

Abstract
Spatiotemporal regulation of cAMP in cardiac myocytes is integral to regulating the diverse functions downstream of β-adrenergic stimulation. The activities of cAMP phosphodiesterases modulate critical and well-studied cellular processes. Recently, in epithelial and smooth muscle cells, it was found that the multi-drug resistant protein 4 (MRP4) acts as a cAMP efflux pump to regulate intracellular cAMP levels and alter effector function, including activation of the cAMP-stimulated Cl− channel, CFTR (cystic fibrosis transmembrane conductance regulator). In the current study we investigated the potential role of MRP4 in regulating intracellular cAMP and β-adrenergic stimulated contraction rate in cardiac myocytes. Cultured neonatal ventricular myocytes were used for all experiments. In addition to wildtype mice, β1-, β2-, β1/β2-adrenoceptor, and CFTR knockout mice were used. MRP4 expression was probed via Western blot, intracellular cAMP was measured by fluorescence resonance energy transfer, while the functional role of MRP4 was assayed via monitoring of isoproterenol-stimulated contraction rate. We found that MRP4 is expressed in mouse neonatal ventricular myocytes. A pharmacological inhibitor of MRP4, MK571, potentiated submaximal isoproterenol-stimulated cAMP accumulation and cardiomyocyte contraction rate via β1-adrenoceptors. CFTR expression was critical for submaximal isoproterenol-stimulated contraction rate. Interestingly, MRP4-dependent changes in contraction rate were CFTR-dependent, however, PDE4-dependent potentiation of contraction rate was CFTR-independent. We have shown, for the first time, a role for MRP4 in the regulation of cAMP in cardiac myocytes and involvement of CFTR in β-adrenergic stimulated contraction. Together with phosphodiesterases, MRP4 must be considered when examining cAMP regulation in cardiac myocytes.
Keywords: MRP4, cAMP, β-adrenergic signaling, cardiac myocytes, CFTR
1. Introduction
The spatial and temporal control of cAMP has become widely accepted as the means whereby a single signaling molecule can lead to a diverse array of cellular responses. This concept has been widely studied in cardiac myocytes and commented on by Kass (2008). Multi-layer compartmentalization of cAMP signaling occurs as a result of differential localization of β-adrenoceptor subtypes, adenylyl cyclase, PKA, Epac (exchange protein activated by cAMP), and phosphodiesterases (PDEs) via anchoring proteins, such as A-kinase anchoring proteins (AKAPs) (Dodge-Kafka et al., 2006). The concentration and spread of cAMP is considered to be largely controlled by PDEs, which are responsible for terminating the signal by breakdown of cAMP. In cardiac myocytes β-adrenergic-stimulated increases in cAMP are predominantly regulated by PDE4 with PDE3 playing a minor role (Jurevicius et al., 2003; Leroy et al., 2008).
Multi-drug resistance protein 4 (MRP4) is a member of the ATP-binding cassette transporter superfamily and has been shown to transport antiviral nucleoside-based drugs (Schuetz et al., 1999) and endogenous molecules involved in cellular communication and signaling, including, but not limited to, organic anions, prostaglandins, conjugated steroids, and cyclic nucleotides (Russel et al., 2008). Of particular interest, MRP4 has been shown to negatively regulate intracellular cAMP levels in both epithelial and smooth muscle cells (Li et al., 2007; Sassi et al., 2008). In colonic epithelial cells, Li et al (2007) showed that inhibition of MRP4 by MK571 caused a compartmentalized increase in cAMP that resulted in activation of the cystic fibrosis transmembrane conductance regulator (CFTR) via PDZK1, which serves as a scaffolding protein for MRP4 and CFTR. In addition to colonic epithelial cells, MRP4 is expressed in prostate tubuloacinar cells, hepatocytes, choroids plexus epithelium, testis, ovary, adrenal gland, various blood cells, and neurons (Ritter et al., 2005; Russel et al., 2008); however, no one has examined its presence in cardiac myocytes.
CFTR, an anion channel permeable to Cl− and HCO3−, is most notably studied in epithelial cells, but is also expressed in other cell types, including cardiac myocytes (Lader et al., 2000; Sellers et al., 2010). Activation of CFTR occurs upon ATP-binding and phosphorylation of its regulatory domain by cAMP-, cGMP-, or Ca2+-dependent proteins (Gadsby and Nairn, 1999; Ostedgaard et al., 2001; Vaandrager et al., 1997). In cardiac myocytes, it has been postulated that as a Cl− channel CFTR is involved in minimizing the depolarizing effect of Ca2+ entry upon β-adrenoreceptor stimulation (Duan, 2009).
In the current study we set out to determine if MRP4 is expressed in cardiac myocytes and if so, examine its involvement in cAMP regulation. We used neonatal ventricular myocytes to monitor changes in contraction rate and fluorescence resonance energy transfer (FRET) to follow changes in cAMP during inhibition of MRP4. Our experiments show that MRP4 is expressed in cardiac myocytes and is involved in the regulation of β-adrenergic stimulated cAMP signaling. Unlike other means of cAMP potentiation, such as PDE4 inhibition, inhibition of MRP4 utilizes CFTR, which is involved in mediating β-adrenergic stimulated contraction rate.
