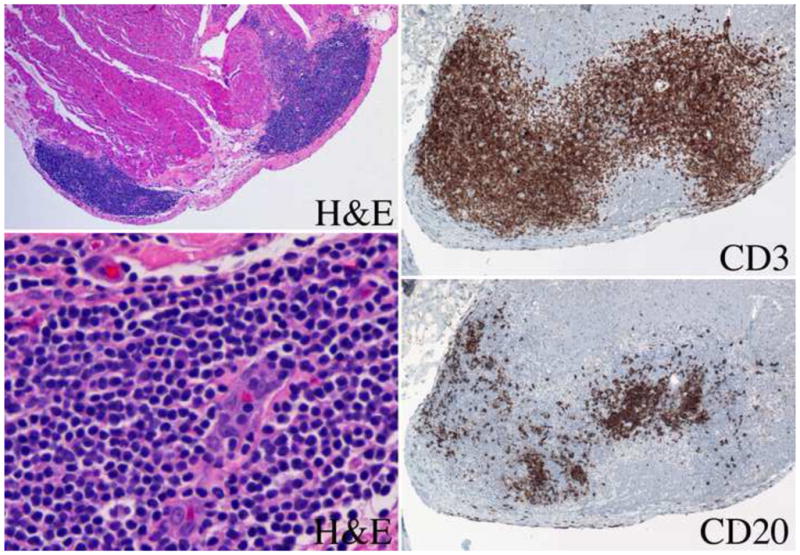
Endocardial nodular lymphoid infiltrate (Quilty effect). Low power (4x original) hematoxylin and eosin (H&E) stain with Quilty A and Quilty B nodular infiltrates. High power (40x) H&E stain demonstrating prominent capillary. Quilt lesion stained for CD3 and CD20 demonstrating T and B cell areas (20x).
More recent histological studies have focused on the size and structure of Quilty lesions. Some reports have ascribed significance to compartmentalization of T and B cells or germinal center structure with follicular dendritic cells (33, 34). These studies were not large enough to determine prognostic significance. Mengel and colleagues (35) probed for molecular signatures of infiltrates in endomyocardial biopsies by microarray analysis. They concluded that the Quilty effect correlated with upregulation of clusters of genes associated with activation of endothelium, macrophages and T cells. The caveat to this work is that the molecular analysis and histological evaluation were performed on two different pieces of the biopsies. This is problematic because Quilty lesions are nodular and often confined to one piece of the biopsy. Nonetheless, molecular analysis of verified Quilty lesions would be valuable. It should be noted, however, that the frequency of the Quilty effect in protocol biopsies has not decreased since the first large surveys even though the incidence of acute rejection has decreased and short-term survival of cardiac transplants has increased greatly. Alternatively, it is possible that Quilty lesions are a manifestation of an immune response related to chronic rejection, which is still a frequent outcome.
4. New Insights into B cells in Epicardial Infiltrates Relative to Chronic Allograft Vasculopathy (CAV)
Chronic rejection of heart transplants arises in the major branches of the coronary arteries that are located on the epicardial surface surrounded by fat. Chronic vasculopathy in allografts is characterized by a diffuse, concentric expansion of the intima in conjunction with inflammation and fibrosis of the adventitia. Microarray profiles of coronary arteries dissected from hearts with CAV indicated that genes for immunoglobulins are highly upregulated compared to coronaries from hearts with atheroslerosis (7). These molecular data are supported by immunohistological stains that identified B cells and plasma cells in mononuclear cells infiltrating the intima and adventitia of coronaries with CAV. In addition, Wehner et al (8) found nodules of lymphocytes in the adventitia and adjacent epicardial fat in most transplants with CAV. Many of these nodules had features of tertiary lymphoid tissue including T cell and B cell compartmentalization and follicular dendritic cells that stained positively for C4d (36). These features suggested that these nodules are responsive to antigen because complement split products are known to localize antigen to complement receptors on follicular dendritic cells.
Tertiary lymphoid neogenesis has been studied extensively in chronic inflammatory diseases such as rheumatoid arthritis. In patients with rheumatoid arthritis, B-cell nodules have been described in coronary arteries as well as the synovium (37). There are many reports of tertiary lymphoid follicles in renal transplants (38) and a few reports in experimental and clinical heart transplants (39). Tertiary lymphoid follicles in transplants are not generally associated with antibodies to HLA in the circulation or C4d deposits in the grafts (8, 40). This does not exclude the possibility that antibodies to HLA are absorbed to the transplant or that the plasma cells produce tissue-specific antibodies or autoantibodies, both of which have been reported in clinical and experimental cardiac transplants.
