

Abstract
Autophagy protects against many infections by inducing the lysosomal-mediated degradation of invading pathogens. However, previous in vitro studies suggest that some enteroviruses not only evade these protective effects, but also exploit autophagy to facilitate their replication. We generated Atg5f/f/Cre+ mice, in which the essential autophagy gene Atg5 is specifically deleted in pancreatic acinar cells, and show that coxsackievirus B3 (CVB3) requires autophagy for optimal infection and pathogenesis. Compared to Cre− littermates, Atg5f/f/Cre+ mice had a ~2000-fold lower CVB3 titer in the pancreas, and pancreatic pathology was greatly diminished. Both in vivo and in vitro, Atg5f/f/Cre+ acinar cells had reduced intracellular viral RNA and proteins. Furthermore, intracellular structural elements induced upon CVB3 infection, such as compound membrane vesicles and highly-geometric paracrystalline arrays, which may represent viral replication platforms, were infrequently observed in infected Atg5f/f/Cre+ cells. Thus, CVB3-induced subversion of autophagy not only benefits the virus, but also exacerbates pancreatic pathology.
Keywords: coxsackievirus, picornavirus, autophagy, Atg5, pancreas, virus, in vivo, acinar
Introduction
Positive-strand RNA viruses often induce dramatic reorganization of host cell cytosolic membranes (Bienz et al., 1992; Kirkegaard and Jackson, 2005; Kopek et al., 2007). The targeted membranes, and the mechanisms by which they are altered, differ greatly from virus to virus (Wileman, 2006), but some of these viruses appear to achieve this outcome, at least in part, by blocking autophagy, a pathway in which vesicular flow is a central feature. Tissue culture studies have shown that some picornaviral infections are accompanied by the accumulation of double-membraned vesicles, termed compound membrane vesicles (CMVs, Jezequel and Steiner, 1966) that appear to be related to the autophagy pathway (Jackson et al., 2005; Miller and Krijnse-Locker, 2008; Schlegel et al., 1996; Suhy et al., 2000; Taylor and Kirkegaard, 2007); these membranes may be used as a two-dimensional scaffold that promotes viral RNA replication and protects newly synthesized viral RNA (Chen et al., 2003; Welsch and Locker, 2010). Additional in vitro studies using immortalized cell lines support the contention that several picornaviruses, including poliovirus, rhinovirus, encephalomyocarditis virus, foot-and-mouth disease virus, and coxsackieviruses, may actively exploit autophagy, and that up- or down-regulation of cellular autophagy increases or decreases, respectively, virus yield from the cells (albeit very modestly, 2- to 4-fold) (Jackson et al., 2005; O’Donnell et al., 2011; Taylor and Kirkegaard, 2007; Wong et al., 2008; Yoon et al., 2008; Zhang et al., 2011). Autophagy also plays an important role in protecting the host against infections by many microbes [reviewed in (Deretic and Levine, 2009; Orvedahl and Levine, 2009)] so, by blocking autophagy, these positive-stranded RNA viruses not only may enhance their replication, but also may evade the inhibitory effects of the pathway.
Our laboratory studies the interactions between autophagy and the picornavirus coxsackievirus B3 (CVB3), an important human pathogen (Gebhard et al., 1998). We have chosen to focus on in vivo infection (i.e., in the living animal), for several reasons. For example, the cells to be studied are in their normal anatomical relationship to other cells within the tissue; they receive normal nutrition; and they are exposed to the natural complement of cytokines and chemokines, both before and after infection. In a previous study we have shown that CVB3 dramatically alters pancreatic autophagy in normal mice (Kemball et al., 2010) and has at least two distinct effects. First, CVB3 increases the abundance of small autophagy-like vesicles and, based largely on the prior studies on poliovirus, we speculated that this may provide CVB3 with a platform for its replication. Second, the virus also blocks a later stage of the pathway, with the consequent formation of extremely large (~10μm) vesicles that we have termed megaphagosomes (Kemball et al., 2010). In the present study, we aimed to extend our in vivo analyses to examine the converse question i.e., how do in vivo alterations in autophagy impact CVB3 replication and pathogenesis? We took a genetic approach to study the effects of autophagy on CVB3 replication and pathogenesis, using mice in which transcription of Cre recombinase was regulated by the elastase-1 promoter, limiting protein expression to the acinar cells (Grippo et al., 2002); these mice were crossed with mice in which the Atg5 gene is flanked by loxP sequences (Hara et al., 2006), selectively deleting, in pancreatic acinar cells, an exon from the Atg5 gene.
Results
The pancreata of Atg5f/f/Cre+ mice are depleted of functional Atg5 protein, but the mice show little sign of constitutive pancreatic dysfunction
Mice were obtained that carry an Atg5 allele in which the second protein-coding exon is flanked by two loxP sequences (Hara et al., 2006). These Atg5f/f mice were bred with transgenic mice in which the rat elastase I (EL) promoter/enhancer drives expression of Cre recombinase specifically in pancreatic acinar cells (EL-Cre mice, Grippo et al., 2002). Three lines of mice were generated and used in the experiments described herein: (i) Atg5f/f/Cre+ mice; (ii) Atg5f/f/Cre− littermates, used as controls in all experiments; and (iii) Atg5f/wt/Cre+ mice, used in some experiments to determine if Atg5 gene dosage affects CVB3 replication and pathogenesis.
