

Abstract
Treatment of base excision repair-proficient mouse fibroblasts with the DNA alkylating agent methyl methanesulfonate (MMS) and a small molecule inhibitor of poly(ADP-ribose) polymerase-1 (PARP-1) results in a striking cell killing phenotype, as previously reported. Earlier studies demonstrated that the mechanism of cell death is apoptosis and requires DNA replication, expression of PARP-1, and an intact S-phase checkpoint cell signaling system. It is proposed that activity-inhibited PARP-1 becomes immobilized at DNA repair intermediates, and that this blocks DNA repair and interferes with DNA replication, eventually promoting an S-phase checkpoint and G2/M block. Here we report studies designed to evaluate the prediction that inhibited PARP-1 remains DNA-associated in cells undergoing repair of alkylation-induced damage. Using chromatin immunoprecipitation with anti-PARP-1 antibody and qPCR for DNA quantification, a higher level of DNA was found associated with PARP-1 in cells treated with MMS plus PARP inhibitor than in cells without inhibitor treatment. These results have implications for explaining the extreme hypersensitivity phenotype after combination treatment with MMS and a PARP inhibitor.
Keywords: PARP inhibitor, base excision repair, PARP-1, alkylation DNA damage, apoptosis
Introduction
Poly(ADP-ribose) polymerase-1 (PARP-1) plays a central and multifaceted role in the array of cellular responses to DNA damage. In the case of the base excision DNA repair (BER) pathway, PARP-1 is considered to be a member of the first group of DNA repair-associated proteins to recognize and bind at the DNA lesion or an initial intermediate of repair (e.g., abasic site or strand break) (1–6). Upon binding to a BER strand breakcontaining intermediate, PARP-1 is activated for synthesis of poly(ADP-ribose) (PAR) and is considered to function as an accessory factor in recruiting and stabilizing the repair complex (2, 7, 8). PARP-1 undergoes automodification with PAR (PARylation) and also PARylates other repair proteins. After auto-PARylation, PARP-1 is proposed to dissociate from DNA (9), yet the precise roles of PARP-1 and PARylation in BER are still obscure and under investigation. Nevertheless, BER is partially blocked when PARP activity is inhibited by treatment of cells with a small molecule inhibitor (7, 10, 11). Despite these observations, repair-proficient cells in culture experiencing endogenous DNA base damage are resistant to PARP inhibitors, presumably because alternative DNA repair pathways can compensate for a block in BER (12–14). In contrast, when double-strand break (DSB) repair by homologous recombination or non-homologous end joining is deficient in cells, they become sensitive to PARP inhibitors (15). In light of this background information, PARP inhibitors are being widely explored in therapeutic approaches for tumors with DSB repair deficiencies (12, 16).
It is important to gain a deeper understanding of cellular mechanisms that will inform cancer therapeutic approaches involving PARP inhibitors. One approach has been the study of cytotoxicity in human and mouse fibroblasts as a function of DNA repair and PARP inhibition. Even wild-type mouse fibroblasts are extremely sensitive to treatment with combinations of PARP inhibitors and alkylating agents that trigger the BER pathway. For example, a low, sub-lethal dose of methyl methanesulfonate (MMS) combined with the PARP inhibitor 4-amino-1,8-naphthalimide (4-AN) results in greater than 99% cell killing after several days of culture (17, 18). This extreme cytotoxicity occurs mainly through apoptotic cell death, requires treatment in replicating cells and involves activation of components of the S-phase cell cycle checkpoint system (18, 19). The effects of treatment with the PARP inhibitor plus alkylating agent combination (MMS + 4-AN) include accumulation of DNA DSBs, as measured by pulsed field gel electrophoresis and immunostaining for phosphorylated histone H2A.X (20, 21). Interestingly, PARP-1 −/− mouse cells are only mildly sensitive to the MMS + 4-AN combination compared to wild-type cells (17) and failed to accumulate DSBs after treatment (20). These results suggested that in wild-type cells, the inhibited PARP-1 protein itself plays a role in the apoptotic cell killing phenotype, since the loss of PARP-1 did not produce the same effect as inhibiting PARP (18).
In human and mouse fibroblasts, the S-phase checkpoint observed following treatment with the MMS + 4-AN combination resulted in activation of ATR and the ATR effector kinase Chk1 (22). ATM was implicated in the checkpoint response also, and loss of either ATM or of its downstream effector kinase, Chk2, limited the S-phase delay (23). The S-phase checkpoint also required NBS1 phosphorylation by ATR, while NBS1 modulated ATM in its response to the combination treatment. NBS1 modulated activation of SMC1, a downstream effector in an alternate S-phase checkpoint mechanism (19).
A working model (Figure 1) for the observed effects following treatment with the MMS + 4-AN combination is as follows: 1) A DNA alkylating agent triggers BER and intermediates with an APE1-incised AP site are generated. These BER intermediates, containing 5′-deoxyribose phosphate (dRP), are bound by the abundant PARP-1 protein in its DNA damage response; 2) Under normal conditions, in the absence of PARP inhibition (Figure 1, left), there is significant PARylation of PARP-1, BER factors are recruited to the BER intermediate, and BER proceeds to completion along with release of PARylated PARP-1 from the DNA. When PARP activity is inhibited by 4-AN (Figure 1, right), PARP-1 becomes frozen at the BER intermediates blocking BER; 3) DNA replication forks collide with the BER intermediate/inhibited PARP-1 complex and stall. Subsequently, the cell signaling system triggers an S-phase checkpoint, accumulation of cells in G2/M, accumulation of DSBs and cell death by apoptosis. Although Figure 1 is consistent with results obtained so far, key information is still lacking at most of the steps, and alternate models for explaining cell sensitivity to PARP inhibitors are possible. Therefore, it is important to further examine the model in Figure 1, along with other models (24, 25), to gain a better understanding of the cytotoxic responses to PARP inhibition during BER in replicating cells (26, 27).
Figure 1.