Organization of lymphoid nodules is promoted by B cell production of lymphotoxin to initiate follicular dendritic cell clusters and CCL21 to attract T cells (36). Many of the lymphoid nodules in cardiac transplants are located in the epicardial fat (Fig 4), which is consistent with data that adipocytes secrete B cell growth factors, such as B cell-activating factor (BAFF) and others (41). It is notable that perivascular adipocytes secrete more proinflammatory cytokines (eg, IL-6, IL-8 and MCP-1) compared with subcutaneous or visceral fat (42, 43). Perivascular adipocytes also secrete less of the anti-inflammatory cytokine adiponectin in comparison to other types of fat. Moreover, obesity and diabetes have been associated with an increased proinflammatory profile of adipocytes and decrease levels of circulating adiponectin. This of particular relevance because a large percentage of recipients become obese and diabetic in the first years after cardiac transplantation (44, 45). Although many recent studies have investigated interactions of perivascular fat with arterial inflammation and smooth muscle proliferation, little has been reported about epicardial fat and CAV (46).
Fig. 4.

Cornary artery branch with advanced vasculopathy and adjacent epicardial fat containing a lymphoid nodule demonstrating compartments of T cells (CD3) and B cells (CD20) as well as plasma cells (CD138). Low (4x) and medium (10x) power.
The use of calcineurin inhibitors may increase the incidence of lymphoid nodules through direct and indirect mechanisms. The diabetogenic effects of calcineurin inhibitors could indirectly bias the inflammatory profile of adipose tissue through decreased levels of adiponectin. In addition, large lymphoid nodules with T and B cell compartmentalization have been found in the kidneys and other organs of mice modified to express only a single gene for the alpha isoform for calcineurin (47).
5. Experimental Models to Test Clinically Relevant Questions
For many years, the dominant effects of T cells in unmodified organ graft rejection deflected attention from B cells in experimental models of transplantation. Interest in B cells resurged with the appreciation that AMR is a major risk of morbidity and mortality in humans. The challenge now is to adapt in vitro and in vivo experimental models to address clinically meaningful questions. We will discuss the experimental models available to test some of these clinical questions.
5.1 How Do Antibodies and B Cells Mediate Graft Injury?
Increasingly sophisticated clinical tests for antibodies have revealed that not all antibodies are equally damaging to transplanted hearts. Clinical tests using beads coated with recombinant HLA molecules are guiding questions regarding parameters of isotype, concentration, avidity and specificity of alloantibodies that mediate graft injury in transplant recipients.
The engagement of antibodies with MHC class I or II molecules on endothelial cells initiates a signaling cascaded that results in exocytosis of P-selectin and vWf, production of cytokines and chemokines, and upregulation of adhesion molecules that are ligands for integrins. Many of the questions dealing with these functional aspects of antibodies can be addressed with human endothelial cells in vitro. For example, it was unclear how antibodies binding to MHC class I molecules, which lack an intracytoplasmic signaling domain, could result in endothelial cell activation. Recent studies by Reed and colleagues have made valuable contributions to understanding this problem by demonstrating that signaling occurs through integrin beta 4 which associates with MHC class I molecules that are crosslinked by alloantibodies (27). As these experiments progress several variables need to be considered. First, it is not clear whether antibodies in the circulation represent accurately antibodies that are bound to the graft. In renal transplants, discrepancies in antigen specificity have been found between antibodies in the circulation and in eluates from rejected transplants. Antibodies eluted from cardiac transplants need to be examined. Second, the endothelial cell substrate should be carefully chosen. Experiments with umbilical vein endothelial cells and cell lines need to be verified with more relevant endothelial cell sources. Important biological differences between arterial and capillary endothelial cells need to be examined (48). Finally, in vitro experiments need to be performed under conditions mimicking blood flow in different vascular beds to account for effects of hydrostatic pressure and sheer stress (49).
Studies of serum from cardiac and renal transplant patients experiencing acute AMR indicate that antibodies to both MHC class I and II antigens can cause graft injury. However, some studies have found that injury mediated by antibodies to MHC class II is more severe and is characterized by more intense infiltrates of neutrophils. What mechanisms underlie the different pathological consequences of antibodies to donor MHC class I and II antigens remain unclear. While antibodies to MHC class I deliver signals indirectly through integrin beta 4, MHC class II molecules posses signaling modules in their intracytoplasmic domains (28).