We first characterized the mice genotypically and phenotypically (Figure 1). PCR of pancreatic DNA using four Atg5f-related primers (termed A-D, sequences provided in Supplemental Materials and Methods) identified the expected bands in all three of the mouse lines mentioned above. Next, we evaluated the presence of functional Atg5 protein in the pancreas, in two ways (Figure 1B). First, active autophagy flux requires the covalent attachment of Atg12 to Atg5 (Mizushima et al., 1998); in the absence of this linkage, autophagy cannot proceed. The Atg12-Atg5 conjugate was abundant in the pancreata of Atg5f/f/Cre− mice, but was absent from their Cre+ littermates. Second, we measured the content of LC3-I, a protein that is rapidly consumed during active autophagy, but which accumulates if the pathway is blocked. When normalized against GAPDH, LC3-I was markedly increased in the pancreata of Cre+ mice, compared to their Cre− counterparts. Despite having autophagy-deficient pancreata, the mice remained healthy: the average weight of the Cre+ mice was comparable to that of their Cre− littermates (Figure 1C), blood glucose levels were within normal limits (Figure 1D), and the quantities of three key pancreatic digestive enzymes, amylase, lipase and elastase 1, were similar in the two strains (Figure 1E). The pancreata of uninfected Atg5f/f/Cre− mice were histologically normal, as expected (Figure 1F, left panel). The Atg5f/f/Cre+ pancreas was largely normal (Figure 1F, right panel); the acinar cells contained abundant zymogen granules, although small vacuoles occasionally were visible and there was mild edema. These minor changes were not present in the pancreata of Atg5f/wt/Cre+ mice (data not shown) indicating that a single copy of the Atg5 gene is sufficient to support normal autophagy. EM studies confirmed the impression that most acinar cells in the Atg5f/f/Cre+ mice were morphologically normal, with fully packed rough endoplasmic reticulum, and abundant zymogen granules in the apical regions close to central ducts; only occasional cells showed vacuolization (Figure 1G, red arrows) or, in rare cases, pathology (acinar cell encircled by dashed red line). In summary, acinar cells in Atg5f/f/Cre+ mice are autophagy-deficient, but retain relatively normal cell morphology and are sufficiently functional to permit normal host development.
Figure 1. The pancreata of Atg5f/f/Cre+ mice are depleted of functional Atg5 protein, but the mice show little sign of constitutive pancreatic dysfunction.
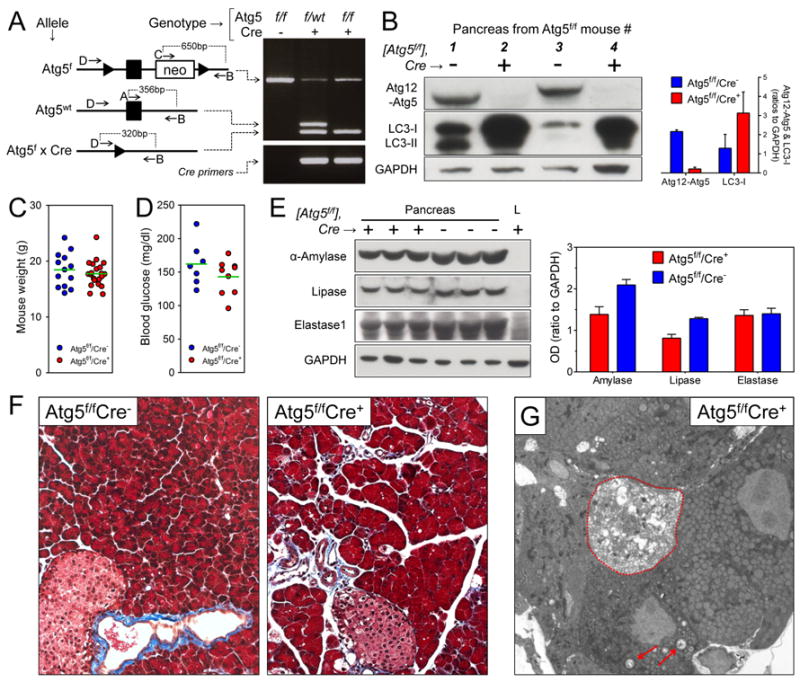
Generation of transgenic mice, and characterization of uninfected animals. (A). Diagram of the three Atg5 genotypes relevant to this study, “floxed”, wt, and deleted (Atg5f × Cre). Black arrowhead = lox p, black rectangle = exon of Atg5. The locations of the four primers (A-D) are shown; for sequences, see Experimental Procedures (online). The results of PCR of the three indicated genotypes are shown. All other panels in the figure show data from only two of the mouse lines: Atg5f/f/Cre+ ± Atg5f/f/Cre− mice. (B). Western blots of representative pancreata (two per mouse line). The right panel shows a graph of the average OD, normalized against GAPDH signal (mean + SE). The body weights (C) and blood glucose levels (D) of age- and gender-matched mice are shown (green lines indicate means). (E). Enzyme contents of pancreata from three mice of each line; L = liver (negative control). The related graph shows the average OD in each mouse line (mean + SE). Histological sections, stained with Masson’s trichrome, are shown in panel F, and G shows an electron micrograph of pancreas from an Atg5f/f/Cre+ mouse; vacuoles are indicated by red arrows, and a severely abnormal acinar cell is encircled by a dashed red line.