2. Materials and Methods
2. 1. Materials
Isoproterenol and genistein came from Sigma-Aldrich (St. Louis, MO). MK571 (L 660711 or (E)-3-[[[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl][[3-(dimethylamino)- 3-oxopropyl]thio]methyl]thio]-propanoic acid) was purchased from Cayman Chemical (Ann Arbor, MI). Rolipram was from Calbiochem (San Diego, CA). CFTRinh-172 was obtained from Calbiochem or the Cystic Fibrosis Foundation Therapeutics (CFFT) compound collection (Rosalind Franklin University of Medicine and Science, Chicago, IL). PG-01 was also obtained from the CFFT compound collection. Drugs were made as 1:1000 stock solutions in DMSO or H2O (isoproterenol) and diluted the day of the experiment.
2. 2. Cardiac myocyte culture
Neonatal ventricular myocytes were cultured from wildtype (FVB), β1-adrenoceptor knockout (KO) (129/SV, C57Bl6/J, DBA2/J) (Rohrer et al., 1996), β2-adrenoceptor knockout (129/SV, FVB/N) (Chruscinski et al., 1999), β1/β2-adrenoceptor double knockout (cross-mating β1 and β2 KO) (Rohrer et al., 1999), or CFTR KO (strain 2515 B6.129S6-Cftrtm1Kth/J) mice (Zhou et al., 1994). All procedures with animals were approved by the University of Illinois IACUC. Cells were isolated and cultured using a similar protocol as previously reported (Xiang et al., 2005). Briefly, freshly born mice were sacrificed and neonatal mouse cardiac tissue devoid of atria were exposed to enzymatic digestion. Subsequently, isolated cells were pipetted onto 35 mm Petri dishes pre-coated with 1.5% gelatin type A or laminin. Cells were plated at high- and low-density for measurement of contraction rate or real-time imaging experiments, respectively. High-density plating allowed for cells to form a syncytium, whereas low-density plating facilitated individual quiescent myocytes. Cells were incubated at 37°C for 2–3 days until they displayed appropriate myocyte morphology or formed a syncytium for contraction rate measurements. Culture media (containing serum and 20 mM HEPES for buffering) was replaced daily and at least 1 h prior to experiments.
2. 3. Western blot
Cultured cardiomyocytes were isolated in 1X dulbecco’s phosphate buffer solution containing the protease inhibitors PMSF (250 μM), NaF (2.5 mM), Na3VO4 (2.5 mM), benzamidine (1 mM) and lysed in PBS with 2% Triton-X 100 and centrifuged prior to protein estimation. For electrophoresis, samples were run out on a 4–15% agarose gel. Following transfer to a PVDF membrane, MRP4 was detected using an IgG anti-MRP4 monoclonal antibody (Li et al., 2007).
2. 4. Myocyte contraction rate
Measurement of myocyte contraction rate was performed as previously reported (Xiang et al., 2002). 35 mm dishes were placed in a temperature/CO2 regulated apparatus allowing for a constant environment of 37° C and 95% O2/5% CO2. Using a CCD camera and MetaMorph imaging software (Molecular Devices, Downington, PA), 5 s video images were obtained every 2 min. Movements in the x and y plane were calculated and number of deflections noted as number of contractions.
2. 5. Fluorescence resonance energy transfer
Fluorescence resonance energy transfer (FRET) was used to measure real-time changes in intracellular cAMP as previously described (De Arcangelis et al., 2010). Following 24 h in culture, neonatal cardiomyocytes plated at low-density were infected with adenovirus (100 multiplicity of infection, 24 h) containing the cAMP sensor ICUE3 (Soto et al., 2009). Between 24 and 48 h post-infection myocytes were washed and stored in DPBS with 1 mM CaCl and 1 mM MgCl for FRET recordings. Myocytes were imaged with a Zeiss Axiovert 200M microscope fitted with a 40x/1.3 NA oil-immersion objective lens and a CCD camera controlled by MetaFlour software (Molecular Devices, Downington, PA). Acquisition was set to 200 millisecond exposure in both channels every 20 s. The binding of cAMP to the Epac site of ICUE3 causes a decrease in FRET (DiPilato et al., 2004). Yellow-to-cyan ratios were calculated at different time points, background corrected, and normalized with data presented as changes in the inverse of YFP/CFP.
2. 6. Data anaylysis
All data are expressed as means ± standard error, except where indicated. For comparison between two groups of data, Student’s unpaired t-test was used to determine significance, while multiple groups were compared by analysis of variance (ANOVA) with Bonferonni post-tests using Prism software (GraphPad, San Diego, CA). Results were considered significant at P < 0.05 and are denoted accordingly. Curve fitting and calculation of EC50 was performed by SigmaPlot (Systat, San Jose, CA).
3. Results
3. 1. Expression of MRP4 in cardiac myocytes
To determine if MRP4 is involved in cardiac myocyte signaling, we first examined the expression of MRP4 protein in mouse neonatal ventricular myocytes using a specific anti-MRP4 monoclonal antibody. As seen in Fig. 1, this antibody recognized MRP4 in MRP4-transfected HEK cells, but not in untransfected HEK cells. Additionally, in two different isolations, this antibody recognized MRP4 protein in wildtype (WT) neonatal ventricular myocytes indicating that MRP4 protein is expressed in neonatal cardiomyocytes.
