

Summary
Disease is often the result of an aberrant or inadequate response to physiologic and pathophysiologic stress. Studies over the last 10 years have uncovered a recurring paradigm in which microRNAs (miRNAs) regulate cellular behavior under these conditions, suggesting an especially significant role for these small RNAs in pathologic settings. Here, we review emerging principles of miRNA regulation of stress signaling pathways and apply these concepts to our understanding of the roles of miRNAs in disease. These discussions further highlight the unique challenges and opportunities associated with the mechanistic dissection of miRNA functions and the development of miRNA-based therapeutics.
Introduction
MicroRNAs (miRNAs) were first discovered in the early 1990s through the analysis of developmental timing mutants in C. elegans (Lee et al., 1993; Wightman et al., 1993). It was not until after 2001, however, that a dedicated field focused on the study of these regulatory RNAs coalesced, following the identification of numerous endogenously-expressed small RNAs in worms, flies, and mammals (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). In the ensuing decade, the study of miRNA biology has attracted remarkable attention, resulting in rapid advances. We have since learned that mammalian genomes encode ~300 conserved miRNA genes and high-throughput sequencing studies have identified ~1000 or more additional loci that produce small RNAs structurally resembling miRNAs. However, since these additional miRNAs tend to be poorly conserved and expressed at low levels, their functional significance is unclear (Chiang et al., 2010; Landgraf et al., 2007). Small RNA cloning and analysis have also revealed the presence of other types of silencing RNAs in mammals, including endogenous short-interfering RNAs (endo-siRNAs) and germline-restricted piwi-interacting RNAs (piRNAs) (Farazi et al., 2008). Nevertheless, among the varied classes of small RNAs in mammals, miRNAs appear to have a uniquely important role in disease phenotypes and we therefore focus our attention on their functions herein.
In this review, we synthesize our current understanding of the physiologic roles of miRNAs in mammalian biology and the manner in which miRNA activities, both normal and aberrant, contribute to disease. Rather than attempting to present a comprehensive survey of the numerous studies that have linked miRNAs to individual disease phenotypes, we emphasize what appear to be the emerging themes of miRNA function gained from their evaluation in vivo. Within this context of normal miRNA biology, we can begin to understand the consequences when miRNA activities go awry. From recent studies, it has become apparent that miRNAs rarely contribute significantly to the establishment of the mammalian body plan and the specification of diverse cell lineages. While at first blush this appears to indicate a lesser role for miRNAs in mammalian biology compared to gene products such as developmentally-regulated transcription factors, their extreme evolutionary conservation would appear to argue otherwise. Accordingly, further study has revealed that miRNAs often profoundly influence the responses of fully-developed tissues to physiologic and pathophysiologic stress (Leung and Sharp, 2010). This functional niche suggests a central role for miRNA-regulated networks in disease states, which often represent an insufficient or aberrant response under conditions of stress or injury. Moreover, this role dictates that, in contrast to classic developmental regulators identified through forward genetic screens, miRNA loss-of-function may rarely result in highly-penetrant phenotypes in controlled laboratory environments. Although somewhat ironic given that the founding miRNA lin-4 was discovered by virtue of its strong developmental phenotype in C. elegans (Lee et al., 1993), an appreciation of this prominent role for mammalian miRNAs in dictating cellular responses in fully-developed tissues is essential for appropriate hypothesis generation, since phenotypes resulting from miRNA deletions may only be revealed in the setting of the appropriate perturbagen.
To provide a foundation for understanding miRNA functions in mammalian systems, we review lessons learned from the study of animals with deletions of individual miRNA-encoding loci. From these results, we derive a series of models that describe the roles of miRNAs in stress responses and then apply these principles to our current understanding of the functions of miRNAs in normal mammalian physiology and disease. Examples from neoplastic and cardiovascular pathology are used to illustrate these concepts since miRNA roles in these settings have been studied extensively, although abnormal miRNA function has been linked to many other disease states. Notable exceptions to these principles, which have important implications for the miRNA contribution to Mendelian and complex genetic disease, are also presented. Finally, we conclude by reviewing the opportunities and obstacles facing translational efforts to utilize and target miRNAs for disease diagnosis and treatment.