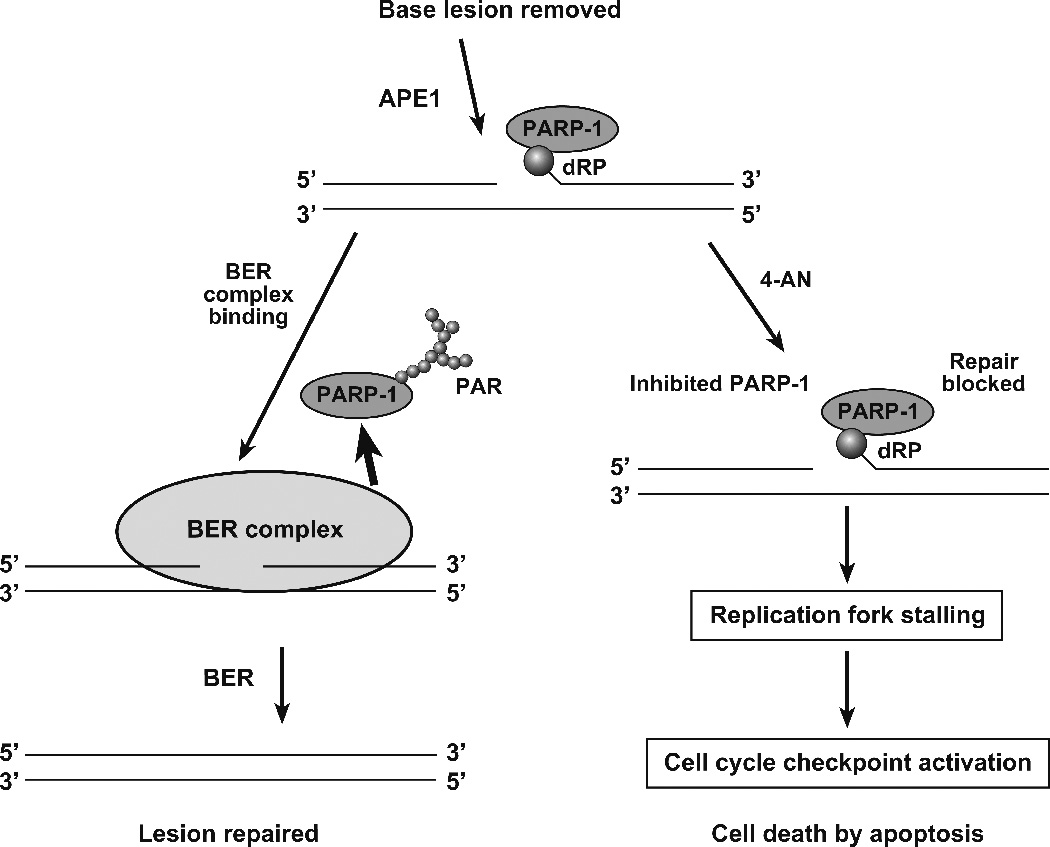
Working model for studies of the effects of PARP inhibition during BER of alkylated DNA bases in replicating cells. After removal of the MMS-induced alkylated base by a monofunctional DNA glycosylase, AP endonuclease (APE1) incises the abasic site leaving a gap with the 5′-deoxyribose phosphate group (dRP) at the margin. In the normal base excision repair (BER) pathway depicted on the left, many BER factors are in a preformed complex that recognizes the BER intermediate shown at the top. Poly(ADP ribose) or PAR-adducted PARP-1 is depicted as releasing from the BER complex during repair. In the presence of the PARP inhibitor 4-AN depicted on the right, inhibited PARP-1 remains bound to the BER intermediate with the 5′ -dRP group. This eventually leads to replication fork stalling, checkpoint activation and apoptosis.
In the present study, we examined a prediction of the model in Figure 1 regarding the mechanism of cytotoxicity after the MMS + 4-AN treatment. The prediction is that upon treatment with this combination, more genomic DNA could be found in association with PARP-1 than in cells treated with MMS alone or in control, untreated cells. We tested this prediction using a chromatin immunoprecipitation (ChIP) procedure with anti-PARP-1 antibody followed by quantification of DNA in the immunoprecipitated samples. The results are discussed in relation to the model in Figure 1.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) and Hank’s balanced salt solution (HBSS) were from HyClone (Logan, UT). GlutaMAX-1, and hygromycin B were from Invitrogen (Carlsbad, CA). MMS and the PARP inhibitor (4-AN) were from Sigma-Aldrich (St. Louis, MO). Restore Western Blot Stripping Buffer and SuperSignal West Pico Chemiluminescent substrate were from Thermo Scientific (Rockford, IL). Western Lightning Plus-ECL Enhanced Chemiluminescent Substrate was from PerkinElmer (Boston, MA). The mouse IgG secondary antibody was goat anti-mouse IgG binding grade affinity purified horseradish peroxidase conjugate, and the rabbit IgG secondary antibody was goat anti-rabbit IgG horseradish peroxidase conjugate; these antibody reagents were from Bio-Rad Laboratories (Hercules, CA), as was the Bio-Rad protein assay kit. Aphidicolin (APH), protein A-sepharose CL-4B and bovine serum albumin (BSA) were from Sigma-Aldrich. Protein G-agarose and protease inhibitor mix Complete© (EDTA-free) were from Roche Diagnostics (Indianapolis, IN). MG-132 protease inhibitor, leupeptin, aprotinin, and phenylmethylsulfonyl fluoride (PMSF) were from EMD Chemicals (Gibbstown, NJ). Primers were from Integrated DNA Technology (Coralville, IA). The Simple Enzymatic Chromatin IP Kit (agarose beads) was from Cell Signaling Technology (Beverly, MA). Rabbit monoclonal anti-PARP-1 antibody, IgG, (46D11, #9532), control (non-immune) normal rabbit IgG (#2729), rabbit monoclonal anti-histone H3 antibody (#4620), and rabbit polyclonal anti-histone H3 antibody (#9715) also were from Cell Signaling Technology. Mouse monoclonal anti-PARP-1 antibody (#556494) was from BD Biosciences Pharmingen (San Diego, CA). Rabbit polyclonal anti-PARP-1 antibody (BML#SA253) was from ENZO Life Sciences (Plymouth Meeting, PA). Mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (G3PDH) antibody (G3PDH11-M) was from Alpha Diagnostic International (San Antonio, TX). Rabbit polyclonal anti-RPA70 antibody (#2267) was from Cell Signaling Technology. Mouse monoclonal anti-NBS1 antibody (#611870) was from BD Biosciences Pharmingen (San Diego, CA). Mouse monoclonal anti-MRE11 antibody (sc-#135992), rabbit polyclonal anti-ATR antibody (sc-#28901) and mouse monoclonal anti-lamin A/C antibody (sc-7293) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The ABI Prism SDS 2.1 (7900 HT) sequence detection system, Power SYBR Green PCR Master mix, and the 96-well optical reaction plate with Barcode (code 128) were from Applied Biosciences Inc. (Foster City, CA). The 60 Sonic Dismembrator was from Fisher Scientific (Pittsburgh, PA). The Peltier Thermal Thermocycler was from Bio-Rad Laboratories.
Methods
Cell culture and synchronization
The wild-type mouse embryonic fibroblasts (MB16.3) have been described (28). Cells were routinely grown at 34 °C in a 10% CO2 incubator in DMEM supplemented with GlutaMAX-1, 10% FBS and hygromycin (80 µg/ml). All cells were routinely tested and found to be free of mycoplasma contamination. Cell synchronization was as described (17). Briefly, cells were seeded at a cell density of 8 × 106 cells/145 mm dish and the following day were washed once with HBSS and once with DMEM containing 0.2% FBS. Then the cells were incubated with DMEM containing 0.2% FBS for 48 h. The medium was then removed and the cells were incubated with 2.5 µM APH in DMEM with 10% FBS for 16 h. The non-cycling cells were washed with HBSS, and drug-free 10% FBS-containing DMEM was added. Culture was continued for 4 h to allow cell cycling to resume.