5.2 Do Autoantibodies Have an Impact on Graft Injury?
There is accumulating evidence in both experimental and clinical transplantation that transplantation may lead to immune responses against autoantigens in addition to alloantigens (50–54). Among the factors implicated in breaking self-tolerance in allograft recipients are the inflammatory environment within the graft, the exposure of neoantigens, and the prolonged responses that eventually can engender epitope spreading. In particular, human heart transplant recipients develop antibodies against numerous self-antigens including cardiac myosin, vimentin, skeletal muscle glycolipid, ICAM-1 and ribosomal protein L7. These antigens vary in their tissue distribution (organ specific versus ubiquitous), in cellular location (intracellular versus cell surface or extracellular matrix), and in biochemical characteristics (proteins vs non-proteins).
As autoimmune responses are delayed and less intense compared to alloresponses, they are more likely to contribute to chronic rejection. In fact, data from both animals and humans indicate that autoimmune responses to vimentin accelerate onset and increase severity of cardiac allograft vasculopathy (53, 54). Notably, experiments testing autoantibodies in experimental models generally generate autoantibodies by direct immunization with autoantigens. It remains to be established whether pathogenic levels of autoantibodies can be induced as a component of the alloimmune response against the graft.
The mechanisms underlying the induction of autoantibodies during the alloimmune response remain to be addressed. IgG autoantibodies require help delivered by CD4 T cells that recognize donor peptides presented by self MHC molecules. This process may be primed in the course of alloimmune responses. In addition, donor-reactive CD4 T cells may recognize polymorphic epitopes of autoantigens or cross-react with recipient MHC molecules presenting self-peptides (55). It is also possible that B cells bearing receptors specific for an autoantigen process large fragments of donor cells released during graft damage and present peptides derived from donor MHC molecules thus soliciting help from CD4 T cells recognizing donor MHC peptides through the indirect pathway. Finally, a recent study demonstrated an intriguing possibility that donor passenger CD4 T cells can induce pathogenic autoantibody by self-reactive B cells of the recipient (56).
While autoantibodies are consistently generated in transplant recipients, questions remain whether and how the signals induced by target molecule ligation in the graft lead to allograft injury, if in fact they do so. It is likely that autoantibodies against cell surface molecules such as ICAM-1 will trigger mechanisms of antibody-mediated tissue damage similar to those for antibodies to MHC antigens. However, the majority of autoantibodies described in transplant recipients are generated against intracellular molecules. Autoantibodies may be produced to antigens exposed by inflammation due to the transplantation procedure (brain death of the donor, ischemia-reperfusion, and surgical trauma). Subsequently, these autoantibodies may react to epitopes exposed during subsequent rejection or infection (52, 53).
5.3 What is the Potential of Manipulating Antibodies and Complement?
In spite of the extensive knowledge of the complement system, many questions remain regarding the anti- and pro-inflammatory effects of critical complement components on solid organ transplants. Some of these questions will be best addressed with human sources of antibodies, complement and tissues because of essential molecular differences among species. These differences do not preclude identifying basic principals in animal models. For example, mouse antibodies have been effectively studied to establish the importance of carbohydrate side chains on antibodies to the binding of C1 and MBL as well as to the binding of different FcgR (9).
In vitro models have been useful to identify some potential mechanisms involved in graft accommodation including the upregulation of certain complement regulators and anti-apoptotic genes, but in vivo models and clinical biopsies will be needed to determine the actual contribution of these mechanisms to transplant survival under particular conditions.
With the introduction of complement inhibitors into clinical practice, new questions arise as to whether the potent anti-inflammatory activity of the early complement components (particularly C1q, MBL and C4) can be exploited. For example, it is possible that C1q mediated clearance of apoptotic bodies induced by low levels of antibodies could induce tolerance. To this end, valuable studies could be performed to examine the function of macrophages and B cells following treatment with C5 inhibitors. Antibodies and small molecule inhibitors of C5 have already been shown to decrease macrophage infiltrates and inhibit acute rejection of cardiac allografts in mice (20). It is not established whether macrophages exposed to C1 or C4 in the absence of stimulation by C5a develop into repair or regulatory macrophages. Some of these questions can be tested with complement deficient mice.
5.4 Can Different Types of Leukocyte Activation be Identified?
Clinically, AMR is now diagnosed and treated long before the catastrophic terminal events of endothelial destruction, hemorrhage and myocyte necrosis (3). Presently, the most advanced pathological findings in AMR are macrophage accumulation and endothelial cell activation. However, both of these pathological findings are incompletely defined. Reed and colleagues have reported that 6RP is phosphorylated in both macrophages and endothelial cells during AMR (28), but there is limited data regarding which receptors or mediators are upregulated in these cells during AMR. Small animal studies indicate that infiltrating macrophages not only respond to C5a, but also produce complement components in the graft (57). It is also likely that when complement activation proceeds to cleavage of C5 that macrophages upregulate FcγRIII and downregulate FcγRIIB (19). This could be a critical indicator of active AMR.