miRNA biogenesis and function
The vast majority of mammalian miRNAs are encoded by RNA polymerase II-transcribed genes that may be tens of kilobases in length and are frequently spliced (Kim and Kim, 2007). Approximately one-third of known miRNAs are embedded within introns of protein-coding genes and are co-transcribed with the host gene, allowing for coordinate regulation of miRNA and protein expression. In some cases, intronic miRNAs have been shown to modulate the same biological process as the protein encoded by that gene. This is exemplified by miR-33 family members, which cooperate with the sterol regulatory element binding protein (SREBP) genes in which they are embedded to reduce cholesterol efflux and increase cholesterol biosynthesis (Najafi-Shoushtari et al., 2010; Rayner et al., 2010). Within miRNA primary transcripts, ~60–80 nucleotide hairpin structures are first released and then further processed by the sequential activity of the RNase III-type endonucleases Drosha and Dicer to produce mature miRNAs of ~21–22 nucleotides in length (Kim et al., 2009). Fully-processed miRNAs associate with, and serve as specificity determinants for, the Argonaute (Ago) family of proteins within the RNA-induced silencing complex (RISC). miRNAs direct Ago proteins to target mRNAs by interacting with sites of imperfect complementarity. Short “seed” sequences at the 5′ ends of miRNAs (nucleotides 2–8) are most critical, and in some cases fully sufficient, for target selection. When directed to mRNAs via these interactions, Ago proteins perform a still incompletely defined activity that results in accelerated turnover and reduced translation of the targeted transcript (Bartel, 2009; Djuranovic et al., 2011).
Many different algorithms exist for the bioinformatic prediction of miRNA targets and all generally predict hundreds of targets for each miRNA (Alexiou et al., 2009). These highly complex target networks pose a significant challenge to the mechanistic dissection of miRNA-mediated phenotypes. The prevailing model posits that miRNAs function by fine-tuning the expression of numerous targets. While each target is regulated subtly (typically less than a 2-fold change in individual target protein abundance results from gain or loss of miRNA function), the additive effect of coordinated regulation of a large suite of transcripts is believed to result in strong phenotypic outputs. Unfortunately, this hypothesis is nearly impossible to test directly since it is not logistically feasible to simultaneously restore the levels of many targets to their natural levels in the setting of miRNA gain- or loss-of-function in vivo. Therefore, any conclusions drawn from these types of target analyses will be correlative.
On the other hand, some miRNA-mediated functions might be driven by the strong regulation of one or a few targets. This is exemplified by the downregulation of the protein-coding gene lin-14 by the founding miRNA lin-4 in C. elegans, which is required for the appropriate timing of larval development (Lee et al., 1993; Wightman et al., 1993). However, such linear miRNA-regulated pathways have rarely been demonstrated in mammals. The demonstration that haploinsufficiency of a target or targets rescues a miRNA deletion phenotype provides the strongest evidence of the importance of a specific miRNA:target interaction in vivo. For example, haploinsufficency of the miR-146a target Stat1 rescues the autoimmune pathology which arises in miR-146a knockout mice, implicating a central role for this target in this miRNA deletion phenotype (Lu et al., 2010). However, published results of this type are scarce, perhaps reflecting the time consuming nature and expense of generating and combining loss-of-function alleles in mice or alternatively reflecting the true complexity of miRNA-regulated networks. The challenges associated with linking specific targets to miRNA-mediated phenotypes underscore the need for new approaches to uncover the mechanisms underlying miRNA-mediated functions in vivo.