In the absence of an intact autophagy pathway, pancreatic titers of infectious CVB3 are dramatically reduced early in infection, and pancreatic pathology is partially mitigated
In wildtype mice, maximal CVB3 titer in the pancreas occurs at 2-3 days post-infection (p.i.) (Mena et al., 2000). To determine the in vivo effect of Atg5 deletion on CVB3 replication, all three strains of mice (Atg5f/f/Cre+, Atg5f/f/Cre− and Atg5f/wt/Cre+) were inoculated intraperitoneally with 104 pfu of wild type (wt) CVB3, and virus titers in the pancreata were measured at days 1, 2, 4 & 7 p.i. The titers of all individual mice, together with the geometric mean for each group (green bars), are shown in Figure 2A & B. In Atg5f/f/Cre− mice (blue circles), in which pancreatic autophagy is normal, CVB3 has already replicated to high titer (>108 pfu/g of pancreas) in most animals by 1 day p.i. In contrast, the titers in Atg5f/f/Cre+ mice (red circles) were substantially lower (p<0.0001), and more variable; the mean was >2000-fold lower in these mice compared to the Cre− animals. This suggests that an intact autophagy pathway may be required for rapid and high-level productive CVB3 infection of acinar cells. Consistent with this, a single copy of wt Atg5 (i.e., in the Atg5f/wt/Cre+ mice, black triangles) very substantially restored virus titers in the day 1 pancreas. To ensure that these observations were not restricted to wt CVB3, a limited study was carried out using a recombinant CVB3 encoding the dsRed protein. As shown in Figure 2C, a similar pattern was observed; DsRed-CVB3 titers were substantially reduced in the Atg5f/f/Cre+ pancreata. Together, these data strongly suggest that active autophagy benefits CVB3, and that a single copy of an intact Atg5 gene is sufficient to support CVB3 replication. Highly-significant differences between the Atg5f/f/Cre+ mice and the other two groups were maintained at day 2 p.i., although the difference in means had been reduced to ~200-fold, and a less dramatic (~10-fold) positive effect of Atg5 on CVB3 replication remained detectable at day 4; by 7 days p.i., virus titers were relatively low (~104 pfu /g) in all three groups, and there was no significant difference between them. Finally, no significant Atg5 gene dosage effect was observed; at each time point, there was no statistically-significant difference in virus titers between the Atg5f/wt/Cre+ mice (black triangles, one intact copy of Atg5) and the Atg5f/f/Cre− mice (blue circles, 2 intact copies). Next, we measured the pathological effects of infection, using two criteria. First, histological examination. Uninfected pancreata of Atg5f/f/Cre− mice are histologically normal, and those of Atg5f/f/Cre+ mice show only minor histological defects (Figure 1). However, following CVB3 infection, a major divergence in appearance was observed (Figure 2D). At day 2 p.i., the pancreata of Cre− mice showed extensive acinar cell hypochromicity and death, and inflammatory cell infiltration. In contrast, the acinar cells in Cre+ animals showed less florid pathology; some cells contained large vacuoles, but the majority appeared histologically normal. These differences in appearance are consistent with the much lower virus titers in the latter group. At 4 days p.i., inflammatory cells were abundant in both mouse strains, but a much greater degree of acinar cell destruction was present in the Cre− mice; only remnants of cells were visible. By 7 days p.i., the pancreas of the Cre− mouse showed widespread inflammation, collagen formation, and an almost complete ablation of acinar cells, as we have previously reported in C57BL/6 mice (Mena et al., 2000). The Cre+ (Atg5-deficient) pancreas also showed some inflammation and scarring at this time point, and acinar cell pathology was both more frequent and more severe than at day 4; however, the number of infiltrating cells was lower than in Cre− pancreata, and acinar cells were readily-detectable, and some appeared histologically-normal. Second, another pathological hallmark of pancreatitis is an increase in serum amylase levels. Prior to infection, the level of serum amylase was similar in all mice, regardless of Cre status (~6 U/ml, not shown). At day 2 p.i., serum amylase averaged ~42 U/ml in Cre− mice, but only ~15 U/ml in Atg5f/f/Cre+ animals (Figure 2E); the difference in amylase level is consistent with the differences in both virus titer and histological appearance. Mice with a single copy of Atg5 (Atg5f/wt/Cre+ mice) showed a level of serum amylase that was slightly lower than that observed in Cre− mice, but the difference was not statistically significant. In summary, the absence of Atg5 appears to protect pancreatic acinar cells from CVB3-induced pathology.
Figure 2. In the absence of an intact autophagy pathway, pancreatic titers of infectious CVB3 are dramatically reduced early in infection, and pancreatic pathology is partially mitigated.