Fig. 1. Expression of MRP4 in mouse neonatal ventricular myocytes.
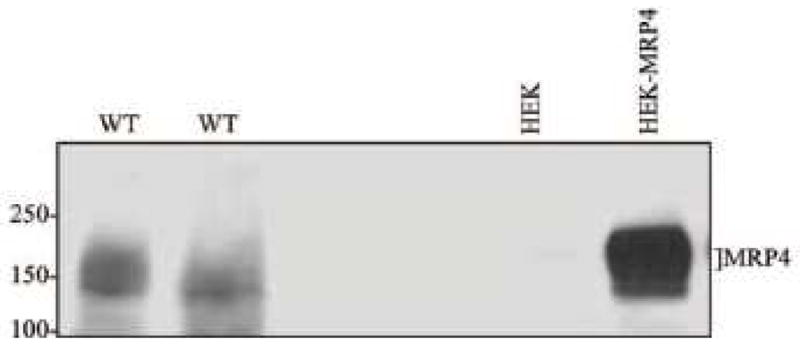
MRP4 protein expression was probed in untransfected HEK cells, MRP4-transfected HEK cells, and two different samples of WT neonatal ventricular myocytes. Cells were cultured and harvested in lysis buffer. 200 μg and 40 μg of cardiomyocytes or HEK cells, respectively, were loaded into agarose gels and MRP4 expression was detected by a MRP4-specific antibody (1:5000) recognizing MRP4 at approximately 159 kDa.
3. 2. Inhibition of MRP4 potentiates β-adrenergic stimulated contraction rate
Subsequently, we examined if MRP4 was functionally active and involved in the regulation of cAMP and β-adrenergic signaling. Similar to De Arcangelis et al (2010) we found that isoproterenol, a non-specific β-adrenoceptor agonist, stimulated dose-dependent increases in cardiomyocyte contraction rate with an EC50 of 1.4 x 10−8 M (Fig. 2A and B). We also found that this response was predominantly mediated by β1-adrenoceptors (Fig. 2C). Subsequently, we examined the effect of MK571, a pharmacological inhibitor of MRP4, on submaximal (10−8 M) or maximal (10−5 M) isoproterenol-stimulated contraction rate. We found that cardiac myocytes pre-treated with MK571 (40 μM) displayed an increased response to submaximal isoproterenol stimulation, compared to DMSO-treated myocytes. This effect was specific to submaximal stimulation, as MK571 had no effect on maximal isoproterenol stimulation (Fig. 3A). This effect of MK571 on contraction rate occurred at 20 and 40 μM, but not 10 μM (Fig. 3B). To determine if MK571-dependent potentiation of submaximal isoproterenol-stimulated contraction rate was β-adrenoceptor subtype specific we investigated the effect of MK571 in β-adrenoceptor KO mice. As seen in Fig. 3C, MK571 only potentiated submaximal isoproterenol-stimulated contraction rate in β2-adrenoceptor KO mice, but not β1- or β1/β2-adrenoceptor KO mice, indicating that MRP4 inhibition specifically alters β1-adrenoceptor stimulated contraction rate in neonatal cardiomyocytes.
Fig. 2. Dose-dependent isoproterenol stimulated contraction rate changes in neonatal cardiomyocytes.

A. Spontaneously beating WT neonatal cardiomyocytes (n = 6) were exposed to incrementally increasing doses of isoproterenol (10−11 to 10−5 M) for 10 min each. Changes in contraction rate from baseline were recorded and plotted. B. Maximal changes in contraction rate for each dose of isoproterenol were plotted and curve-fitted with calculation of EC50 via SigmaPlot software. C. Following a 10 min baseline, cardiomyocytes from β1-, β2-, and β1/β2 KO mice were exposed to 10−8 M isoproterenol, then 10−5 M isoproterenol for 10 min each. Peak responses to each concentration were subtracted from values prior to isoproterenol addition. *, P < 0.05; ***, P < 0.001 vs. β1- and β1/β2 KO mice by ANOVA. ++, P < 0.01 vs. 10−8 M by Student’s t-test.
Fig. 3. Pharmacological inhibition of MRP4 by MK571 potentiates submaximal isoproterenol-stimulated contraction rate via β1-adrenoceptors.

A. WT syncytial cardiomyocytes were exposed to DMSO (n = 8) or MK571 (40 μM, n = 5) for 20 min prior to isoproterenol stimulation. Subsequently, myocytes were stimulated with 10−8 M, then 10−5 M isoproterenol for 20 min each. B. Similarly, additional experiments were carried out with 10 or 20 μM MK571 (n ≥ 5) and peak changes in contraction rates were compared to DMSO. **, P < 0.01 vs. DMSO by Student’s t-test. C. Spontaneously beating neonatal ventricular myocytes from β1-, β2-, and β1/β2 KO mice were exposed to DMSO (n ≥ 6) or MK571 (40 μM, n ≥ 6) in a similar manner as above with peak responses to submaximal isoproterenol stimulation (10−8 M) represented. ***, P < 0.001 vs. DMSO for each group by Student’s t-test.