Lessons from miRNA loss-of-function studies in model organisms
Over the last 10 years, the consequences of loss-of-function of numerous miRNAs in worms, flies, and mice have been reported. In addition to classic genetic deletion techniques, the ability to suppress miRNA function using inhibitory antimiRs has facilitated functional analyses in cultured cells and animal models of disease and has provided unique opportunities for therapeutic modulation of miRNAs, as discussed in detail later in this review. AntimiRs are chemically modified oligonucleotides containing antisense sequences against mature miRNAs. A variety of chemical modifications enhance the stability, cellular uptake, and efficacy of antimiRs. “Antagomirs” contain a 2′-0-methyl modification of the sugar moieties and a phosphorothioate backbone to prevent nuclease degradation and enhance binding affinity, as well as a cholesterol moiety at the 3′ end to promote cellular uptake (Krutzfeldt et al., 2005). Antagomirs associate with target miRNAs in the RISC, preventing their association with mRNA targets and promoting miRNA degradation (Ameres et al., 2010). Antisense oligonucleotides containing a locked nucleic acid (LNA) modification, in which a covalent bridge connects the 2′-oxygen and the 4′ carbon of the ribose moiety of the nucleotide, creating a rigid bicycle, form especially strong duplexes with target miRNAs and are readily taken up by cells in vivo and in vitro (Stenvang et al., 2008). LNA-modified oligonucleotides are thought to sequester miRNAs but not to promote their degradation. Because of the high affinity of LNA-modified antisense oligonucleotides for their targets, it has been possible to use short, so-called “tiny” LNAs, to target just the seed regions of miRNAs, thereby allowing for inhibition of miRNA families that share homology in this region (Hullinger et al., 2011; Obad et al., 2011).
Given the high degree of evolutionary conservation of many miRNAs chosen for loss-of-function studies and the fact that the first miRNAs, lin-4 and let-7, were discovered through classic forward genetic screens and result in potent developmental defects when deleted (Lee et al., 1993; Reinhart et al., 2000), the expectation was that miRNA deletion or inhibition would frequently produce overt developmental phenotypes. Although there have been important exceptions, this has not been the case for the vast majority of miRNAs. This has been most thoroughly documented in C. elegans where individual deletion of nearly all known miRNAs has been shown to have little effect on viability or development (Miska et al., 2007). Similarly, in mice, the majority of the ~25 miRNA knockouts that have been examined thus far do not exhibit defects in embryonic development.
Initially, it seemed likely that redundancy among related miRNAs might account for the apparent non-essential role for individual miRNA genes during development. Within all animal species, numerous miRNAs share common seed sequences and are on this basis grouped into families. From a bioinformatic standpoint, miRNAs within a given family should be largely redundant since they are expected to have highly overlapping sets of predicted targets. Nevertheless, bona fide intra-family redundancy has rarely been documented through loss-of-function studies in vivo. For example, deletion of multiple or all members of 16 miRNA families in C. elegans results in overt phenotypes in only 4 cases (the let-7, miR-35, miR-51, and miR-58 families) (Alvarez-Saavedra and Horvitz, 2010). In mice, the majority of miRNAs that have been knocked out do not have identifiable paralogs, essentially ruling out a contribution of intra-familial redundancy to the absence of developmental abnormalities in these animals. Thus far, intra-familial redundancy in mice has been documented only for the miR-17-92 and miR-106b-25 clusters, which act together to regulate multiple aspects of embryonic development (Ventura et al., 2008), miR-133a-1 and miR-133a-2, which together are required for appropriate cardiac and skeletal muscle development (Liu et al., 2011; Liu et al., 2008), and miR-208b and miR-499 which co-regulate muscle fiber type identity (van Rooij et al., 2009). While other examples of intra-familial redundancy will undoubtedly be uncovered, this mechanism is unlikely to be the major cause of the apparent tolerance of animals to individual miRNA deletion.