Chromatin immunoprecipitation (ChIP)
For the ChIP assay, synchronized cells in 145 mm dishes were subjected to cell treatment protocols as follows. For MMS alone, MMS was added directly to the medium to a final concentration of 0.25 mM, and after 1 h the cells were washed with HBSS. Fresh medium was added, and culture was continued for 1 h. For 4-AN alone, 4-AN was added directly to the medium to a final concentration of 10 µM, and culture was continued for 2 h. For combination treatment with MMS + 4-AN, both MMS and 4-AN were added to final concentrations of 0.25 mM and 10 µM, respectively. After 1 h, the medium was removed and fresh medium with 10 µM 4-AN was added. Culture then was continued for 1 h. Synchronized cells referred to as “control” were mock-treated and cultured at each step, but with drug-free medium.
The ChIP procedure involved an in vivo protein/DNA cross-linking treatment of cells with formaldehyde, nuclei preparation, micrococcal nuclease digestion of chromatin and then immunoprecipitation. ChIP was performed with the Chromatin IP Kit described above using 5 dishes of cultured cells. Briefly, cells were grown to ~80 to 90% confluence, cross-linked with 1% formaldehyde in medium for 10 min at room temperature, and then the medium was adjusted to 125 mM glycine. The cells then were held for 5 min at room temperature, to quench the formaldehyde cross-linking. The cells were washed twice with PBS at 4 °C. Finally, 2 ml PBS containing 5 mM PMSF at 4 °C was added to each dish. Cells were scraped, and cells from the five dishes were combined into a 15 ml conical tube. The cells were pelleted by centrifugation, then resuspended in 10 ml Buffer A (provided by the manufacturer) at 4 °C and held at 4 °C for 10 min. Nuclei were obtained by centrifugation, and the nuclear pellets were resuspended in 10 ml Buffer B (provided by the manufacturer) at 4 °C. The suspension was digested with 5 µl of micrococcal nuclease (2000 gel units/µl) with incubation at 37 °C for 20 min. The nuclease reaction was stopped by adding EDTA and placing the mixture at 4 °C. Nuclei were then collected by centrifugation. Nuclear pellets were resuspended in 1×ChIP Buffer (provided by the manufacturer). Each tube was sonicated with several pulses with a Sonic Dismembrator; the samples were held for 30 sec in wet ice between pulses. The lysate, i.e., chromatin preparation, was clarified by centrifugation, divided into aliquots and stored at −80 °C.
The chromatin preparations from cells treated with the MMS + 4-AN combination, MMS or 4-AN alone, and control were used in immunoprecipitation incubations. For each chromatin preparation, the sample was thawed and diluted to 400 µl with 1×ChIP Buffer containing protease inhibitor cocktail (provided by the manufacturer). A portion corresponding to 2% of the diluted chromatin preparation from each cell treatment was transferred to a microfuge tube and stored at −20 °C. Next, the samples were thawed and mixed with either monoclonal antibody against PARP-1, monoclonal antibody against histone H3 (as a positive control), or non-immune normal rabbit IgG (as a negative control). After overnight incubation at 4 °C, ChIP-grade protein G-agarose beads were added, and the mixture was incubated for 4 h at 4 °C. The beads were collected by centrifugation and washed with 1×ChIP Buffer. The precipitated proteins were digested by proteinase K treatment, and DNA was obtained according to the manufacturer. Real-time quantitative-PCR (qPCR) was used to measure the DNA content of the immunoprecipitated material. As a positive control for the DNA quantification, ChIP samples were prepared from each type of cell treatment with anti-histone H3 antibody.
Real-time quantitative-PCR (qPCR) analysis of DNA in the ChIP samples
The PCR program was set up in the ABI Prism SDS 2.1 (with 7900 HT Sequence detection system). The PCR reaction was conducted in a 96-well clear plate with a final reaction volume of 25 µl containing 12.5 µl of Power SYBR Green Master mix, 10 µM each of forward and reverse isochore primers (described below), and 1.5 µl of DNA extracted from the ChIP sample; the final volume was made up to 25 µl with water. Each reaction mixture was set up in triplicate. The cycling conditions comprised 50 °C for 2 min (1 cycle), 95 °C for 10 min (1 cycle), and 40 cycles of 95 °C for 15 sec and 60 °C for 1 min. The amount of immunoprecipitated DNA in each sample was determined relative to input chromatin DNA. Each ChIP experiment was conducted a minimum of three times with independent chromatin isolates. ChIP samples prepared with histone H3 antibody served as a positive control and DNA was quantified with isochore primers. A similar amount of DNA was found in these anti-histone H3 ChIP samples isolated after the different types of cell treatment (data not shown).
For quantification of DNA by qPCR, we first chose primers targeted for GC-rich isochores (29–32). Many different primer sets were surveyed, but most were found to be problematic, including a line sequence NCBI: NM_013627 and isochore primer NCBI: NM_018779. However, the isochore primer set NCBI: NM_019426 gave excellent results and was used in the DNA quantification experiments. Sequence information on this mouse oligonucleotide isochore primer set is described in NCBI, Ref. Seq.: NM 019426, Mus musculus Chromosome:Ch6. The sequences used were as follows:
Sense primer: 5′-CGT AGG AAG GAA CTC GGG TC
Anti-sense primer: 5′-CTC ATT GTT TTC CGA GCC TT
DNA polymerase β primers were similarly found to give excellent results in the survey of different primer sets noted above, and DNA quantification experiments also were conducted with the DNA polymerase β primer set shown here:
Sense primer: 5′-GCA GAT TTG ATC TAC GGC TAA CTC AC
Anti-sense primer: 5′-GGG TCT TAC CGG TGG AAC ATT G
Before use in real-time qPCR experiments, these primers were analyzed for efficiency using the thermocycler.
Analysis of PARP-1 protein in the ChIP samples
For the analysis of PARP-1 in the ChIP samples, the cross-linked immunoprecipitated samples were not digested with proteinase K, but instead were subjected to a treatment protocol to reverse the cross-linking before analysis on SDS-PAGE gels. The washed beads containing the ChIP sample were suspended in 1×ChIP Buffer. The mixture was adjusted to 200 mM NaCl and incubated for 2 h at 65 °C. The mixture was combined with SDS-PAGE gel loading solution and heated at 95 °C for 6 min. The mixture was centrifuged and the supernatant fraction was loaded onto a 4 to 12% gradient gel. In some experiments, the gel was subjected to analysis by mass spectrometry (33), and in others proteins were transferred to a nitrocellulose membrane, and the membrane was probed with anti-PARP-1 mouse monoclonal antibody and G3PDH as a loading control (34). Purified PARP-1, used as a positive control, was prepared as described (35).