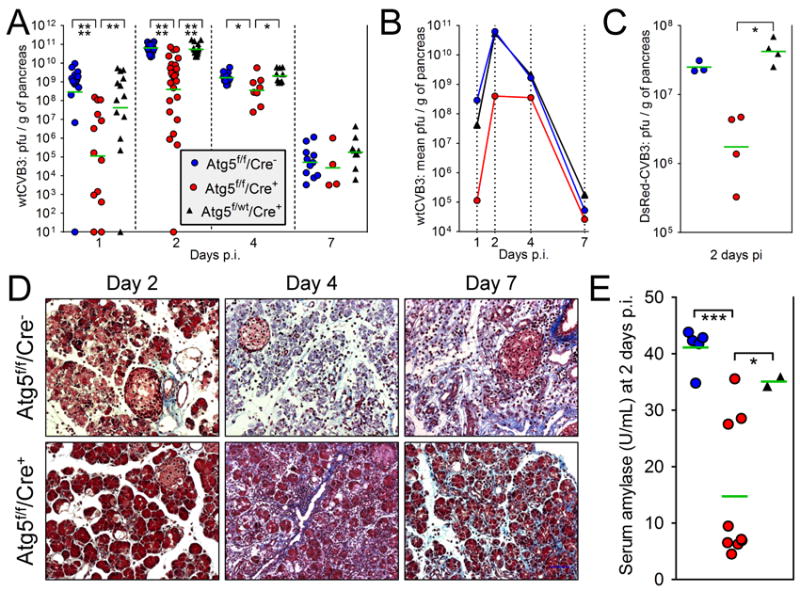
The key in panel A, showing the three lines of mice, applies also to panels B, C & E. (A & B). Mice were infected with wtCVB3 (104 pfu i.p.) and, at days 1, 2, 4 and 7 p.i. the pancreata were harvested and virus titers of pancreatic homogenates were determined by plaque assay. In each group, the total number of mice used was: Atg5f/f/Cre−, 52; Atg5f/f/Cre+, 50; Atg5f/wt/Cre+, 47. (A). Horizontal green bars indicate the geometric means, which are re-plotted in (B) to more clearly present the viral replication kinetics in each group. (C). Mice were infected with a recombinant CVB3, DsRed-CVB3 (107 pfu i.p.), and pancreatic titers were measured at 2 days p.i. (D). At the indicated times p.i., histological sections were prepared from pancreata of Atg5f/f/Cre− or Atg5f/f/Cre+ mice and stained with Masson’s trichrome. (E). Serum amylase levels were measured at 2 days p.i. in the indicated mouse strains. Prior to infection, the average level was ~6 U/mL.
No single feature of the CVB3 lifecycle can be identified as being acutely Atg5-dependent in acinar cells, and responsible for the reduced in vivo replication and pathogenesis
Tissue culture studies of poliovirus (Jackson et al., 2005), CVB (Wong et al., 2008; Yoon et al., 2008), human rhinoviruses (Brabec-Zaruba et al., 2007; Klein and Jackson, 2011) and enterovirus 71 (Huang et al., 2009) have found only minor changes in virus titer when autophagy is modulated, and there is no clear understanding of precisely which stage(s) of the enterovirus lifecycle are affected. The data in Figure 2 reveal a far greater effect in vivo; in the absence of intact Atg5, pancreatic CVB3 titers early in infection are reduced by ~3 logs. Therefore, we next asked if in vivo Atg5 deficiency caused a marked disruption at any one particular step of the CVB3 life-cycle in vivo. These studies were carried out both in vivo, and in tissue culture. For the in vivo analyses, mice (four Atg5f/f/Cre− and nine Atg5f/f/Cre+) were inoculated with CVB3 and, at day 2 p.i., the pancreata were recovered, homogenized, and assayed. For the tissue culture studies, we evaluated CVB3 infection using isolated primary pancreatic acinar cells, which were infected with wt CVB3 at an approximate MOI of 100.
Acinar cell cultures are shown in Figure 3A. The percentage of infected cells was similar, regardless of Atg5 status (Figure 3A, graph); this argues against a requirement for autophagy in cell attachment or entry, although these steps in the virus lifecycle have not been directly measured. Quantitative PCR was carried out and, as shown in Figure 3B, viral RNA replication proceeded in both cell types; the overall kinetics of genome amplification were similar, although genomic RNA accumulation was reduced by ~10-fold in the Cre+ cells. Next, virus protein levels in Cre+ and Cre− pancreata were compared (Figure 3C). Pancreatic samples from Cre− and Cre+ mice were analyzed by Western blot using antibodies specific for four viral proteins, VP1, 3A/3AB, 3Dpol, and 2C (Figure 3C). These proteins were more abundant in the Cre− group at 2 days p.i., and the average difference in abundance of the assayed CVB3 proteins was ~6-fold. The relative signal of each of the virus proteins in the Cre+ tissue was very similar to that observed in the tissue from the Cre− mouse, indicating that the co- and post-translational processing of the CVB3 polyprotein had proceeded normally in the Cre+ acinar cells. Next, we evaluated viral genomic RNA content in the pancreata and, for each individual mouse, we compared this with the quantity of infectious virus (Figure 3D); we reasoned that, if there were a defect in RNA encapsidation in Atg5-deficient pancreata, this might be revealed by a change in the ratio of genome to infectious virus. As was noted previously (Figure 2), there was more variability in the Cre+ group (red circles) than in the Cre− group (blue). However, there was no wholesale block in RNA replication in Cre+ acinar cells in vivo, because viral genomic RNA was abundant (>1011 copies per gram of tissue) in most Atg5f/f/Cre+ animals (Figure 3D, x axis). Genomic RNA content was ~1-2 logs higher in the Cre− mice, consistent with the higher virus titers that we had observed in the larger study group (Figure 2A & B). There was a highly-significant correlation between RNA genome content and infectious virus titer (p<0.0001), and almost all of the animals (both Cre− and Cre+) showed the same ratio of genome to infectious virus (~160:1), suggesting that the efficiency of encapsidation of RNA into infectious particles is not markedly affected by the autophagy status of the cell. Finally, it has been proposed that autophagy may play a part in the release of enteroviruses from cells (Jackson et al., 2005), so we evaluated virus egress from infected acinar cells by comparing the quantities of intracellular (cell) and extracellular (sup) virus during a one-step growth curve (Figure 3E). As expected, the titers of both cell and sup virus were higher in Atg5-expressing acinar cells. However, the cell:sup ratios were almost identical in both cell types, indicating that Atg5 deficiency does not have any obvious effect on viral egress.