Another source of redundancy may come from miRNAs that do not share seed sequences yet have acquired overlapping sets of targets. Interestingly, many miRNA deletions in C. elegans exhibit synthetic phenotypes when combined with loss-of-function of argonaute like 1 (alg-1), which globally reduces miRNA levels (Brenner et al., 2010). These findings are consistent with the hypothesis that reduced activity of unrelated compensatory miRNAs may sensitize animals to the effects of individual miRNA deletion. However, it is important to consider that the global reduction in miRNA pathway activity that occurs in alg-1-mutant worms results in a broad range of defects and likely induces a state of organismal stress that may sensitize animals to individual miRNA loss-of-function. Indeed, whereas the majority of miRNA knockout mice analyzed thus far lack developmental phenotypes, many have been found to exhibit profoundly abnormal responses to various stress conditions. In these cases, the absence of a miRNA may enhance or diminish the organismal response to stress, thereby augmenting or diminishing a pathologic process. For example, deletion of miR-208a has no effect on cardiovascular development or baseline function but abrogates stress-responsive cardiac remodeling (van Rooij et al., 2007). Deletion of the miR-143/145 cluster does not influence vasculogenesis but prevents neointima formation in response to vascular injury (Xin et al., 2009). The endocrine pancreas develops normally in miR-375−/− mice yet these animals fail to appropriately expand pancreatic β-cell mass in the setting of obesity-induced insulin resistance, resulting in severe diabetes (Poy et al., 2009). These findings have important implications for the study of miRNA functions in animals and the relevance of these functions to various disease states. A prominent role for miRNAs in stress responses dictates that many miRNA loss-of-function phenotypes will be difficult to detect or absent under controlled laboratory conditions. Therefore, when evaluating the consequences of miRNA deletion, a battery of stress paradigms should be applied to the tissue of interest to reveal otherwise undetectable defects. Furthermore, it can be expected that the stressors that reveal robust miRNA-mediated phenotypes will provide insight into the disease settings in which a given miRNA plays a prominent role. Elucidation of the underlying mechanisms through which miRNAs participate in these responses are likely to provide new understanding of disease pathophysiology and uncover new opportunities for therapeutic intervention.
Potential mechanisms of miRNA action in stress responsive pathways
In Figure 1, we provide a conceptual framework for understanding the roles of miRNAs in stress response pathways. Five mechanisms through which miRNAs can influence stress signaling are proposed and the predicted consequences of miRNA loss-of-function on pathway activity are schematized. It is important to note that these mechanisms are not mutually exclusive and a single miRNA could potentially act through several of these modes, depending on the cellular or functional context. After describing each model below, specific examples of miRNAs that function through these mechanisms are provided in the context of cancer and cardiovascular disease in the sections that follow.
Figure 1.
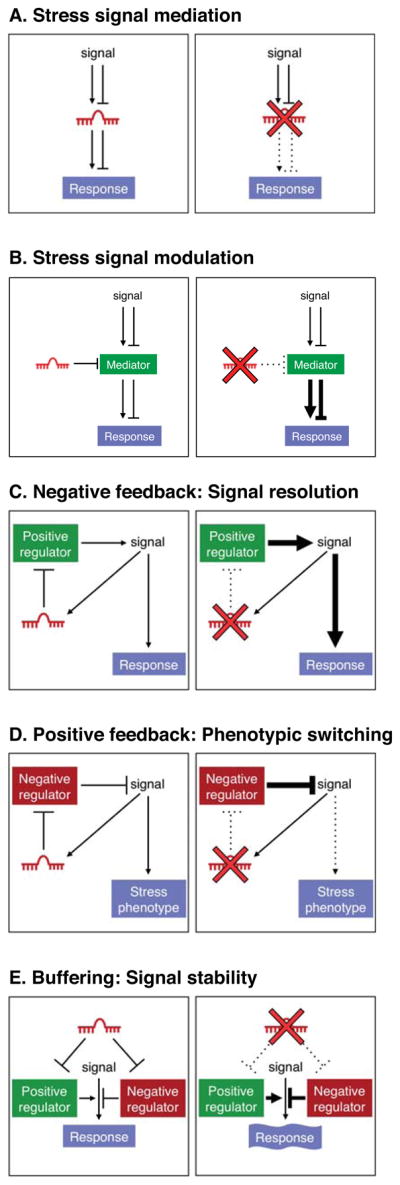
miRNAs can regulate stress signaling pathways and thereby modify stress-related phenotypes through a number of distinct mechanisms, some of which are schematized here. A miRNA can perform a stress signal mediation function (A) in which it acts as a critical intermediate in a signaling pathway or it may act as a stress signal modulator (B) in which it titrates a signaling intermediate. A miRNA may participate in a negative (C) or positive (D) feedback loop which serves to dampen or amplify a signal, respectively. Lastly, a miRNA may target both positive and negative regulators of a pathway (E), thereby buffering pathway activity from stochastic fluctuation. For each mode of regulation, the consequences of deletion of the miRNA on pathway activity are schematized on the right.