Mass spectrometry verification of PARP-1 in anti-PARP-1 ChIP samples
Whole lanes from a polyacrylamide gel were digested using a Progest robotic digester (Genomic Solutions, Ann Arbor, MI) where each lane was cut into 24 slices. These slices were incubated twice for 15 min in 100 µl of 50:50 (v:v) 25 mM ammonium bicarbonate:acetonitrile. The gel was then dehydrated by incubating for 20 min in 100 µl of acetonitrile followed by drying under a nitrogen stream for 18 seconds. 250 ng of trypsin (Promega, Madison, WI) was added and the gel pieces incubated for 12 h at 37 °C. The supernatant solutions from the digests were saved and combined with the supernatants from re-extractions. The gels were re-extracted three times: once with 50 µl of water for 20 min and twice with 20 min incubations in 50 µl of 45:50:5 (v:v:v) water:acetonitrile:formic acid. The pooled supernatants were lyophilized and resuspended in 40 µl of 0.1% formic acid.
NanoLC-ESI-MS/MS analyses were performed using an Agilent 1200 nanoLC system on-line with an Agilent 6340 ion trap mass spectrometer with the chip cube interface. Buffer A was 99.9:0.1 (v:v) water:formic a cid a nd buffer B was 99.9:0.1 (v:v) a cetonitrile:formic a cid. Briefly, 20 µl of the peptide digest was loaded onto an Agilent ProtID chip (75 µm × 43 mm) followed by a 15 -min wash with 95% A, 5% B. Peptides were eluted with a linearly increasing gradient of buffer B as follows: 0–45 min, 5–50% B, 45–50 min, B increased to 95%, 50–60 min maintained at 95% B. The mass spectrometer was used in the positive ion, standard enhanced mode and included settings of a mass range from 200 to 2200 m/z, an ionization potential of 1.9 kV, an ICC smart target (number of ions in the trap prior to scan out) of 200,000 or 200 ms of accumulation, and a 1.0-V fragmentation amplitude. MS/MS data were acquired using a data-dependent acquisition format with the six most abundant ions from each MS scan further interrogated by MS/MS. The automated switching for MS/MS required a threshold of 10,000 counts.
Peak lists were generated from the data obtained from each nanoLC-ESI-MS/MS analysis using the data extractor feature of the Spectrum Mill MS Proteomics Workbench from Agilent. The data extractor settings included limiting the data search to ions observed between 300 and 5000 Da and a retention time between 10 min and 55 min. MS scans with the same precursor mass (±1.5 m/z) and retention time within 30 s were merged. Additionally, of the remaining MS/MS spectra, only spectra that contained sequence tag information greater than 2 residues were submitted for database searching. The resulting extracted data were searched against the human and rodent limited International Protein Index database (IPI_human_rodent) using the MS/MS Search function in the Spectrum Mill software. Search settings included a trypsin specificity with up to a single missed cleavage allowed, a precursor ion mass tolerance of 1.5 Da, a product ion mass tolerance of 1.0 Da, variable methionine oxidation, and a minimum matched spectral intensity of 80%. PARP-1 peptides found in the ChIP samples are summarized in Table 1.
Table 1.
Verification of PARP-1 in ChIP samples: Number of authentic PARP-1 peptides identified in immunoprecipitates by mass spectrometry
| ChIP Antibodya | ||
|---|---|---|
| Cell Treatment | IgG | anti-PARP-1 |
| Control (mock) | 0 | 18 |
| MMS | 0 | 14 |
| 4-AN | 0 | 10 |
| MMS + 4-AN | 1 | 16 |
Non-immune IgG was used at the same concentration as the anti-PARP-1 IgG.
Analysis of PARP-1 in the chromatin-associated and nuclear soluble protein fractions
The isolation of the nuclear fraction and the separation of soluble and chromatin-associated nuclear proteins were based on the protocol described by Mendez and Stillman (36), with modifications from Ritzi et al. (37). To inhibit proteases, all buffers were supplemented with 25 µM MG-132; a 10 mM stock solution was prepared in DMSO. Cells in one 145 mm dish were synchronized and treated as described above. Cells were then harvested, washed with PBS, and resuspended in 250 µl hypotonic Buffer A (10 mM Hepes, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, and protease inhibitor mix Complete© [according to the manufacturer]). Next, for cell lysis, the suspension was adjusted to 0.1% Triton X-100 and incubated for 5 min at 4 °C. The nuclei were separated by centrifugation at 1300 g for 4 min at 4 °C and washed once with Buffer A. The nuclei were then lysed by suspension in low-stringency solution (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT) for 10 min at 4 °C. The insoluble chromatin fraction and the nuclear soluble protein fraction were separated by centrifugation at 1700 g for 4 min, and the pelleted chromatin fraction was washed once with low-stringency solution. This final pellet fraction was used to assess the chromatin-associated proteins. These proteins were extracted by addition of 250 µl buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% NP40) at 4 °C and incubation for 30 min at 4 °C. The mixture was centrifuged at 16,000 g for 10 min at 4 °C. The supernatant fraction (extract) was removed, and the protein concentration was measured using the Bio-Rad protein assay with BSA as standard. Equal “cell equivalents” of nuclear soluble protein fraction and chromatin-associated protein fraction were analyzed by immunoblotting for PARP-1 and histone H3 or Lamin A, as a control (38) (see Figure. 4A). The amount of PARP-1 in the chromatin-associated protein fraction also was analyzed by immunoblotting as described above (see Figures. 4B and C).
Figure 4.