Figure 3. No single feature of the CVB3 lifecycle can be identified as being acutely Atg5-dependent in acinar cells, and responsible for the reduced in vivo replication and pathogenesis.
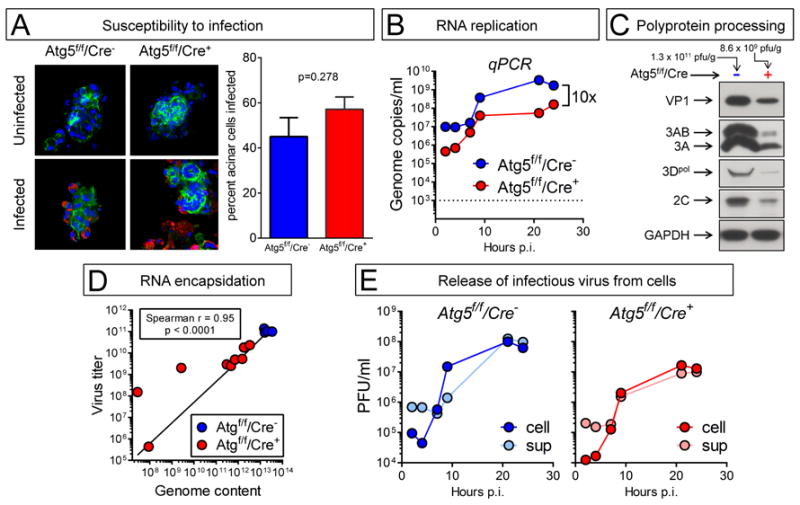
Panels A, B, E: Acinar cells were isolated from the pancreata of uninfected Atg5f/f/Cre− and Atg5f/f/Cre+ mouse lines, and were infected in vitro with wtCVB3 (MOI ~100). Panels C & D: Atg5f/f/Cre+ and Atg5f/f/Cre− mice were infected with CVB3, and were sacrificed two days later. The pancreata were recovered and homogenized. (A). F-actin was detected using Alexa Fluor 488-phalloidin (green), and CVB3 VP1 using a specific antibody (red) The graph shows the proportion of each cell type that was infected (mean + SE). (B). Viral RNA content in isolated acinar cells was followed over a 24-hour period by qPCR. The dashed line shows the lower limit of detection; uninfected cells scored below this line. (C). Western immunoblot analysis of viral protein content at 2 days p.i. in the pancreata of two representative mice. The virus titer in each mouse is shown above each column. (D). Infectious virus titers in pancreata were measured by plaque assay, and genomic CVB3 RNA content was determined by qPCR. Each symbol represents a single mouse, and a Spearman correlation line for the relationship between virus titer (y axis) and viral genomic RNA content (x axis) is shown. (E) One-step growth curve in both cell types (Cre− and Cre+); cell-associated (cell) and extracellular (sup) infectious virus titers were measured.
Electron microscopy of CVB3-infected Atg5f/f/Cre− and Atg5f/f/Cre+ acinar cells
CVB3 infection causes a number of changes that are detectable by EM, including: (i) swelling and remodelling of the rough endoplasmic reticulum (RER); (ii) the development CMVs; and (iii) the formation of highly-geometric lattices, which we have previously proposed may be sites of viral RNA replication (Kemball et al., 2010). Therefore, we used EM to evaluate the effects of Atg5 deficiency. At day 2 p.i., the pancreata of Atg5f/f/Cre− mice contained infected acinar cells in which the RER was swollen and extensively remodeled, with frequent multilamellar swirls (Figure 4A). These cells also contained abundant CMVs (an example is shown Figure 4B; the double-membraned nature of these small autophagy-like vesicles are clearly visible). In contrast, neither extensive multilamellar RER swirling nor CMVs were frequent in the pancreata of Atg5f/f/Cre+ mice at 2 days p.i. (representative cell is shown in Figure 4C). Furthermore, lattices were frequently observed in infected Atg5f/f/Cre− isolated acinar cells (Figure 4D). These paracrystalline structures very often are adjacent to RER/ribosomes and comprise an array of subunits, each with an approximate diameter of 8 nm, which we have speculated may be the viral RNA 3D (polymerase) protein (Kemball et al., 2010). These lattices were frequently observed in infected Atg5f/f/Cre− acinar cells, but not in Atg5f/f/Cre+ cells, even though the latter cells were infected, as shown by extensive vacuolization (Figure 4E), and by the presence of virions (Figure 4F, red arrow).