Stress signal mediation
The most conceptually simple mechanism through which a miRNA can influence a stress response is the scenario in which the miRNA is directly regulated by a signaling pathway and functions as an essential downstream component within the pathway (Figure 1A). Stress-dependent regulation can involve up-regulation or down-regulation of miRNA expression with consequent effects on downstream mRNA targets and cellular responses. In the absence of such a miRNA, cellular responsiveness to the pathway is impaired.
Stress signal modulation
A miRNA may be expressed constitutively in a cell but may modulate the response of the cell to a stress stimulus by titrating a critical component of a signal transduction pathway (Figure 1B). In the absence of the miRNA, its target is hyperactive, leading to an excessive or insufficient response, depending upon whether the mediator is a positive or negative regulator.
Negative feedback: Signal resolution
A miRNA may be activated by, and function as a negative regulator of, a signaling pathway (Figure 1C). Under stress conditions, the pathway may be excessively driven thus increasing the requirement for miRNA-mediated negative feedback to prevent pathologic hyperactivity or to restore homeostasis once stress resolves.
Positive feedback: Phenotypic switching
In this mode of miRNA action, the miRNA is activated by, and inhibits negative regulators of, the signaling pathway (Figure 1D). In this capacity, the miRNA would promote the irreversible activation of the pathway and thereby the stable switching of the cellular state under the stress condition, which can be important for restoring homeostasis but can also contribute to disease.
Buffering: Signal stability
Given the multiplicity of miRNA targets within complex biological pathways, a miRNA may function to buffer pathway activity by simultaneously dampening expression of both positive and negative regulators (Figure 1E). In this capacity, the miRNA would prevent stochastic fluctuations in signaling. This model is similar to the concept of signal robustness previously put forward by Cohen, Brennecke, and Stark (Cohen et al., 2006). Under normal conditions or in controlled laboratory environments, this type of buffering may not be critical to maintain normal function. However, in stress states, pathways may need to be transiently activated to a high level thus increasing the requirement for buffering to avoid runaway pathway activation or a failure to achieve the appropriate level of activation. There are documented examples in which gain- and loss-of-function of a specific miRNA result in similar phenotypes, which may reflect the perturbation of a buffering function.
MicroRNAs in Cancer
The initial indication that miRNAs play important roles in human disease came from studies of their functions in cancer cells. Several factors contributed to an early appreciation of their significance in this pathologic setting. First, the founding miRNAs discovered in the 1990’s in C. elegans, lin-4 and let-7, exhibit loss-of-function phenotypes that evoke some aspects of tumor biology. In lin-4 and let-7 mutants, specific stem cell lineages fail to differentiate at the appropriate larval stages, instead reiterating divisions characteristic of earlier stages and undergoing extra cell divisions in adults (Lee et al., 1993; Reinhart et al., 2000). miRNAs were subsequently identified in Drosophila that regulate cell proliferation and apoptosis, further linking these regulatory RNAs to cancer-relevant pathways (Brennecke et al., 2003; Xu et al., 2003). At the same time, a seminal study by Croce and colleagues showed that the miR-15a/16-1 cluster is frequently deleted in chronic lymphocytic leukemia (CLL), implicating these miRNAs as tumor suppressors (Calin et al., 2002). Together, these findings launched nearly a decade of intensive study of the roles of miRNAs in cancer.