3. 3. Inhibition of MRP4 increases β-adrenergic stimulated cAMP
To provide further evidence for MRP4 involvement in β-adrenergic signaling in cardiac myocytes we used FRET to measure real-time changes in intracellular cAMP following isoproterenol stimulation in the presence or absence of MRP4 inhibition. Addition of 10−8 M isoproterenol caused a substantial increase in intracellular cAMP that was characterized by an initial rapid, but transient, increase followed by a secondary slow, but steady, increase over time (Fig. 4A). In contrast, stimulation of cardiomyocytes with 10−5 M isoproterenol caused a significantly increased amount of cAMP compared to 10−8 M, due to potentiation of the secondary cAMP response to stimulation (Fig. 4B). When we simultaneously added MK571 (40 μM) and 10−8 M isoproterenol, intracellular cAMP significantly increased compared to isoproterenol alone, both in magnitude and duration. In contrast, the level of cAMP activity stimulated by MK571 and 10−5 M isoproterenol was similar to 10−5 M isoproterenol alone, indicating that MK571 potentiates cAMP activity stimulated by submaximal, but not maximal isoproterenol stimulation.
Fig. 4. MK571 increases intracellular cAMP in response to submaximal, but not maximal, isoproterenol stimulation.

WT cardiomyocytes were cultured at low-density and incubated with an ICUE3 adenovirus for 24–48 h. Myocytes were exposed to DMSO (n ≥ 20) or MK571 (40 μM, n ≥ 23) for 20 min prior to data acquisition. Intracellular cAMP was measured for a 3 min baseline, after which cells were exposed to 10−8 M (A) or 10−5 M (B) isoproterenol for 20 min. Data are expressed as changes from initial measurements made during baseline.
3. 4. CFTR mediates MRP4-dependent alterations in contraction rate
Based on previous findings that MRP4 activity can influence CFTR activity in epithelial cells (Li et al., 2007), we next examined if MRP4-dependent potentiation of contraction rate involved CFTR in neonatal cardiac myocytes. While we have previously shown that CFTR is involved in un-stimulated neonatal mouse contraction rate (Sellers et al., 2010), there is currently no evidence of its involvement in β-adrenergic stimulated contraction rate. Therefore, in similar experiments to those done in WT mice (Fig. 2), we examined the effect of various doses of isoproterenol on cardiomyocyte contraction rate in CFTR KO mice. Elimination of CFTR expression resulted in a preferential loss of response to isoproterenol stimulation at doses less than 10−7 M with no effect on higher doses (Fig. 5A and C). Pharmacological inhibition of CFTR in WT mice with CFTRinh-172 (20 μM) produced similar results when stimulated with 10−8 and 10−5 M isoproterenol (Fig. 5C). This inhibitory effect of CFTRinh-172 on submaximal isoproterenol stimulatiuon also occurred in β2-adrenoceptor KO mice (DMSO: 10.5 ± 1.5 vs. CFTRinh-172: 4.7 ± 1.7 Δbeats/min, P < 0.05, n = 6 each), confirming involvement of CFTR in β1-stimulated contraction rate. Conversely, pre-activation of CFTR with genistein (50 μM) specifically potentiated contraction rates stimulated with isoproterenol at doses less than 10−7 M, with no effect on contraction rate at maximal doses (Fig. 5B and C). These results were confirmed with the CFTR activator PG-01 (Pedemonte et al., 2005) (Fig. 5C). Thus, positive or negative modulation of CFTR preferentially altered contraction rates in response to submaximal isoproterenol stimulation.
Fig. 5. Effect of CFTR modulation on isoproterenol stimulated contraction rate.

A. Neonatal ventricular myocytes from CFTR KO mice (n = 4) were exposed to graded doses of isoproterenol (10−11 to 10−5 M) in a similar manner as Fig. 2. Responses were compared to those in WT mice (control, same as Fig. 2). B. In a similar manner, WT cardiomyocytes were exposed to genistein (50 μM, n = 4) for 20 min prior to stimulation with isoproterenol. C. Comparison of submaximal (10−8 M) and maximal (10−5 M) isoproterenol stimulated contraction rate responses with CFTR disruption (CFTR KO mice or 20 min pre-incubation with CFTRinh-172 (20 μM) (n ≥ 5 for each)) or pre-activation (20 min pre-incubation with genistein or PG-01 (5 μM) (n ≥ 4 for each)). Data are expressed as peak responses over baseline. *, P < 0.05; **, P < 0.001 vs. control + 10−8 M isoproterenol by Student’s t-test. ++, P < 0.01 vs. control + 10−5 M isoproterenol Student’s t-test.
When we examined the effect of MK571 in CFTR KO mice we found that MK571 failed to increase submaximal (10−8 M) isoproterenol stimulated contraction rate, such as seen in WT mice, with no affect on maximal (10−5 M) stimulation (Fig. 6A).
Fig. 6. MRP4-, but not PDE4-dependent potentiation of isoproterenol stimulated contraction rate, is CFTR-dependent.

CFTR KO cardiomyocytes were incubated with MK571 (40 μM, n = 6, A) or rolipram (10 μM, n = 8, B) for 20 min prior to stimulation with 10−8 and 10−5 M isoproterenol. Data are presented with data from WT mice (taken from Fig. 3).