5.5 Does Platelet Activation Contribute to AMR?
Platelets can greatly extend the effects of antibodies on endothelial cells (18). Antibody induced release of vWf provides a ligand for GP1b and GPIIb/IIIa on platelets. In experimental models, antibody-mediated rejection is associated with release of vWf and attachment of platelets to vascular endothelium in cardiac allografts (58). In MHC incompatible skin grafts a transfer of alloantibodies causes release of vWf and initiates platelet rolling in the capillaries of the skin graft but not in the adjacent recipient skin (59). Complement is not necessary for the antibody-induced interaction of platelets with endothelial cells in this model, but complement activating antibodies do induce more prolonged rolling of platelets and leukocytes. Moreover, the retention of leukocytes by antibodies is in part dependent on platelets because platelet depletion abbreviates leukocyte rolling.
In models of vascular inflammation, platelets attract mononuclear cells by secretion of chemokines, such as MIP-1alpha (CCL3) and RANTES (CCL5). Human monocytes are also stimulated by platelet factor 4 (PF4; CXCL4), which is a prototypical product of platelet activation, to express TNF, IL-6, CCL22, MHC class II and CD86. Interestingly, CCL22 can feed back on platelets to induce expression of P-selectin. In addition to P-selectin, activated platelets express CD40 and CD154 on their surface. These receptors and ligands promote adhesion and signaling interactions among platelets, neutrophils, monocytes, lymphocytes and endothelial cells. They also can modulate the maturation and function of macrophages and dendritic cells.
Clearly, platelets could augment many aspects AMR and animal models will be useful to probe platelet function and potential interventions.
5.6 What is the Role of Donor Reactive Antibody in the Pathogenesis of CAV?
The pathogenesis of CAV is difficult to establish in humans because the lesions develop in the major branches of the coronary arteries, which are on the epicardial surface of the heart. These are anatomically distant from the right interventricular septum where endomyocardial biopsies are obtained. Occasionally the right free wall is perforated and a sample of the coronary is obtained. Otherwise, histological evaluation of CAV is limited to advanced lesions. Therefore, animal models could be pertinent to understanding pathogenic factors in CAV. Unfortunately there are anatomical differences between the coronary arteries of humans and of rats and mice. Among the differences are the location and size of the coronaries. Human coronary arteries are located on the epicardial surface of the heart surrounded by adipose tissue. Mouse and rat coronaries are mainly intramyocardial arteries. Human coronaries also have thick media and adventitia that require vasa vasorum for a blood supply.
Despite these anatomical differences, rodent models have been used to demonstrate that antibodies can promote the development of CAV. Initial studies in rats and mice simply correlated levels of donor reactive antibodies in the serum of cardiac allograft recipients with the development of CAV. These studies were followed by experiments demonstrating the ability of passively transferred antibodies to promote the development of CAV in cardiac allografts in RAG-1−/− recipients (60). In one study complement was not required to produce these lesions (24). It should be noted that, similar to models of atherosclerosis in mice, these lesions were limited to the origins of the coronary arteries where they are epicardial and surrounded by adipose tissue.
The anatomical differences between rodents and humans also make modeling of the epicardial B cell infiltrates observed in humans problematic in mice and rats. Baddoura et al (61) described lymphoid neogenesis on the epicardial surface of mouse cardiac allografts that underwent chronic rejection. Some of these structures were described as lymph node-like and contained medullary sinuses that are not typical of tertiary lymphoid nodules (36). Epicardial B cell nodules and infiltrates are worthwhile modeling because they may provide insights to the high incidence of CAV in human cardiac transplants. These B cells may be sustained by cytokines produced in the epicardial fat, and the B cells may promote T cell function or fibrosis. Adventitial infiltrates of B cells are often embedded in fibrosis, and recent data from experimental models indicate that B cells can promote fibrosis (62).
It is clear that the induction of antibody responses to allo- and autoantigens is a predisposing for the development of CAV. However, the short and longterm consequences of donor reactive antibodies on grafts remain poorly understood. Specifically, does the induction of an antibody response to donor antigens commit a program of graft injury leading to AMR and/or CAV or is there a window in which depletion of B cells at the time or shortly after detection of circulating antibody will at least attenuate the injury leading to acute or chronic rejection? In a primate model, Pierson and colleagues have provided evidence that preemptory treatment with Rituximab inhibits the development of CAV (63).