It is now clear, based on hundreds of expression profiling studies, that tumors ubiquitously exhibit dysregulated miRNA expression patterns relative to corresponding normal tissue. Patterns of miRNA expression provide useful information for tumor classification and prognosis (Calin and Croce, 2006). Nevertheless, since miRNA expression is in many cases highly cell-type specific, the results of such profiling studies must be interpreted cautiously since apparent dysregulation of miRNAs in cancer cells can be a manifestation of the distinct cell populations represented in tumors versus normal tissue. This caveat highlights the critical importance of rigorous functional experiments to directly assess the consequences of miRNA gain- and loss-of-function on tumor cell behavior. Large numbers of such experiments have now been performed using both human cancer cell lines and genetically engineered mice and in numerous cases, miRNA activity has been shown to dramatically influence tumorigenesis. For example, transgenic expression of miR-155 (Costinean et al., 2006) or miR-21 (Medina et al., 2010) is sufficient to initiate lymphomagenesis in mice. Likewise, expression of the miR-17-92 cluster in mouse hematopoietic progenitor cells accelerates Myc-induced B-cell lymphomagenesis (He et al., 2005) and deletion of this miRNA locus induces apoptosis in Myc-driven lymphoma cells in vitro (Mu et al., 2009). Conversely, systemic delivery of selected miRNAs including let-7, miR-26a, miR-34a, and miR-143/145 inhibits tumor progression in vivo (Kota et al., 2009; Pramanik et al., 2011; Trang et al., 2011). Coupled with the expression data documenting dysregulation of these miRNAs in various tumor types and genomic data linking these miRNA genes to recurrent amplifications or deletions in cancer cells, the case has convincingly been made that specific miRNAs act as oncogenes and tumor suppressors.
The potent effects of miRNA gain- and loss-of-function in cancer cells starkly contrast with the results obtained from miRNA deletion studies in worms and mice which, as discussed above, rarely result in overt phenotypes in unstressed animals. Moreover, in many cases, delivery of anti-tumorigenic miRNAs appears to selectively affect tumor cell growth and survival while sparing normal cells from these effects (Kota et al., 2009; Pramanik et al., 2011; Trang et al., 2011). These findings suggest that neoplastic transformation represents a state of heightened sensitivity to miRNA-mediated activity. Why is cancer a setting where the phenotypic consequences of miRNA dysregulation are so robust and what distinguishes the activity of miRNAs in cancer cells from their activity in non-transformed cells? A parsimonious explanation is suggested by the observation that the phenotypic effects of miRNA-mediated regulation are enhanced under stress conditions. There is, in fact, a growing appreciation that cancer itself represents a state of cellular stress. Cancer cells experience chronic stress induced by DNA damage, aneuploidy, widespread protein misfolding and aggregation due to imbalances in the stoichiometry of protein complex components, and metabolic reprogramming which can fuel accelerated growth but lead to the accumulation of reactive oxygen species and other toxic intermediates (Luo et al., 2009). The resulting dependency on the compensatory activity of stress-induced signaling pathways, as well as the requirement for sustained activation of mitogenic and pro-survival signaling arising from oncogene activation or tumor suppressor loss, distinguishes cancer cells from non-transformed cells and provides an opportunity for miRNAs to drive strong and selective phenotypic outputs in this pathologic setting. In this regard, the five models of miRNA activity in stress signaling proposed earlier in this review (Figure 1) provide a useful framework for mechanistically understanding the consequences of miRNA dysregulation in cancer.
The integration of miRNAs into signaling pathways that play crucial roles in cancer cells has been extensively documented. This signal mediation function (Figure 2A) is perhaps best represented by members of the miR-34 family, which are important components of the p53 tumor suppressor network. From the perspective of cancer biology, p53 may well be the most important stress sensor discovered thus far. Upon its activation in the setting of genotoxic stress or oncogene hyperactivity, p53 directly transactivates the miR-34a and miR-34b/miR-34c transcription units which then are able to mediate some aspects of the cellular response to p53 activation, including cell-cycle arrest and apoptosis (Bommer et al., 2007; Chang et al., 2007; He et al., 2007; Raver-Shapira et al., 2007; Tarasov et al., 2007). Oncogenic pathways also utilize miRNAs as effectors of their pro-tumorigenic programs, as exemplified by the induction of the miR-17-92 cluster by the MYC oncogene (Figure 2A), which results in enhanced cellular proliferation, survival, and tumor angiogenesis (Mendell, 2008; O’Donnell et al., 2005). The gain or loss of these miRNAs therefore enhances or impairs the activity of these critical pathways in cancer cells.
Figure 2.