Figure 4. Electron microscopy of CVB3-infected Atg5f/f/Cre− and Atg5f/f/Cre+ acinar cells.
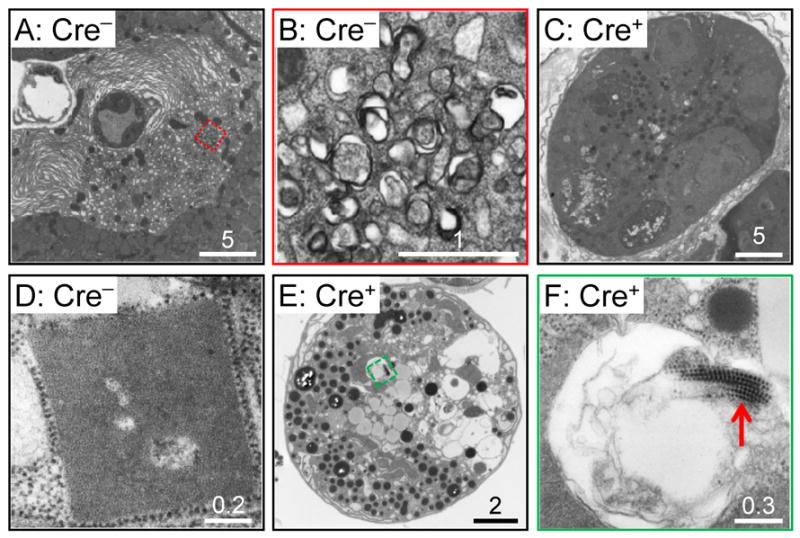
A-C: Pancreata were studied at 2 days p.i. A-B, Atg5f/f/Cre− mice; C, Atg5f/f/Cre+ animal. (A) Infected acinar cell in a Cre− pancreas, showing extensive RER remodelling. The region enclosed in a red dashed square is magnified in B, and contains compound membrane vesicles. (C) Infected cell in a Cre+ pancreas; vacuolization is present, but extensive RER remodelling, and CMVs, are absent. D-F: Acinar cells were isolated from Atg5f/f/Cre− mice (D) or from Atg5f/f/Cre+ animals (E,F). The cells were infected with CVB3 (MOI = ~100) and, 18 hours later, the cells were evaluated by EM. Paracrystalline arrays, adjacent to ribosomes, and comprising subunits of ~8 nm diameter, were present in Cre− cells (D) but were less frequently observed in Cre+ cells, even though the latter cells were infected, as indicated by the vacuolization that is visible at low magnification (E), and by the presence of virions, revealed at high magnification (F, red arrow). Scale bars are show for each image, and the number immediately above each bar indicates its size (microns).
Discussion
Herein, we have investigated the effects of autophagy on CVB replication in vivo, and on viral pathogenesis. To this end, we generated mice in which pancreatic acinar cells – a key target of CVB3 in vivo – were deficient in autophagy. Prior to CVB3 infection, acinar cell function and health in these mice appeared almost indistinguishable from those in normal (Cre−) animals (Figure 1). However, several dramatic differences were observed after CVB3 infection in vivo. The disruption of autophagy in pancreatic acinar cells led to a profound reduction in their capacity to support productive CVB3 infection in vivo (Figure 2). A single intact copy of Atg5 (in Atg5f/wt/Cre+ mice) mitigated this effect, permitting near-normal levels of virus replication. The effect of Atg5 deficiency was most pronounced early post-infection, and gradually reduced over the course of infection, as virus titers in autophagy-competent mice fell. Published tissue culture studies have not clearly identified any one stage of the enteroviral lifecycle that is strongly dependent on autophagy. Herein, working in vivo and with primary acinar cells, we have found that the Atg5 status appears not to affect the cells’ susceptibility to infection (Figure 3A), but the subsequent synthesis of genomic RNA appears to be reduced in Atg5-deficient cells (Figure 3B). There is a concomitant decrease in viral protein content, although viral polyprotein processing appears to be largely normal (Figure 3C). A comparison of genome copy number and infectious virus titer in the pancreata of individual animals revealed a very tight correlation between the two, and the ratio (~160:1) was similar in both lines of mice (Figure 3D), suggesting that, in autophagy-deficient acinar cells in vivo, the proportion of viral genomes that is ultimately encapsidated is similar to that observed in autophagy-competent cells. Others have proposed that autophagy may play a role in the escape of virions from the infected cell (Jackson et al., 2005), but we see little indication of such an effect (Figure 3E). During EM analyses of infected pancreata (Figure 4) there was a marked reduction in the frequency with which CMV clusters were observed in Atg5f/f/Cre+ acinar cells, consistent with the notion that a lack of intracellular membranes may contribute to the reduction in CVB3 replication. In addition, the analyses of isolated acinar cells showed that paracrystalline lattices – putative sites of RNA synthesis (Kemball et al., 2010; Lyle et al., 2002; Tellez et al., 2011) – were much more readily identified in acinar cells from Atg5f/f/Cre− mice. This is consistent with the lower yield of infectious virus (Figure 2), and the lower levels of genomic RNA and protein (Figure 3). However, from the data presented herein, we cannot exclude the possibility that another mechanism, in addition to autophagy, may contribute to the observed effects of Atg5 deficiency on CVB3 replication and pathogenesis in vivo; for example, it has been reported that Atg5 also may be involved in triggering apoptosis (Yousefi et al., 2006). Thus, it is important that more detailed studies be carried out to identify the precise molecular mechanism(s) that underlie the in vivo effects reported herein.