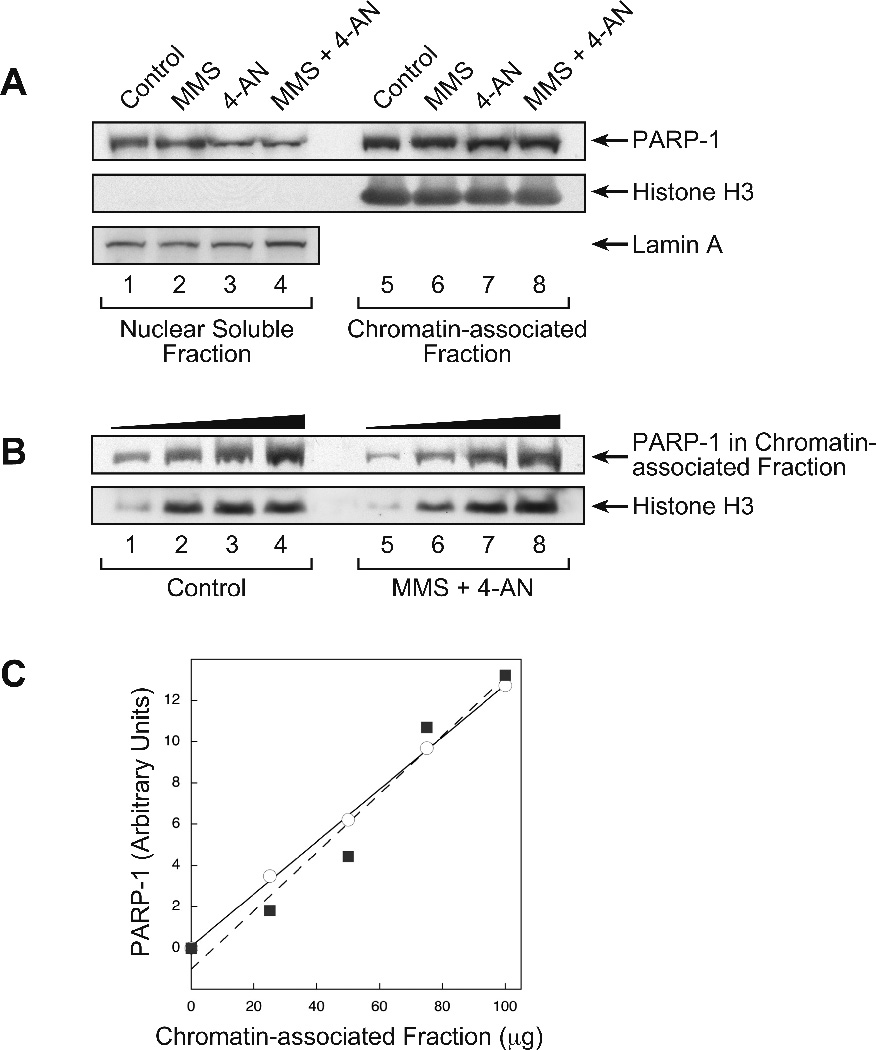
Analysis of PARP-1 protein in the nuclear fraction isolated from cells after the various cell treatments. Experiments were conducted as described under Methods. A, The nuclear soluble protein fraction and chromatin-associated protein fraction obtained from cells, treated as indicated, by a biochemical fractionation procedure were analyzed by immunoblotting with monoclonal antibody against PARP-1 and polyclonal antibody against histone H3 or Lamin A, as a control. Lanes 1 and 5, control cells; lanes 2 and 6, MMS treated cells; lanes 3 and 7, 4-AN treated cells; lanes 4 and 8, MMS + 4-AN treated cells. B, Increasing amounts of chromatin-associated protein fraction (lanes 1 and 5, 25 µg; lanes 2 and 6, 50 µg; lanes 3 and 7, 75 µg; lanes 4 and 8, 100 µg) from control and MMS + 4-AN-treated cells were analyzed for PARP-1 by immunoblotting. The solid symbols at the top of the immunoblot indicate increasing amounts of protein added. Histone H3 was probed as an internal loading control. C, Quantification of PARP-1 in the chromatin-associated protein fraction from control cells (circles) or cells treated with the MMS + 4-AN combination (squares) shown in panel B. The lines, solid and dashed, represent a curve fitting of the respective data points.
Results
We explored a prediction of the model in Figure 1. Briefly, when BER occurs in a replicating cell, PARP inhibition will cause PARP-1 to freeze at BER intermediates, and eventually a replication fork will collide with the inhibited PARP-1/DNA complex. Replication fork progression will stall, leading to S-phase checkpoint signaling and DSBs. The prediction to be tested is that more DNA will be found in association with PARP-1 when cells are treated with the MMS + 4-AN combination than when cell are untreated. For evaluating this prediction, we used the ChIP procedure with anti-PARP-1 antibody. After in vivo cross-linking of DNA/protein complexes and antibody pull-down, the cross-linking was reversed and genomic DNA in the samples was quantified by real-time qPCR using isochore primers for amplification of general genomic DNA. Mouse fibroblasts that had been synchronized in S-phase were used and the treatment protocol is summarized in Figure 2A.
Figure 2.
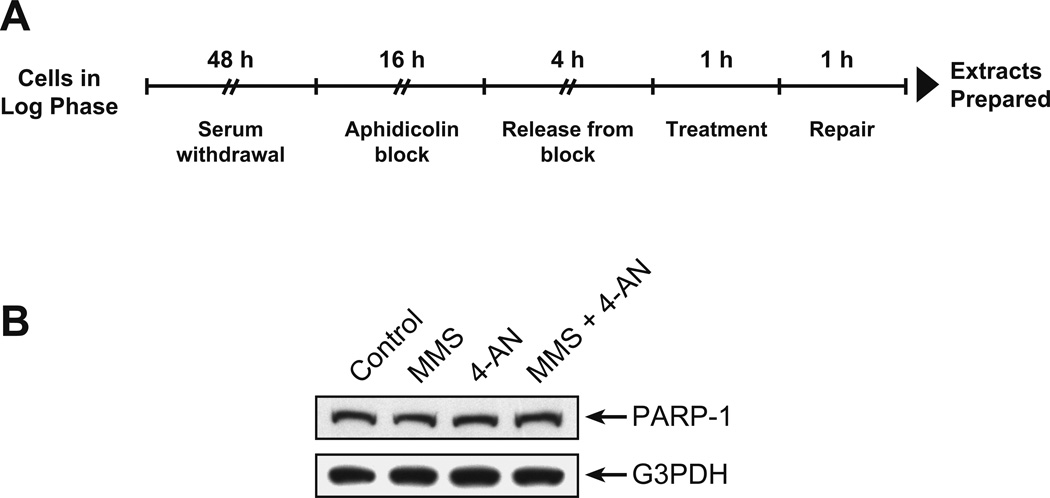
Mouse fibroblast synchronization, treatment and characterization. All experiments were conducted as described under Methods. A, Synchronization and treatment protocol. Mouse fibroblasts in log phase were grown for 48 h with medium containing only 0.2% FBS (serum withdrawal), followed by culture with 2.5 µM APH in DMEM with 10% FBS for 16 h. Cells were released from the APH block by washing then addition of DMEM containing 10% FBS. After 4 h, the cells were treated with 0.25 mM MMS or 10 µM 4-AN or the combination in complete medium for 1 h. The medium was removed and the cells were cultured for 1 h (repair) +/− 4-AN in complete medium. Control cells were mock-treated in parallel at each step. B, Assessment of PARP-1 protein level in the cell extracts as a function of treatment. After treatment as illustrated in Panel A, cell extracts were immunoblotted with anti-PARP-1 monoclonal antibody as described; the membrane was stripped and probed with monoclonal antibody against G3PDH, used as loading control.