miRNAs that promote or inhibit tumorigenesis provide diverse functions within oncogenic and tumor suppressor signaling pathways. (A) miR-34 family members and the miR-17-92 cluster function as signal mediators for the p53 and Myc pathways, respectively. (B) miR-16 negatively regulates multiple components of mitogenic pathways and thereby provides an inhibitory signal modulation function. (C) miR-146a is activated by NF-κB signaling and negatively feeds back on the pathway by repressing upstream activators of NF-κB. In this capacity, miR-146a restrains excessive NF-κB activity which can lead to tumorigenesis. (D) Let-7, miR-21, and the miR-143/145 cluster participate in positive feedback loops which function to stably enforce cellular transformation programs upon activation of oncogenes such as NF-κB, Kras, and Myc. (E) miR-26 can repress both pro-tumorigenic targets (cyclins D2 and E2) and anti-tumorigenic targets (PTEN). These opposing activities endow miR-26 with context-dependent positive or negative effects on tumorigenesis.
miRNAs also influence tumor biology through their action as signal modulators in which they titrate important components of cancer-relevant pathways. This role is exemplified by the miR-15a/miR-16-1 cluster, which targets several factors that promote cell-cycle progression including CDK6, CARD10, and CDC27 (Figure 2B) (Linsley et al., 2007). Deletion of these miRNAs, as is known to occur in hematopoietic and solid malignancies, would therefore be expected to enhance the proliferative response of cancer cells to a variety of mitogenic stimuli.
Participation in negative and positive feedback loops is another common mechanism of miRNA action in cancer cells. This is well illustrated by miR-146a which is transactivated by the NF-κB pathway and negatively feeds back on this signaling cascade by targeting two upstream activators of the pathway, TRAF6 and IRAK1 (Figure 2C) (Taganov et al., 2006). Deletion of miR-146a in mice results in basally increased activity of NF-κB in splenocytes and the consequent development of NF-κB-dependent myeloid sarcomas (Zhao et al., 2011). The NF-κB pathway also has been shown to employ a miRNA-mediated positive feedback circuit in some contexts (Figure 2D). NF-κB was demonstrated to transactivate LIN28B, which encodes an RNA binding protein that blocks processing and accelerates turnover of let-7 precursors (Iliopoulos et al., 2009). Lin28B-dependent downregulation of let-7 causes upregulation of IL-6, a let-7 target, which further stimulates NF-κB. Once activated, this circuit enforces a stable conversion to a transformed state since the continued production of IL-6 activates pro-tumorigenic STAT3 signaling and the downregulation of let-7 de-represses its oncogenic targets that include MYC, KRAS, and HMGA2. MYC also directly transactivates LIN28B and thus reduces expression of its negative regulator let-7 (Chang et al., 2009), constituting an overlapping positive feedback loop (Figure 2D). Other miRNAs that appear to participate in similar positive feedback pathways to stably enforce oncogenic signaling programs include miR-21, which is activated by MAPK/ERK signaling and represses negative regulators of this signaling cascade (Hatley et al., 2010), and the miR-143/145 cluster, which is repressed when its target, Kras is activated (Kent et al., 2010) (Figure 2D). In these examples, restoration of normal miRNA activity would reinstate or interrupt these negative and positive feedback circuits, respectively, thereby diminishing oncogenic signaling.
Finally, we consider miRNAs that perform signal buffering functions. Through their ability to target both positive and negative regulators of a pathway, these miRNAs have the potential to promote or inhibit tumorigenesis in different cellular contexts depending on which targets are most critical for pathogenesis of a given tumor type. The miR-26 family appears to conform to this paradigm (Figure 2E). miR-26a and miR-26b levels are frequently reduced in hepatocellular carcinoma (HCC) and low expression of these miRNAs correlates with shorter patient survival (Ji et al., 2009). In liver cancer cell lines, expression of miR-26a induces cell-cycle arrest in part through inhibition of multiple G1 cyclins and delivery of miR-26a to a mouse model of HCC potently inhibits tumorigenesis (Kota et al., 2009). In contrast to these findings which convincingly support a tumor suppressor role for miR-26 in liver cancer, a pro-tumorigenic function for this miRNA family has been uncovered in the context of glioma. miR-26a accelerates PDGF-induced gliomagenesis in a mouse model, most likely through its ability to directly target PTEN, a critical tumor suppressor in many types of cancer (Huse et al., 2009). Additional study is clearly needed to further characterize the contexts in which miR-26 pro- or anti-tumorigenic functions predominate and the full set of targets which underlie these opposing activities. Nevertheless, this example highlights the disparate phenotypic outputs that may result from aberrant miRNA-mediated control of a complex network of targets composed of positive and negative regulators.