Others have carried out tissue culture studies with enteroviruses, using a variety of immortalized cell lines, and have reported that autophagy status can alter enterovirus yield. In almost all cases the reported changes were quite modest (often, ~2- to 4-fold). Our in vitro studies with isolated primary pancreatic acinar cells (Figure 3B, E) are consonant with the published work, and strongly suggest that the viral requirement for autophagy is far from absolute. Notably, however, we show here that the extent of the Atg5KO effect is substantially greater in vivo (~1000-fold at 1 day p.i., Figure 2) than in vitro (~10-fold, Figure 3B, E). What might explain the profoundly greater impact of autophagy on CVB3 when evaluated in vivo? First, our in vitro data come from a one-step growth curve, while in vivo titers often are the result of multiple rounds of virus infection and multiplication; this would amplify any difference in yield, at least until the virus titers had peaked. For example, in the latter situation, a 10-fold difference in yield, when applied to N rounds of cellular infection, would result in a 10N-fold difference in total viral titer. Second, viruses have been honed by evolution to multiply in vivo, and it is not unreasonable to suggest that in vivo studies allow the viruses to best exploit their hard-won evolutionary advantages. For example, target cells in vivo will be in their normal anatomical location, interacting with neighboring cells, and receiving normal nutrition, etc.; removing them to the tissue culture dish interrupts these interactions, with unpredictable effects on cellular gene expression.
In conclusion, our data suggest that, in autophagy-deficient pancreata, CVB3 RNA replication and translation are reduced, but subsequent steps in the virus life-cycle, including polyprotein processing and encapsidation/release, are qualitatively and quantitatively close to normal. The molecular reason(s) for the reduction remain undetermined, but we speculate that it may be related to the diminished availability of double-membraned scaffolds upon which the virus can replicate. Nevertheless, despite the inactivation of autophagy, CVB3 still replicates to a significant extent in the Atg5f/f/Cre+ pancreata / acinar cells, so it will be interesting to identify the other cellular pathways and organelles that may be exploited by CVB3, and other enteroviruses, in the absence of intact autophagy. For example, a recent study has suggested that, even in the absence of Atg5, there may be some residual autophagy executed by a non-conventional, alternative, pathway (Nishida et al., 2009). Finally, we show here that the genetic disruption of acinar cell autophagy conferred upon the pancreas some degree of protection from CVB3-induced disease. Consequently, we speculate that the temporary pharmacological inhibition of acinar cell autophagy may represent one possible therapeutic approach to mitigate the harmful effects of CVB3 in this tissue.
Experimental procedures
More detailed procedures can be found in Supplemental Materials.
Generation of pancreatic acinar Atg5-deficient mice
Homozygous mice bearing “floxed” Atg5 alleles, in which the second protein-coding exon was flanked by loxp sequences (Atg5f/f mice), were crossed with a transgenic line in which Cre recombinase is expressed in pancreatic acinar cells, under the control of the elastase-1 promoter.
Viruses and plaque assays
The wild-type (wt) CVB3 used in these studies is a plaque purified isolate (designated H3) of the myocarditic Woodruff variant of CVB3. Plaque assays were performed on sub-confluent HeLa cell monolayers, and the virus titers (pfu/g or pfu/ml) were calculated for each sample.
Quantitative PCR
Pancreatic tissue was collected, weighed, and RNA was isolated using an RNeasy Mini Kit (QIAGEN), following the manufacturer’s instructions. 0.25 μg total RNA from the samples was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s protocol, followed by Taqman quantitative real time PCR.
Serum amylase assay
Serum was collected, and the level of amylase was determined using the EnzChek® Ultra Amylase Assay Kit (Invitrogen) according to the manufacturer’s instructions.
Isolation of dispersed pancreatic acini
Dispersed pancreatic acini were prepared using a modified collagenase digestion protocol previously published by the Gukovsky laboratory at UCLA.
Measuring susceptibility to infection
Isolated acini were incubated with CVB3 (MOI=100) for 1 hour and, 21 hours later, were stained for the presence of the viral VP1 protein (rabbit polyclonal VP1, Dr. Karin Klingel, University Hospital Tübingen) and F-actin was stained using Alexa Fluor 488 phalloidin (Invitrogen). Confocal images were captured using a Zeiss LSM 710 Laser Confocal Scanning Microscope.
Analysis of protein expression by Western blot
Western blots were carried out as previously described (Kemball et al., 2010). The primary antibodies that were used are described in the Supplemental Materials.
Statistical analyses
Virus titers in the various strains of mice were compared using non-parametric tests [(Mann-Whitney and/or Anova (Kruskal-Wallis with Dunn’s post-test for multiple comparisons); GraphPad Prism v6.0]. In the figures, p values are indicated by asterisks as follows: **** p < 0.0001; *** 0.0001 < p < 0.001; ** 0.001 < p < 0.01; * 0.01 < p < 0.05.