3. 5. PDE4-dependent increases in contraction rate are CFTR-independent
Rolipram, an inhibitor of PDE4, has been shown to increase cAMP and contraction rates in response to 10−9 M isoproterenol (De Arcangelis et al., 2010). Therefore, we examined if rolipram-dependent increases in contraction rate were also CFTR-dependent. We found that in the presence of rolipram 10−8 M isoproterenol stimulated a significant increase in contraction rate that was comparable in magnitude to that during maximal stimulation (10−5 M) (Fig. 6B). It was also significantly increased from that of MK571 (P < 0.01). In contrast to MK571, in CFTR KO cardiomyocytes, rolipram continued to elicit a significant increase in contraction rate that was similar to that seen in WT mice (Fig. 6B). These results indicate that MK571-induced potentiation of contraction rate is CFTR-dependent, whereas rolipram-induced potentiation is CFTR-independent.
4. Discussion
Cardiac myocytes utilize a vast network of membrane-bound and intracellular proteins to regulate contraction. Amongst these, β-adrenoceptors (β1, β2, β3) are central to modulating dynamic changes in cardiomyocyte contraction rate and force generation (Devic et al., 2001). Simplistically, β1-adrenoceptor activation leads to activation of protein kinase A (PKA) via increased production of cAMP by adenylyl cyclase. Subsequently, PKA directly phosphorylates and enhances the activity of a host of ion channels and transporters, which have been summarized by Kuzumoto et al (2008), but include the sarcolemmal L-type Ca2+ current, the slowly activating component of delayed rectifier K+ current, the plasma membrane Ca2+-ATPase, the ryanodine receptor (Huke and Bers, 2008), and phospholemman and phospholamban, which regulate the Na+/K+ ATPase and sarcoplasmic reticulum Ca2+ pump (SERCA), respectively (Despa et al., 2005).
Of central importance to our study, cAMP and PKA regulation are critical to the generation and regulation of spontaneous beating neonatal cardiac myocytes, as loss of either one of these markedly blunts the ability of these cells to contract (Sellers et al., 2010). Unlike adult ventricular myocytes, neonatal ventricular myocytes have the ability to beat as a single syncytial unit. This property allowed us to examine myocyte function without exogenous perturbation. While it may be tempting to compare these cells to adult pacemaker or atrial cells, which also spontaneously contract, it is more likely that neonatal ventricular myocytes are analogous to adult ventricular myocytes. In a comparison of their electrophysiological properties, Nuss and Marban (1994) showed that mouse neonatal and adult ventricular myocytes express similar ion channels and have comparable action potential durations.
Widely studied as an ATP-dependent drug efflux pump, MRP4 has also been shown to regulate intracellular cAMP levels via transport out of the cell. Our use of the FRET probe ICUE3 shows, for the first time, that MRP4 is involved in negatively regulating cAMP levels in cardiac myocytes. This data is in agreement with prior studies in epithelial (Li et al., 2007) and smooth muscle cells (Sassi et al., 2008) which showed that inhibition of MRP4 with the pharmacological MRP4 inhibitor MK571 (Reid et al., 2003) or siRNA targeting MRP4 caused a decrease in the efflux of cAMP, resulting in accumulation of intracellular cAMP and activation of downstream effectors. In our study we utilized the pharmacological inhibitor MK571 to examine the role of MRP4 in cardiac myocytes. The limitation of this approach is that like many other drugs, MK571 has also been shown to affect other targets, such as MRP1 (Mueller et al., 2008), MRP5 (Reid et al., 2003) and cysteinyl leukotriene receptors (CystLT1) (Lynch et al., 1999). While we did not examine these other targets, we do not anticipate that they confound our results. MRP1 has been implicated in the transport of glutathione (Mueller et al., 2008), thus making it unlikely to be involved in our short-term experiments. MRP5 transports cyclic nucleotides, but with a high affinity for cGMP (Jedlitschky et al., 2000). In contrast to the stimulatory effect of cAMP, cGMP negatively regulates cardiomyocyte contraction through reduction of myofilament Ca2+ responsiveness. CysLT1 receptors are coupled to Gαq receptors resulting in mobilization of intracellular Ca2+. Thus, one would expect that inhibition of MRP5 or CysLT1 would lead to a decrease in contraction rate through increased negative stimulus or a lack of a positive stimulus, respectively. On the contrary, we found that MK571 caused an increase in the intracellular accumulation of isoproterenol-stimulated cAMP and caused an increase in isoproterenol-stimulated cardiomyocyte contraction rate; both of which are supported by an inhibitory effect of MRP4.