In summary, the recognition that B cells are important components of the immune response to cardiac transplants has opened a wide range of questions that need to be addressed with clinically relevant experimental approaches.
Highlights.
The effects of antibodies and B cells on cardiac transplants are controversial
Antibody-mediated rejection is now accepted but questions of diagnosis and mechanisms remain
Antibodies cause graft injury by activation of complement, leukocytes and endothelial cells
Complement is a therapeutic target for antibody-mediated rejection
Nodular endo- and epicardial B cell infiltrates are frequent in cardiac grafts
Acknowledgments
WMB, AV and RLF are supported by grants from the ROTRF and by P01AI087586 from the National Institutes of Health (NIH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stewart S, Winters GL, Fishbein MC, Tazelaar HD, Kobashigawa J, Abrams J, et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J Heart Lung Transplant. 2005;24(11):1710–1720. doi: 10.1016/j.healun.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 2.Kobashigawa J, Crespo-Leiro MG, Ensminger SM, Reichenspurner H, Angelini A, Berry G, et al. Report from a consensus conference on antibody-mediated rejection in heart transplantation. J Heart Lung Transplant. 2011;30(3):252–269. doi: 10.1016/j.healun.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berry GJ, Angelini A, Burke MM, Bruneval P, Fishbein MC, Hammond E, et al. The ISHLT working formulation for pathologic diagnosis of antibody-mediated rejection in heart transplantation: Evolution and current status (2005–2011) J Heart Lung Transplant. 2011;30(6):601–611. doi: 10.1016/j.healun.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Kucirka LM, Maleszewski JJ, Segev DL, Halushka MK. Survey of North American pathologist practices regarding antibody-mediated rejection in cardiac transplant biopsies. Cardiovasc Pathol. 2010:19. doi: 10.1016/j.carpath.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Reed EF. Effect of antibodies on endothelium. Am J Transplant. 2009;9(11):2459–2465. doi: 10.1111/j.1600-6143.2009.02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joshi A, Masek MA, Brown BW, Jr, Weiss LM, Billingham ME. “Quilty” revisited: a 10-year perspective. Human pathology. 1995;26(5):547–557. doi: 10.1016/0046-8177(95)90252-x. [DOI] [PubMed] [Google Scholar]
- 7.Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM., 3rd Antibody and complement in transplant vasculopathy. Circulation research. 2007;100(2):191–203. doi: 10.1161/01.RES.0000255032.33661.88. [DOI] [PubMed] [Google Scholar]
- 8.Wehner JR, Fox-Talbot K, Halushka MK, Ellis C, Zachary AA, Baldwin WM., 3rd B cells and plasma cells in coronaries of chronically rejected cardiac transplants. Transplantation. 2010;89(9):1141–1148. doi: 10.1097/TP.0b013e3181d3f271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anthony RM, Nimmerjahn F. The role of differential IgG glycosylation in the interaction of antibodies with FcgammaRs in vivo. Current opinion in organ transplantation. 2011 doi: 10.1097/MOT.0b013e328342538f. [DOI] [PubMed] [Google Scholar]
- 10.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science (New York, NY. 2006;313(5787):670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 11.Van Beneden K, Coppieters K, Laroy W, De Keyser F, Hoffman IE, Van den Bosch F, et al. Reversible changes in serum immunoglobulin galactosylation during the immune response and treatment of inflammatory autoimmune arthritis. Ann Rheum Dis. 2009;68(8):1360–1365. doi: 10.1136/ard.2008.089292. [DOI] [PubMed] [Google Scholar]
- 12.Fan X, Ang A, Pollock-Barziv SM, Dipchand AI, Ruiz P, Wilson G, et al. Donor-specific B-cell tolerance after ABO-incompatible infant heart transplantation. Nature medicine. 2004;10(11):1227–1233. doi: 10.1038/nm1126. [DOI] [PubMed] [Google Scholar]
- 13.Mason JC, Yarwood H, Sugars K, Morgan BP, Davies KA, Haskard DO. Induction of decay-accelerating factor by cytokines or the membrane-attack complex protects vascular endothelial cells against complement deposition. Blood. 1999;94(5):1673–1682. [PubMed] [Google Scholar]
- 14.Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. The Journal of biological chemistry. 1989;264(15):9053–9060. [PubMed] [Google Scholar]