Supplementary Material
Highlights.
Mice with autophagy-deficient pancreatic acinar cells were generated
Upon coxsackievirus infection, viral titers were lower in autophagy-deficient pancreata
Pancreatic pathology was dramatically reduced in infected autophagy-deficient animals
Putative virus replication platforms were infrequent in autophagy-deficient pancreata
Acknowledgments
We thank: Drs. Noburu Mizushima (Tokyo Medical and Dental University) and Herbert W. “Skip” Virgin (Washington University) for providing Atg5f/f mice; Dr. Eric P. Sandgren (University of Wisconsin-Madison) for providing EL-Cre transgenic mice; Dr. Karin Klingel (University Hospital Tübingen) & Dr. Karla Kirkegaard (Stanford University) for providing, respectively, 3Dpol and VP1 antibodies; and Ilya Gukovsky & Anna S. Gukovskaya and the members of their laboratory (UCLA) for training us in the preparation of isolated acinar cells. We also thank Dr. Chris Kemball for helpful discussions, and Sheila Silverstein for excellent secretarial support. This work was supported by NIH R01 awards AI042314 and HL093177 (to J.L.W.). This is manuscript number 20701 from the Scripps Research Institute.
Footnotes
The authors have no conflicting financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bienz K, Egger D, Pfister T, Troxler M. Structural and functional characterization of the poliovirus replication complex. J Virol. 1992;66:2740–2747. doi: 10.1128/jvi.66.5.2740-2747.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabec-Zaruba M, Berka U, Blaas D, Fuchs R. Induction of autophagy does not affect human rhinovirus type 2 production. J Virol. 2007;81:10815–10817. doi: 10.1128/JVI.00143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Noueiry A, Ahlquist P. An alternate pathway for recruiting template RNA to the brome mosaic virus RNA replication complex. J Virol. 2003;77:2568–2577. doi: 10.1128/JVI.77.4.2568-2577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhard JR, Perry CM, Harkins S, Lane T, Mena I, Asensio VC, Campbell IL, Whitton JL. Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol. 1998;153:417–428. doi: 10.1016/S0002-9440(10)65585-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grippo PJ, Nowlin PS, Cassaday RD, Sandgren EP. Cell-specific transgene expression from a widely transcribed promoter using Cre/lox in mice. Genesis. 2002;32:277–286. doi: 10.1002/gene.10080. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J Med Virol. 2009;81:1241–1252. doi: 10.1002/jmv.21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezequel AM, Steiner JW. Some ultrastructural and histochemical aspects of Coxsackie virus-cell interactions. Lab Invest. 1966;15:1055–1083. [PubMed] [Google Scholar]
- Kemball CC, Alirezaei M, Flynn CT, Wood MR, Harkins S, Kiosses WB, Whitton JL. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J Virol. 2010;84:12110–12124. doi: 10.1128/JVI.01417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard K, Jackson WT. Topology of double-membraned vesicles and the opportunity for non-lytic release of cytoplasm. Autophagy. 2005;1:182–184. doi: 10.4161/auto.1.3.2065. [DOI] [PubMed] [Google Scholar]
- Klein KA, Jackson WT. Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J Virol. 2011;85:9651–9654. doi: 10.1128/JVI.00316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 2007;5:e220. doi: 10.1371/journal.pbio.0050220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle JM, Bullitt E, Bienz K, Kirkegaard K. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science. 2002;296:2218–2222. doi: 10.1126/science.1070585. [DOI] [PubMed] [Google Scholar]
- Mena I, Fischer C, Gebhard JR, Perry CM, Harkins S, Whitton JL. Coxsackievirus infection of the pancreas: evaluation of receptor expression, pathogenesis, and immunopathology. Virology. 2000;271:276–288. doi: 10.1006/viro.2000.0332. [DOI] [PubMed] [Google Scholar]
- Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol. 2008;6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- O’Donnell V, Pacheco JM, LaRocco M, Burrage T, Jackson W, Rodriguez LL, Borca MV, Baxt B. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology. 2011;410:142–150. doi: 10.1016/j.virol.2010.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 2009;16:57–69. doi: 10.1038/cdd.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel A, Giddings TH, Jr, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy DA, Giddings TH, Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MP, Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J Virol. 2007;81:12543–12553. doi: 10.1128/JVI.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellez AB, Wang J, Tanner EJ, Spagnolo JF, Kirkegaard K, Bullitt E. Interstitial contacts in an RNA-dependent RNA polymerase lattice. J Mol Biol. 2011;412:737–750. doi: 10.1016/j.jmb.2011.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S, Locker JK. Hijacking cellular garbage cans. Cell Host Microbe. 2010;7:424–426. doi: 10.1016/j.chom.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and autophagy generate sites for virus replication. Science. 2006;312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol. 2008;82:9143–9153. doi: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, Lee JY, Kim DH. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J Virol. 2008;82:11976–11978. doi: 10.1128/JVI.01028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li Z, Xinna G, Xin G, Yang H. Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy. 2011;7:613–628. doi: 10.4161/auto.7.6.15267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.