PDE4 is a well-established modulator of cAMP signaling in cardiac myocytes, which we have also shown by the ability of rolipram to potentiate submaximal isoproterenol stimulated contraction rate. These findings are similar to De Arcangelis et al (2010) who examined the effect of rolipram at another submaximal concentration (10−9 M). When stimulated with submaximal concentrations of isoproterenol, rolipram significantly increases intracellular cAMP by potentiating the magnitude and duration of cAMP (De Arcangelis et al., 2010). These effects are similar to what we found, as inhibition of MRP4 with MK571 increased both the initial and sustained phases of cAMP accumulation during submaximal isoproterenol stimulation, which led to increases in contraction rates. Thus, both PDE4 and MRP4 inhibition increase intracellular cAMP, resulting in downstream changes in contraction rates. Despite theses similarities, we found that inhibition of MRP4 elicits its effects in a functionally distinct manner from that of PDE4 inhibition. First, inhibition of PDE4 resulted in a more robust increase in contraction rates than inhibition of MK571. This is not likely due to differences in the magnitude of cAMP generation as we found MK571 to increase cAMP to maximal levels, similar to what has been found with rolipram (De Arcangelis et al., 2010). While rolipram increases isoproterenol-stimulated contraction rate via β1- and β2-adrenoceptors (De Arcangelis et al., 2010; Xiang et al., 2005), we found that MK571 potentiates only β1-adrenoceptor stimulation. Thus, we cannot rule out that recruitment of additional β-adrenoceptors could account for the difference in contraction rates between MRP4 and PDE4 inhibition. Second, we found that MRP4-dependent potentiation of isoproterenol stimulated contraction rate was dependent on CFTR expression, but PDE4-dependent potentiation was not. With this latter finding, we hypothesize that the differences in contraction rates between PDE4 and MRP4 inhibition occur as a result of their distinct spatiotemporal resolutions, not their absolute magnitude.
Compartmentalization of G-protein coupled receptor complexes and cAMP in response to their activation accounts for distinct functional responses to different stimuli that produce increases in cAMP. De Arcangelis et al (2010) have proposed a model whereby submaximal β-adrenoceptor stimulation leads to localized increases in cAMP, with spread constrained by PDE4. Inhibition of PDE4 or maximal activation of β-adrenoceptors eliminates this “brake” and cAMP spreads and exerts farther-reaching effects. Additionally, others have shown that inhibition of PDE3 leads to spatially confined subsarcolemmal increases in cAMP, whereas inhibition of PDE4 results in broad cytosolic cAMP accumulation (Leroy et al., 2008). While we were not able to measure discrete microdomains of cAMP, in epithelial cells Li et al (2007) showed that inhibition of MRP4 leads to increased cAMP in a spatially confined microdomain at or near the plasma membrane. Therefore, we propose that in cardiac myocytes inhibition of MRP4 leads to an increase in subsarcolemmal cAMP, which accounts for its ability to alter contraction rates in a distinct manner from that of PDE4 (Fig. 7). Further investigation of the way in which modulation of MRP4 activity affects cAMP compartmentalization in cardiac myocytes will lead to an increased understanding of the complex events that occur downstream of G-protein coupled receptor activation.
Fig. 7. Proposed involvement of MRP4 and CFTR in β1-adrenergic stimulation.

A. 10−8 M isoproterenol stimulation. B. 10−5 M isoproterenol stimulation. C. 10−8 M isoproterenol stimulation in the presence of MRP4 inhibition by MK571. D. 10−8 M isoproterenol stimulation in the presence of PDE4 inhibition by rolipram. Additional abbreviations: β1-AR, β1-adrenoceptors; AC, adenylyl cyclase; NBD, nucleotide binding domain of CFTR; R, regulatory domain of CFTR; SR, sarcoplasmic reticulum; CaCC, Ca2+-activated Cl− channels.
Another important discovery from our study is the role of CFTR in the regulation of β-adrenergic stimulated contraction of cardiac myocytes. In previous work we found that CFTR is involved in the spontaneous contraction of un-stimulated neonatal ventricular myocytes; however, CFTR loss could be compensated for by activation of Ca2+-dependent proteins (Sellers et al., 2010). In the current study, we found that modulation of CFTR alters cardiomyocyte contraction rate in response to β-adrenergic stimulation. Cardiac Cl− channels have generally been thought to be involved in minimizing action potential duration in response to β-adrenoceptor stimulation (Duan, 2009). Genistein, a known activator of CFTR, has been found to potentiate subthreshold isoproterenol activation of cardiac Cl− currents to maximal levels (Hool et al., 1998). Additionally, simulation studies predict that increases in CFTR Cl− current will lead to shortening of the myocyte action potential during β1-adrenoceptor activation (Kuzumoto et al., 2008). Our findings that pre-activation of CFTR, with genistein or PG-01, increases submaximal isoproterenol stimulated contraction rate support these findings. Interestingly, we found that the effect of CFTR only occurred at submaximal concentrations of isoproterenol (<10−7 M). Based on the evidence presented above, we hypothesize that CFTR is primarily involved in mediating functional effects caused by local changes in cAMP at the sarcolemmal membrane. When cAMP signals are constricted to the plasma membrane, they are confined to a microdomain that contains CFTR, leading to its activation and modulation of myocyte contraction. In contrast, a global increase of cAMP by high concentrations of isoproterenol or inhibition of PDE4 would allow, or increase, phospholamban phosphorylation and activation of other ion channels, including Ca2+-activated Cl− channels through sarcoplasmic reticulum release of Ca2+ (De Arcangelis et al., 2010) (Fig. 7). To this effect, preliminary studies show that Ca2+-activated Cl− channels are important in mediating contraction rate increases stimulated by 10−5 M isoproterenol (Z.M. Sellers, unpublished results).