- 15.Benzaquen LR, Nicholson-Weller A, Halperin JA. Terminal complement proteins C5b-9 release basic fibroblast growth factor and platelet-derived growth factor from endothelial cells. J Exp Med. 1994;179:985–992. doi: 10.1084/jem.179.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. The Journal of experimental medicine. 1997;185(9):1619–1627. doi: 10.1084/jem.185.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saadi S, Platt JL. Transient perturbation of endothelial integrity induced by natural antibodies and complement. The Journal of experimental medicine. 1995;181(1):21–31. doi: 10.1084/jem.181.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirk AD, Morrell CN, Baldwin WM., 3rd Platelets influence vascularized organ transplants from start to finish. Am J Transplant. 2009;9(1):14–22. doi: 10.1111/j.1600-6143.2008.02473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. The Journal of clinical investigation. 2002;110(12):1823–1830. doi: 10.1172/JCI200216577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Jiang J, Liu W, Kubelik D, Chen G, Gies D, et al. Prevention of acute vascular rejection by a functionally blocking anti-C5 monoclonal antibody combined with cyclosporine. Transplantation. 2005;79(9):1121–1127. doi: 10.1097/01.tp.0000161218.58276.9a. [DOI] [PubMed] [Google Scholar]
- 21.Loupy A, Cazes A, Guillemain R, Amrein C, Hedjoudje A, Tible M, et al. Very Late Heart Transplant Rejection Is Associated with Microvascular Injury, Complement Deposition and Progression to Cardiac Allograft Vasculopathy. Am J Transplant. 2011:11. doi: 10.1111/j.1600-6143.2011.03563.x. [DOI] [PubMed] [Google Scholar]
- 22.Baldwin WM, III, Kasper EK, Zachary AA, Wasowska BA, Rodriguez ER. Beyond C4d: other complement related diagnostic approaches to antibody-mediated rejection. Am J Transplant. 2004;4:311–318. doi: 10.1111/j.1600-6143.2004.00348.x. [DOI] [PubMed] [Google Scholar]
- 23.Minami K, Murata K, Lee C-Y, Fox-Talbot K, Wasowska B, Pescovitz M, et al. C4d deposition and clearance in cardiac transplants correlates with alloantibody levels and rejection in rats. Am J Transplant. 2006;6(5):923–932. doi: 10.1111/j.1600-6143.2006.01281.x. [DOI] [PubMed] [Google Scholar]
- 24.Hirohashi T, Uehara S, Chase CM, DellaPelle P, Madsen JC, Russell PS, et al. Complement independent antibody-mediated endarteritis and transplant arteriopathy in mice. Am J Transplant. 2010;10(3):510–517. doi: 10.1111/j.1600-6143.2009.02958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee C-Y, Reynolds M, Garyu J, Baldwin WM, III, Wasowska BA. The involvement of FcR mechanisms in antibody-mediated rejection. Transplantation. 2007;84:1324–1334. doi: 10.1097/01.tp.0000287457.54761.53. [DOI] [PubMed] [Google Scholar]
- 26.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, et al. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(4):1301–1306. doi: 10.1073/pnas.0602035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Rozengurt E, Reed EF. HLA class I molecules partner with integrin beta4 to stimulate endothelial cell proliferation and migration. Science signaling. 2011;3(149):ra85. doi: 10.1126/scisignal.2001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lepin EJ, Zhang Q, Zhang X, Jindra PT, Hong LS, Ayele P, et al. Phosphorylated S6 ribosomal protein: a novel biomarker of antibody-mediated rejection in heart allografts. Am J Transplant. 2006;6(7):1560–1571. doi: 10.1111/j.1600-6143.2006.01355.x. [DOI] [PubMed] [Google Scholar]
- 29.Meehan SM, Limsrichamrern S, Manaligod JR, Junsanto T, Josephson MA, Thistlethwaite JR, et al. Platelets and capillary injury in acute humoral rejection of renal allografts. Human pathology. 2003;34(6):533–540. doi: 10.1016/s0046-8177(03)00189-8. [DOI] [PubMed] [Google Scholar]
- 30.Yamani MH, Ratliff NB, Starling RC, Tuzcu EM, Yu Y, Cook DJ, et al. Quilty lesions are associated with increased expression of vitronectin receptor (alphavbeta3) and subsequent development of coronary vasculopathy. J Heart Lung Transplant. 2003;22(6):687–690. doi: 10.1016/s1053-2498(02)01181-6. [DOI] [PubMed] [Google Scholar]