Cells treated with MMS alone or 4-AN alone were prepared along with control (mock-treated) cells and cells treated with the MMS + 4-AN combination. In each case, the same number of cells was subjected to the cross-linking and ChIP procedure. In initial experiments, to rule out any differences in PARP-1 level in these cells, we quantified PARP-1 expression in the various cell extracts by immunoblotting. The amount of PARP-1 protein was similar in the control and three types of treated cells (Figure 2B, top panel) using G3PDH as a loading control (lower panel). In addition, we verified that PARP-1 had been immunoprecipitated in our ChIP samples, and this was accomplished using mass spectrometric analysis. In each case where the ChIP procedure included anti-PARP-1 antibody, the presence of PARP-1 in the immunoprecipitate was verified (Table 1). ChIP samples prepared with the control non-immune IgG failed to yield significant PARP-1 (Table 1).
Results of DNA quantification with the anti-PARP-1 ChIP samples are shown in Figure 3. Considerably increased levels (> 10-fold) of DNA were found in samples prepared from MMS + 4-AN treated cells than from the control cells. With cells treated either with MMS or 4-AN alone, there were increases in DNA associated with PARP-1, but these were considerably less than with the MMS + 4-AN combination. Next, we designed confirmatory experiments with PCR primers other than the isochore primers used in the experiments in Figure 3A. The DNA quantification of the ChIP samples was conducted using primers against an expressed gene (pol β). The results with independently prepared ChIP samples are shown in Figure 3B. Again, increased levels of DNA were found in association with PARP-1 in the samples from MMS + 4-AN treated cells compared with control cells. In contrast to the results with isochore primers, samples from cell treated either with MMS or 4-AN alone had significantly less DNA associated with PARP-1. We conclude from the experiments in Figure 3 that more genomic DNA was associated with PARP-1 in the MMS + 4-AN treated cells than in the control or single agent-treated cells. Thus, our initial prediction was confirmed.
Figure 3.
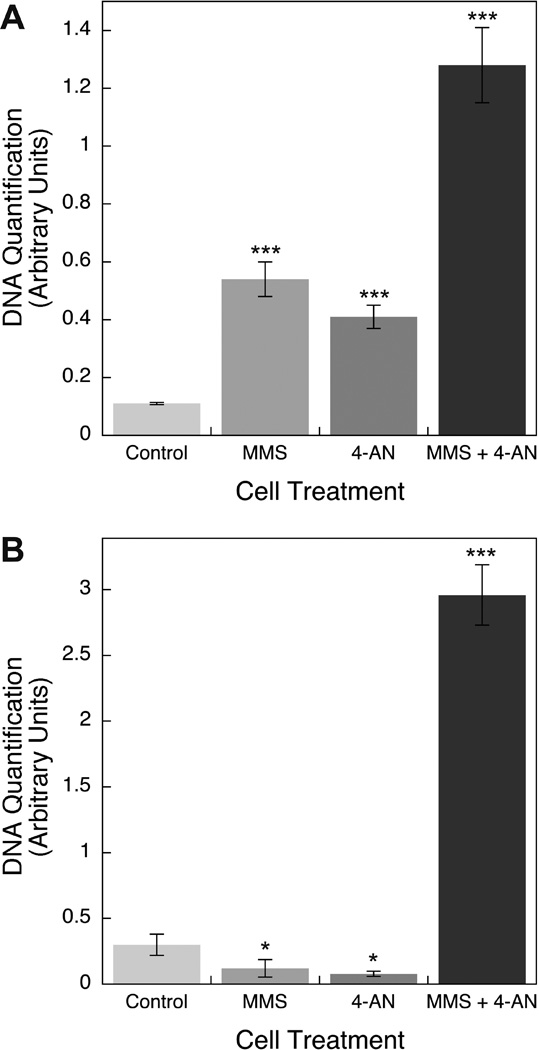
DNA associated with PARP in ChIP samples from control and treated cells. Experiments were conducted as described under Methods. DNA was purified from the various ChIP samples and measured by qPCR using A, isochore primers and B, DNA polymerase β primers. The amount of DNA in each type of ChIP sample is represented as the qPCR signal relative to a constant amount of input chromatin DNA. The data represent average values (with S.D. bars) from 3 independent ChIP samples, each one measured in triplicate. In panels A and B, the triple asterisk symbols indicate difference from the respective control samples at p < 0.001 using the two-sample t-test, and in panel B the single asterisk symbols indicate difference from the control samples at p < 0.05 using the two-sample t-test. The results with the MMS + 4-AN samples in panels A and B are different from all other samples at p < 0.001 using the two-sample t-test.
In view of the results shown in Figure 3, indicating increased DNA in association with PARP-1 in the MMS + 4-AN treated cells, we were curious to know if these cells also contained more chromatin-associated PARP-1 than cells subjected to the other treatments studied here, i.e., control, MMS treated and 4-AN treated. Control and the three types of treated cells were prepared as described in Figure 2A, and nuclei were isolated. The nuclei were then separated into a nuclear soluble fraction and a chromatin-associated fraction, and the amount of PARP-1 in each was quantified by immunoblotting. For the control cells and all cell treatments, the amount of PARP-1 in the chromatin-associated fraction was greater than in the nuclear soluble fraction (Figure 4A). In the sample from MMS + 4-AN treated cells, there was a modest increase in the ratio of chromatin-associated PARP-1 vs. nuclear soluble fraction PARP-1 (compare lanes 4 and 8, Figure 4A). Yet, there was no difference in the amount of chromatin-associated PARP-1 as a function of cell treatment (Figure 4A). This was confirmed in an immunoblotting quantification experiment of chromatin-associated PARP-1, using increasing amounts of chromatin-associated fraction from the control and MMS + 4-AN treated cells (Figure 4B). Interestingly, the results revealed no difference in the amount of chromatin-associated PARP-1, as a function of the combination treatment (Figure 4C).
Finally, another prediction of the model in Figure 1 is that S-phase checkpoint signaling is associated with the inhibited PARP-1/DNA complex. It is known that an S-phase checkpoint response occurs in these MMS + 4-AN treated cells (22) and that this involves phosphorylation of ATR and NBS1 as well as other cell signaling proteins (19). We asked whether the ChIP sample with anti-PARP-1 antibody also contained ATR, NBS1 and related factors, such as RPA70 and MRE11. The results of immunoblotting analysis of the ChIP samples prepared with anti-PARP-1 antibody and control IgG are shown in Figure 5. PARP-1 was present, as expected, and NBS1, MRE11, RPA70 and ATR also were detected. These results are consistent with the prediction that a protein/DNA complex containing inhibited PARP-1 is in close proximity to these S-phase checkpoint response factors.
Figure 5.