In epithelial cells CFTR is physically and functionally linked to MRP4 through interactions with PDZK1 providing a compartment where MRP4-dependent increases in cAMP can lead to activation of CFTR (Li et al., 2007). Additionally, in lung epithelial cells, β2-adrenoceptors form a complex with PKA and CFTR through PDZ interactions with ezrinradixinmoesin-binding phosphoprotein 50 (EBP50, also referred to as NHERF) (Naren et al., 2003). He et al (2006) have shown that β1-adrenoceptors interact with multiple PDZ scaffold proteins in HEK-293 and COS-7 cells, while Valentine and Haggie (2011) found that rat H9c2 cells (derived from embryonic myocardium) utilize PDZ and AKAPs to spatially organize β1-adrenoceptors. Further work is necessary to explore compartmentalized interactions between β1-adrenoceptors, CFTR, and MRP4 in cardiac myocytes, however, our finding that MRP4-dependent potentiation of contraction rate is β1-adrenoceptor-dependent and requires CFTR, supports this notion. Subsequent examination of the role of CFTR in β-adrenergic stimulated heart function will lead to a more complete understanding regarding the physiologic control of heart function.
5. Conclusions
This is the first report of MRP4 expression or activity in cardiac myocytes. Likewise, we have also provided the first experimental evidence for involvement of CFTR in β-adrenergic cardiomyocyte contraction. Our findings suggest that both MRP4 and CFTR are involved in regulating and mediating the function of local, compartmentalized increases in cAMP. The involvement of these membrane proteins in β-adrenoceptor stimulated cAMP regulation and function make them important factors to consider in future studies examining the regulation of β-adrenergic signaling and contraction in the heart.
Acknowledgments
Dr. Deborah Nelson (University of Chicago) kindly provided us with CFTR KO mice. We thank Dr. Robert Bridges (Rosalind Franklin University of Medicine and Science) and Cystic Fibrosis Foundation Therapeutics (CFFT) for the gift of CFTRinh-172 and PG-01 from CFFT’s compound collection.
This study was supported by the American Heart Association (0815670G to Z.M.S.; 0635331N to Y.X.), the National Institutes of Health (DK-074996, DK-080834 to A.P.N.; HL082646 to Y.X.), and the University of Illinois (P.M.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of β2-adrenergic gene. J Biol Chem. 1999;274:16694–16700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- De Arcangelis V, Liu S, Zhang D, Soto D, Xiang YK. Equilibrium between adenylyl cyclase and phosphodiesterase patterns adrenergic agonist dose-dependent spatiotemporal cAMP/PKA activities in cardiomyocytes. Mol Pharmacol. 2010;78:340–349. doi: 10.1124/mol.110.064444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, Tucker AL, Bers DM. Phospholemman-phosphorylation mediates the beta-adrenergic effects on Na/K pump function in cardiac myocytes. Circ Res. 2005;97:252–259. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- Devic E, Xiang Y, Gould D, Kobilka B. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol. 2001;60:577–583. [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci USA. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Langeberg L, Scott JD. Compartmentation of cyclic nucleotide signaling in the heart: the role of A-kinase anchoring proteins. Circ Res. 2006;98:993–1001. doi: 10.1161/01.RES.0000218273.91741.30. [DOI] [PubMed] [Google Scholar]
- Duan D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J Physiol. 2009;587:2163–2177. doi: 10.1113/jphysiol.2008.165860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev. 1999;79:S77–S107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- He J, Bellini M, Inuzuka H, Xu J, Xiong Y, Yang X, Castleberry AM, Hall RA. Proteomic analysis of beta1-adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem. 2006;281:2820–2827. doi: 10.1074/jbc.M509503200. [DOI] [PubMed] [Google Scholar]
- Hool LC, Middleton LM, Harvey RD. Genistein increases the sensitivity of cardiac ion channels to beta-adrenergic receptor stimulation. Circ Res. 1998;83:33–42. doi: 10.1161/01.res.83.1.33. [DOI] [PubMed] [Google Scholar]
- Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlitschky G, Burchell B, Keppler D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J Biol Chem. 2000;275:30069–30074. doi: 10.1074/jbc.M005463200. [DOI] [PubMed] [Google Scholar]
- Jurevicius J, Skeberdis VA, Fischmeister R. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa,L in frog ventricular myocytes. J Physiol. 2003;551:239–252. doi: 10.1113/jphysiol.2003.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass DA. Message delivered: how myocytes control cAMP signaling. Circ Res. 2008;102:1002–1004. doi: 10.1161/CIRCRESAHA.108.176420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzumoto M, Takeuchi A, Nakai H, Oka C, Noma A, Matsuoka S. Simulation analysis of intracellular Na+ and Cl− homeostasis during beta 1-adrenergic stimulation of cardiac myocyte. Prog Biophys Mol Biol. 2008;96:171–186. doi: 10.1016/j.pbiomolbio.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Lader AS, Wang Y, Jackson GR, Jr, Borkan SC, Cantiello HF. cAMP-activated anion conductance is associated with expression of CFTR in neonatal mouse cardiac myocytes. Am J Physiol Cell Physiol. 2000;278:C436–450. doi: 10.1152/ajpcell.2000.278.2.C436. [DOI] [PubMed] [Google Scholar]
- Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res. 2008;102:1091–1100. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- Li C, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, Zaccolo M, Jalink K, Li M, Nelson DJ, Schuetz JD, Naren AP. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell. 2007;131:940–951. doi: 10.1016/j.cell.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KR, O’Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, Connolly BM, Bai C, Austin CP, Chateauneuf A, Stocco R, Greig GM, Kargman S, Hooks SB, Hosfield E, Williams DL, Jr, Ford-Hutchinson AW, Caskey CT, Evans JF. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- Mueller CF, Wassmann K, Widder JD, Wassmann S, Chen CH, Keuler B, Kudin A, Kunz WS, Nickenig G. Multidrug resistance protein-1 affects oxidative stress, endothelial dysfunction, and atherogenesis via leukotriene C4 export. Circulation. 2008;117:2912–2918. doi: 10.1161/CIRCULATIONAHA.107.747667. [DOI] [PubMed] [Google Scholar]
- Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan J, Clancy JP. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci USA. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. J Physiol. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard LS, Baldursson O, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by its R domain. J Biol Chem. 2001;276:7689–7692. doi: 10.1074/jbc.R100001200. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, Robins LI, Dicus CW, Willenbring D, Nantz MH, Kurth MJ, Galietta LJ, Verkman AS. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol. 2005;67:1797–1807. doi: 10.1124/mol.105.010959. [DOI] [PubMed] [Google Scholar]
- Reid G, Wielinga P, Zelcer N, De Haas M, Van Deemter L, Wijnholds J, Balzarini J, Borst P. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol Pharmacol. 2003;63:1094–1103. doi: 10.1124/mol.63.5.1094. [DOI] [PubMed] [Google Scholar]
- Ritter CA, Jedlitschky G, Meyer zu Schwabedissen H, Grube M, Kock K, Kroemer HK. Cellular export of drugs and signaling molecules by the ATP-binding cassette transporters MRP4 (ABCC4) and MRP5 (ABCC5) Drug Metabol Rev. 2005;37:253–278. doi: 10.1081/dmr-200047984. [DOI] [PubMed] [Google Scholar]
- Rohrer DK, Chruscinsky A, Schauble EH, Bernstein D, Kobilka BK. Cardiovascular and metabolic alterations in mice lacking both β1- and β2-adrenergic receptors. J Biol Chem. 1999;274:16701–16708. doi: 10.1074/jbc.274.24.16701. [DOI] [PubMed] [Google Scholar]
- Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP, Jr, Barsh GS, Bernstein D, Kobilka BK. Targeted disruption of the mouse β-adrenergic receptor gene: developmental and cardiovascular effects. Proc Natl Sci USA. 1996;93:7375–7380. doi: 10.1073/pnas.93.14.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel FG, Koenderink JB, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008;29:200–207. doi: 10.1016/j.tips.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Sassi Y, Lipskaia L, Vandecasteele G, Nikolaev VO, Hatem SN, Cohen Aubart F, Russel FG, Mougenot N, Vrignaud C, Lechat P, Lompre AM, Hulot JS. Multidrug resistance-associated protein 4 regulates cAMP-dependent signaling pathways and controls human and rat SMC proliferation. J Clin Invest. 2008;118:2747–2757. doi: 10.1172/JCI35067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz JD, Connelly MC, Sun D, Paibir SG, Flynn PM, Srinivas RV, Kumar A, Fridland A. MRP4: A previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nature Med. 1999;5:1048–1051. doi: 10.1038/12487. [DOI] [PubMed] [Google Scholar]
- Sellers ZM, Arcangelis VD, Xiang Y, Best PM. Cardiomyocytes with disrupted CFTR function require CaMKII and Ca2+-activated Cl− channel activity to maintain contraction rate. J Physiol. 2010;588:2417–2429. doi: 10.1113/jphysiol.2010.188334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto D, De Arcangelis V, Zhang J, Xiang Y. Dynamic protein kinase a activities induced by beta-adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res. 2009;104:770–779. doi: 10.1161/CIRCRESAHA.108.187880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaandrager AB, Tilly BC, Smolenski A, Schneider-Rasp S, Bot AG, Edixhoven M, Scholte BJ, Jarchau T, Walter U, Lohmann SM, Poller WC, de Jonge HR. cGMP stimulation of cystic fibrosis transmembrane conductance regulator Cl− channels co-expressed with cGMP-dependent protein kinase type II but not type Ibeta. J Biol Chem. 1997;272:4195–4200. doi: 10.1074/jbc.272.7.4195. [DOI] [PubMed] [Google Scholar]
- Valentine CD, Haggie PM. Confinement of β(1)- and β(2)-adrenergic receptors in the plasma membrane of cardiomyocyte-like H9c2 cells is mediated by selective interactions with PDZ domain and A-kinase anchoring proteins but no caveolae. Mol Biol Cell. 2011;22:2970–2982. doi: 10.1091/mbc.E11-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Devic E, Kobilka B. The PDZ binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem. 2002;277:33783–33790. doi: 10.1074/jbc.M204136200. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci USA. 2005;102:909–914. doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Dey CR, Wert SE, DuVall MD, Frizzell RA, Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994;266:1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]