- 31.Chu KE, Ho EK, de la Torre L, Vasilescu ER, Marboe CC. The relationship of nodular endocardial infiltrates (Quilty lesions) to survival, patient age, anti-HLA antibodies, and coronary artery disease following heart transplantation. Cardiovasc Pathol. 2005;14(4):219–224. doi: 10.1016/j.carpath.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Truell JS, Fishbein MC. Case report of a Quilty lesion within a coronary artery. Cardiovasc Pathol. 2006;15(3):161–164. doi: 10.1016/j.carpath.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Sattar HA, Husain AN, Kim AY, Krausz T. The presence of a CD21+ follicular dendritic cell network distinguishes invasive Quilty lesions from cardiac acute cellular rejection. The American journal of surgical pathology. 2006;30(8):1008–1013. doi: 10.1097/00000478-200608000-00012. [DOI] [PubMed] [Google Scholar]
- 34.Di Carlo E, D’Antuono T, Contento S, Di Nicola M, Ballone E, Sorrentino C. Quilty effect has the features of lymphoid neogenesis and shares CXCL13-CXCR5 pathway with recurrent acute cardiac rejections. Am J Transplant. 2007;7(1):201–210. doi: 10.1111/j.1600-6143.2006.01584.x. [DOI] [PubMed] [Google Scholar]
- 35.Mengel M, Sis B, Kim D, Chang J, Famulski KS, Hidalgo LG, et al. The molecular phenotype of heart transplant biopsies: relationship to histopathological and clinical variables. Am J Transplant. 2010;10(9):2105–2115. doi: 10.1111/j.1600-6143.2010.03182.x. [DOI] [PubMed] [Google Scholar]
- 36.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6(3):205–217. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 37.Aubry MC, Riehle DL, Edwards WD, Maradit-Kremers H, Roger VL, Sebo TJ, et al. B-Lymphocytes in plaque and adventitia of coronary arteries in two patients with rheumatoid arthritis and coronary atherosclerosis: preliminary observations. Cardiovasc Pathol. 2004;13(4):233–236. doi: 10.1016/j.carpath.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Zarkhin V, Li L, Sarwal M. “To B or not to B?” B-cells and graft rejection. Transplantation. 2008;85(12):1705–1714. doi: 10.1097/TP.0b013e318177793e. [DOI] [PubMed] [Google Scholar]
- 39.Thaunat O, Field AC, Dai J, Louedec L, Patey N, Bloch MF, et al. Lymphoid neogenesis in chronic rejection: evidence for a local humoral alloimmune response. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(41):14723–14728. doi: 10.1073/pnas.0507223102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thaunat O. Pathophysiologic Significance of B-Cell Clusters in Chronically Rejected Grafts. Transplantation. 2011 doi: 10.1097/TP.0b013e31821f74fe. [DOI] [PubMed] [Google Scholar]
- 41.Alexaki VI, Notas G, Pelekanou V, Kampa M, Valkanou M, Theodoropoulos P, et al. Adipocytes as immune cells: differential expression of TWEAK, BAFF, and APRIL and their receptors (Fn14, BAFF-R, TACI, and BCMA) at different stages of normal and pathological adipose tissue development. J Immunol. 2009;183(9):5948–5956. doi: 10.4049/jimmunol.0901186. [DOI] [PubMed] [Google Scholar]
- 42.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6(10):772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 43.Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circulation research. 2009;104(4):541–549. doi: 10.1161/CIRCRESAHA.108.182998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grady KL, Naftel D, Pamboukian SV, Frazier OH, Hauptman P, Herre J, et al. Postoperative obesity and cachexia are risk factors for morbidity and mortality after heart transplant: multi-institutional study of post-operative weight change. J Heart Lung Transplant. 2005;24(9):1424–1430. doi: 10.1016/j.healun.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 45.Williams JJ, Lund LH, LaManca J, Kunavarapu C, Cohen DJ, Heshka S, et al. Excessive weight gain in cardiac transplant recipients. J Heart Lung Transplant. 2006;25(1):36–41. doi: 10.1016/j.healun.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 46.Wehner JR, Baldwin WM., 3rd Cardiac allograft vasculopathy: do adipocytes bridge alloimmune and metabolic risk factors? Current opinion in organ transplantation. 2010;15(5):639–644. doi: 10.1097/MOT.0b013e32833deaee. [DOI] [PubMed] [Google Scholar]