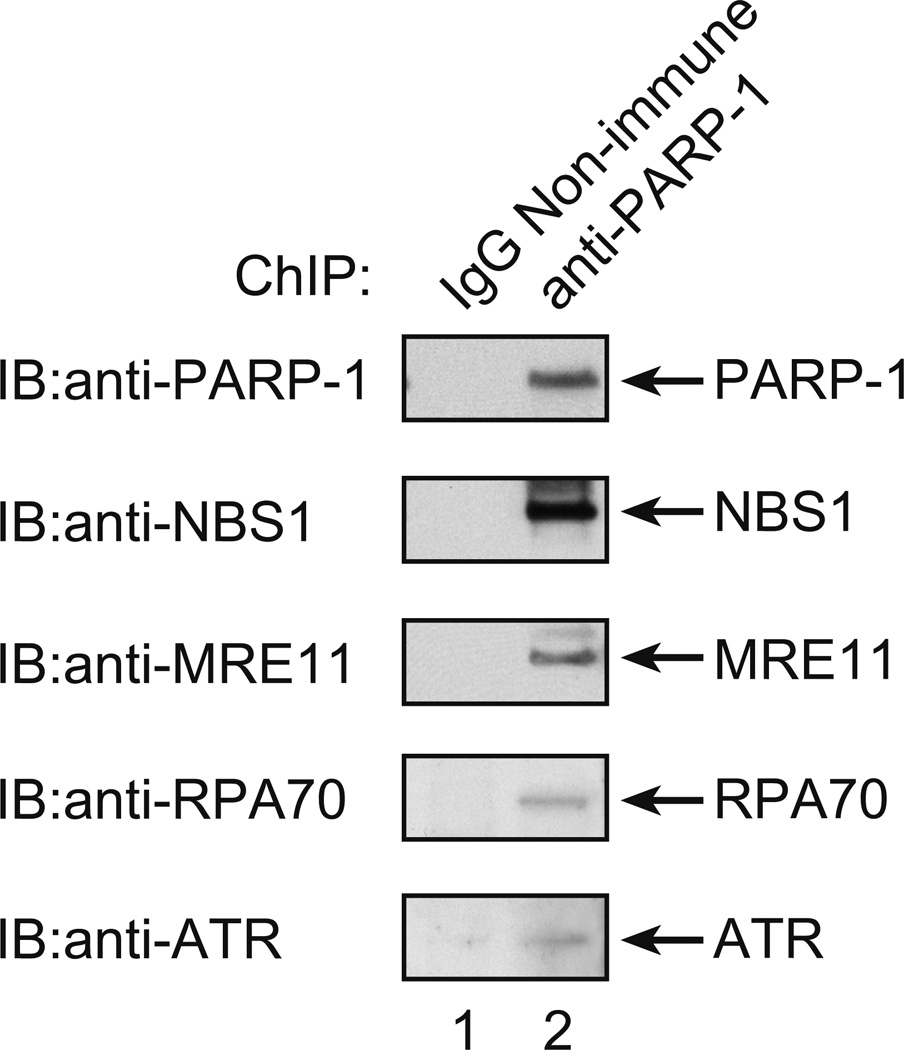
Co-immunoprecipitation analysis of proteins in ChIP samples prepared from cells treated with MMS + 4-AN. Experiments were conducted as described under Methods and as illustrated in Figure 2. Two types of ChIP samples were prepared after treatment of cells as described; one with the anti-PARP-1 antibody, as usual; and the other with negative control non-immune IgG. For immunoblotting analysis (IB), proteins in the two ChIP samples were separated by 4–12% SDS-PAGE and transferred to a membrane. Then, the membrane was probed with antibodies against PARP-1, NBS1, MRE11, RPA70, and ATR, as illustrated. IB probing with non-immune IgG was negative corresponding to the each of these proteins (not shown).
Discussion
In vivo cross-linking and ChIP analysis has been widely used to identify DNA sequence elements associated with a protein of interest. In the experiments described here, the method was used to measure general genomic DNA associated with the PARP-1 protein. The relative amount of DNA in association with PARP-1 was found to be greater in PARP inhibitor treated cells undergoing MMS-induced repair than in cells without MMS treatment or without inhibitor treatment (Figure 3). These results are consistent with a prediction from the working model in Figure 1 and represent the first direct measurements of DNA-associated PARP-1 as a function of inhibitor treatment. In addition, S-phase checkpoint response factors were found to co-immunoprecipitate in the anti-PARP-1 ChIP samples. This finding is also consistent with a prediction of the model in Figure 1 and suggests that these response factors are in proximity to PARP-1 in the MMS + 4-AN treated cells.
The model in Figure 1 resembles models proposed by others (7, 39, 40), and also is in line with an original proposal on DNA association of inhibited PARP-1 by Satoh and Lindahl (9). As summarized above, accumulation of DSBs in mouse fibroblasts cells treated with the MMS + 4-AN combination had been demonstrated earlier (20), and these cells exhibited single-strand DNA-mediated and DSB-mediated cell cycle checkpoint signaling (22, 23). Yet, these results are also consistent with other models where PARP-1 becomes DNA-associated and is frozen in complex with DNA upon inhibition, and different models explaining the cytotoxicity of PARP inhibitors and DNA damaging agents have been discussed by others (15, 24, 41).
PARP-1 is abundantly expressed in mouse fibroblasts and almost all of it is found in nuclei (Prasad et al., unpublished observations). The results presented here on quantification of the cellular level of PARP-1 as a function of MMS + 4-AN treatment indicated that the PARP-1 protein level did not fluctuate as a function of the treatments (Figure 2B). Much of the nuclear PARP-1 was found in the chromatin-associated fraction, as opposed to the nuclear soluble fraction, and this picture failed to change significantly with the various cell treatments (Figure 4A). Similarly, in the chromatin immunoprecipitates with anti-PARP-1 antibody, the amount of PARP-1 immunoprecipitated did not change as a function of the cell treatments. In contrast, the amount of DNA associated with PARP-1 in the MMS + 4-AN treated cells was greater than that for the other treatments (Figure 3). Thus, a larger portion of the chromatin-bound PARP-1 was associated with DNA and found cross-linked by the ChIP procedure. The lower level of PARP-1/DNA complex found in control cells implies that PARP-1 is bound to chromatin proteins rather than the DNA.
Finally, regarding therapeutic approaches with PARP inhibitors, it may be interesting to test strategies for inhibiting additional factors in the BER pathway (42, 43). A phenomenon of increased cytotoxicity of this type has already been observed in mouse fibroblasts that are deficient in pol β (8) or XRCC1 (10). Thus, inhibition of an enzyme in BER, in addition to PARP-1, may enhance accumulation of BER intermediates and consequently provide more sites for trapping inhibited PARP-1. Increased cytotoxicity would be anticipated with the combination of a BER enzyme deficiency and a PARP inhibitor while triggering BER. Hence, strategies for increasing the cytotoxicity of combination treatment with alkylating agents and PARP inhibitors may include small molecule inhibitors against the BER enzymes and co-factors including AP endonuclease1, pol β, polynucleotide kinase, tyrosyl-DNA phosphodiesterase, aprataxin, DNA ligases and XRCC1. Similarly, genetic deficiencies in these BER factors in tumor cells may render them hypersensitive to combination treatment (42).
Acknowledgments
We thank Jason Williams and the Protein Microcharacterization Core for assistance with the mass spectrometry analyses, Grace Kissling for assistance with the statistical analyses, Yuan Liu for assistance with analysis of the ChIP results, and especially Melissa Adkins for assistance with qPCR experiments when using the API Prism SDS 2.1 sequence detection system. We thank Bonnie Mesmer for editorial assistance Lois Wyrick for assistance with the figures and Payal Chokshi for technical assistance. We thank Esther Hou and Rajen Prasad for samples of purified PARP-1 used as positive controls in the experiments.
Grant Support
This research was supported by Research Project Number Z01-ES050159 in the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
The authors declare no conflict of interest.
References
- 1.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, et al. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci USA. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 3.Dantzer F, de La Rubia G, Ménissier-de Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 4.Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol. 2007;27:5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodhouse BC, Dianova II, Parsons JL, Dianov GL. Poly(ADP-ribose) polymerase-1 modulates DNA repair capacity and prevents formation of DNA double strand breaks. DNA Repair. 2008;7:932–940. doi: 10.1016/j.dnarep.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate: Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 7.Godon C, Cordelières FP, Biard D, Giocanti N, Mégnin-Chanet F, Hall J, et al. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008;36:4454–4464. doi: 10.1093/nar/gkn403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horton JK, Wilson SH. Hypersensitivity phenotypes associated with genetic and synthetic inhibitor-induced base excision repair deficiency. DNA Repair. 2007;6:530–543. doi: 10.1016/j.dnarep.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 10.Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masaoka A, Horton JK, Beard WA, Wilson SH. DNA polymerase β and PARP activities in base excision repair in living cells. DNA Repair. 2009;8:1290–1299. doi: 10.1016/j.dnarep.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008;8:363–369. doi: 10.1016/j.coph.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 13.Ellisen LW. PARP inhibitors in cancer therapy: promise, progress, and puzzles. Cancer Cell. 2011;19:165–167. doi: 10.1016/j.ccr.2011.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandhu SK, Yap TA, de Bono JS. Poly(ADP-ribose) polymerase inhibitors in cancer treatment: a clinical perspective. Eur J Cancer. 2010;46:9–20. doi: 10.1016/j.ejca.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 15.Loser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9:1775–1787. doi: 10.1158/1535-7163.MCT-09-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferraris DV. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem. 2010;53:4561–4584. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- 17.Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J Biol Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 18.Horton JK, Stefanick DF, Wilson SH. Involvement of poly(ADP-ribose) polymerase activity in regulating Chk1-dependent apoptotic cell death. DNA Repair. 2005;4:1111–1120. doi: 10.1016/j.dnarep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Horton JK, Stefanick DF, Zeng JY, Carrozza MJ, Wilson SH. Requirement for NBS1 in the S phase checkpoint response to DNA methylation combined with PARP inhibition. DNA Repair. 2011;10:225–234. doi: 10.1016/j.dnarep.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heacock ML, Stefanick DF, Horton JK, Wilson SH. Alkylation DNA damage in combination with PARP inhibition results in formation of S-phase-dependent double-strand breaks. DNA Repair. 2010;9:929–936. doi: 10.1016/j.dnarep.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 22.Horton JK, Stefanick DF, Kedar PS, Wilson SH. ATR signaling mediates an S-phase checkpoint after inhibition of poly(ADP-ribose) polymerase activity. DNA Repair. 2007;6:742–750. doi: 10.1016/j.dnarep.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrozza MJ, Stefanick DF, Horton JK, Kedar PS, Wilson SH. PARP inhibition during alkylation-induced genotoxic stress signals a cell cycle checkpoint response mediated by ATM. DNA Repair. 2009;8:1264–1272. doi: 10.1016/j.dnarep.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol Oncol. 2011;5:387–393. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011;39:3166–3175. doi: 10.1093/nar/gkq1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang JB, Goellner EM, Wang XH, Trivedi RN, St Croix CM, Jelezcova E, et al. Bioenergetic metabolites regulate base excision repair-dependent cell death in response to DNA damage. Mol Cancer Res. 2010;8:67–79. doi: 10.1158/1541-7786.MCR-09-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sobol RW, Horton JK, Kühn R, Gu H, Singhal RK, Prasad R, et al. Requirement of mammalian DNA polymerase β in base excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 29.Oliver JL, Carpena P, Hackenberg M, Bernaola-Galvan P. IsoFinder: computational prediction of isochores in genome sequences. Nucleic Acids Res. 2004;32:W287–W292. doi: 10.1093/nar/gkh399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavlicek A, Paces J, Clay O, Bernardi G. A compact view of isochores in the draft human genome sequence. FEBS Lett. 2002;511:165–169. doi: 10.1016/s0014-5793(01)03283-5. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe Y, Abe T, Ikemura T, Maekawa M. Relationships between replication timing and GC content of cancer-related genes on human chromosomes 11q and 21q. Gene. 2009;433:26–31. doi: 10.1016/j.gene.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Matassi G, Labuda D, Bernardi G. Distribution of the mammalian-wide interspersed repeats (MIRs) in the isochores of the human genome. FEBS Lett. 1998;439:63–65. doi: 10.1016/s0014-5793(98)01338-6. [DOI] [PubMed] [Google Scholar]
- 33.Choi JH, Williams J, Cho J, Falck JR, Shears SB. Purification, sequencing, and molecular identification of a mammalian PP-InsP5 kinase that is activated when cells are exposed to hyperosmotic stress. J Biol Chem. 2007;282:30763–30765. doi: 10.1074/jbc.M704655200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kedar PS, Kim S-J, Robertson A, Hou E, Prasad R, Horton JK, et al. Direct interaction between mammalian DNA polymerase β and proliferating cell nuclear antigen. J Biol Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- 35.Lavrik OI, Prasad R, Sobol RW, Horton JK, Ackerman EJ, Wilson SH. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J Biol Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 36.Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, et al. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J Cell Sci. 2003;116:3971–3984. doi: 10.1242/jcs.00708. [DOI] [PubMed] [Google Scholar]
- 38.Dai J, Sultan S, Taylor SS, Higgins JMG. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2011;19:472–488. doi: 10.1101/gad.1267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Shi Y, Guan R, Donawho C, Luo Y, Palma J, et al. Potentiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitor ABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks. Mol Cancer Res. 2008;6:1621–1629. doi: 10.1158/1541-7786.MCR-08-0240. [DOI] [PubMed] [Google Scholar]
- 40.Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADP-ribosyl)ation: a promising approach in cancer therapy. Trends Mol Med. 2005;11:456–463. doi: 10.1016/j.molmed.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20:294–302. doi: 10.1016/j.ceb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelley MR, Fishel ML. DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem. 2008;8:417–425. doi: 10.2174/187152008784220294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, et al. Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS One. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]