- 47.Kelly FM, Reddy RN, Roberts BR, Gangappa S, Williams IR, Gooch JL. TGF-beta upregulation drives tertiary lymphoid organ formation and kidney dysfunction in calcineurin A-alpha heterozygous mice. Am J Physiol Renal Physiol. 2009;296(3):F512–520. doi: 10.1152/ajprenal.90629.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circulation research. 2007;100(2):174–190. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 49.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, et al. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(41):14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jurcevic S, Ainsworth ME, Pomerance A, Smith JD, Robinson DR, Dunn MJ, et al. Antivimentin antibodies are an independent predictor of transplant-associated coronary artery disease after cardiac transplantation. Transplantation. 2001;71(7):886–892. doi: 10.1097/00007890-200104150-00011. [DOI] [PubMed] [Google Scholar]
- 51.Fedoseyeva EV, Kishimoto K, Rolls HK, Illigens BM, Dong VM, Valujskikh A, et al. Modulation of tissue-specific immune response to cardiac myosin can prolong survival of allogeneic heart transplants. J Immunol. 2002;169(3):1168–1174. doi: 10.4049/jimmunol.169.3.1168. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka M, Zwierzchoniewska M, Mokhtari GK, Terry RD, Balsam LB, Robbins RC, et al. Progression of alloresponse and tissue-specific immunity during graft coronary artery disease. Am J Transplant. 2005;5(6):1286–1296. doi: 10.1111/j.1600-6143.2005.00880.x. [DOI] [PubMed] [Google Scholar]
- 53.Leong HS, Mahesh BM, Day JR, Smith JD, McCormack AD, Ghimire G, et al. Vimentin autoantibodies induce platelet activation and formation of platelet-leukocyte conjugates via platelet-activating factor. Journal of leukocyte biology. 2008;83(2):263–271. doi: 10.1189/jlb.0607339. [DOI] [PubMed] [Google Scholar]
- 54.Mahesh B, Leong HS, Nair KS, McCormack A, Sarathchandra P, Rose ML. Autoimmunity to vimentin potentiates graft vasculopathy in murine cardiac allografts. Transplantation. 2010;90(1):4–13. doi: 10.1097/TP.0b013e3181dfa694. [DOI] [PubMed] [Google Scholar]
- 55.Win TS, Pettigrew GJ. Humoral autoimmunity and transplant vasculopathy: when allo is not enough. Transplantation. 2010;90(2):113–120. doi: 10.1097/TP.0b013e3181e25a59. [DOI] [PubMed] [Google Scholar]
- 56.Win TS, Rehakova S, Negus MC, Saeb-Parsy K, Goddard M, Conlon TM, et al. Donor CD4 T cells contribute to cardiac allograft vasculopathy by providing help for autoantibody production. Circ Heart Fail. 2009;2(4):361–369. doi: 10.1161/CIRCHEARTFAILURE.108.827139. [DOI] [PubMed] [Google Scholar]
- 57.Qian Z, Wasowska BA, Behrens E, Brody JR, Kadkol SS, Cangello DL, et al. C6 produced by macrophages contributes to cardiac allograft rejection. Amer J Pathol. 1999;155:1293–1302. doi: 10.1016/S0002-9440(10)65231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ota H, Fox-Talbot K, Hu W, Qian Z, Sanfilippo F, Hruban RH, et al. Terminal complement components mediate release of von Willebrand factor and adhesion of platelets in arteries of allografts. Transplantation. 2005;79(3):276–281. doi: 10.1097/01.tp.0000146195.76904.d3. [DOI] [PubMed] [Google Scholar]
- 59.Morrell CN, Murata K, Swaim AM, Mason E, Martin TV, Thompson LE, et al. In vivo platelet-endothelial cell interactions in response to major histocompatibility complex alloantibody. Circulation research. 2008;102(7):777–785. doi: 10.1161/CIRCRESAHA.107.170332. [DOI] [PubMed] [Google Scholar]
- 60.Uehara S, Chase C, Cornell L, Madsen J, Russell P, Colvin R. Chronic cardiac transplant arteriopathy in mice: Relationship of alloantibody, C4d deposition and neointimal fibrosis. Am J Transpl. 2007;7:57–65. doi: 10.1111/j.1600-6143.2006.01599.x. [DOI] [PubMed] [Google Scholar]
- 61.Baddoura FK, Nasr IW, Wrobel B, Li Q, Ruddle NH, Lakkis FG. Lymphoid neogenesis in murine cardiac allografts undergoing chronic rejection. Am J Transplant. 2005;5(3):510–516. doi: 10.1111/j.1600-6143.2004.00714.x. [DOI] [PubMed] [Google Scholar]
- 62.Yoshizaki A, Iwata Y, Komura K, Ogawa F, Hara T, Muroi E, et al. CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma. The American journal of pathology. 2008;172(6):1650–1663. doi: 10.2353/ajpath.2008.071049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kelishadi SS, Azimzadeh AM, Zhang T, Stoddard T, Welty E, Avon C, et al. Preemptive CD20+ B cell depletion attenuates cardiac allograft vasculopathy in cyclosporine-treated monkeys. The Journal of clinical investigation. 2010;120(4):1275–1284. doi: 10.1172/JCI41861. [DOI] [PMC free article] [PubMed] [Google Scholar]