MicroRNAs in Cardiovascular Disease
The cardiovascular system has been an especially rich source of miRNAs with roles in disease, perhaps reflecting the susceptibility of the heart and blood vessels to injury and the dependence of mammals on persistent cardiovascular function (Small and Olson, 2011). miRNA profiling in mouse models of cardiovascular disease and in human biopsies has revealed signature patterns of miRNAs diagnostic for numerous cardiovascular disorders including heart failure, cardiomyopathy, myocardial infarction, atherosclerosis, ischemia, and angiogenesis. Gain- and loss-of-function studies in mice have also validated the importance of specific miRNAs in the pathophysiology of many of these disorders.
As in cancer, numerous miRNAs function as mediators of pathogenic stress-related signaling pathways in settings of cardiovascular disease (Figure 3A). For example, cardiac stress commonly results in fibrosis due to excessive extracellular matrix (ECM) production and collagen deposition, leading to stiffening of the ventricular chambers and cardiac arrhythmias (Hill and Olson, 2008). miR-29 is down-regulated under conditions of cardiovascular disease associated with fibrosis and excessive ECM production (Boon et al., 2011; Cushing et al., 2011; van Rooij et al., 2008), implicating this miRNA as a repressive mediator of fibrotic disease (Figure 3A). This miRNA targets a broad collection of mRNAs encoding multiple collagen isoforms and other ECM proteins, such that its downregulation during cardiovascular disease enhances fibrosis. TGF-β signaling in fibroblasts, a key driver of fibrosis, triggers miR-29 down-regulation (van Rooij et al., 2008). Due to the ability of miR-29 to impede excessive ECM production, delivery of this miRNA may represent an effective therapeutic approach for fibrosis in the cardiovascular system and elsewhere. However, there are settings where miR-29 overexpression appears to be detrimental. Aneurysms, characterized by ballooning of the vasculature, arise from a loss of ECM components and therefore may be exacerbated by miR-29 activity. Accordingly, inhibition of miR-29 with antisense oligonucleotides stimulates ECM production and diminishes aortic dilatation in the setting of aortic aneurysm in mice (Boon et al., 2011).
Figure 3.

Examples of miRNAs that function in cardiac stress signaling pathways. (A) miR-29 and miR-15 family members act as mediators of stress signaling pathways that regulate fibrosis and cardiomyocyte proliferation and survival, respectively. (B) miR-208a and miR-126 titrate regulators of cardiac remodeling and angiogenesis and thereby function as stress signal modulators. (C) miR-133a directly targets its activator SRF and in this manner restrains excessive SRF activity in adult cardiomyocytes which can lead to heart failure. (D) miR-21, miR-199a, and the miR-23a/27a/24-2 cluster participate in positive feed back loops which serve to stably activate signaling pathways that lead to pathologic cardiac remodeling and angiogenesis. (E) The miR-143/145 cluster targets both positive and negative regulators of smooth muscle differentiation. Through this buffering activity, these miRNAs maintain the characteristic phenotypic plasticity of this cell type, allowing smooth muscle cells to proliferate in response to injury.
Members of the miR-15/16 family also serve as important stress signal mediators through their roles in pathways that regulate cardiomyocyte proliferation and survival in response to injury (Figure 3A). During neonatal development, the miR-15 family is upregulated in the heart, coinciding with irreversible withdrawal of cardiomyocytes from the cell cycle and loss of regenerative potential (Porrello et al., 2011). This family of miRNAs is further upregulated following myocardial infarction (MI), causing irreversible loss of cardiomyocytes and cardiac dysfunction (Cimmino et al., 2005; Hullinger et al., 2011; Linsley et al., 2007; van Rooij et al., 2008). In keeping with a critical role for these miRNAs in ischemic cardiac pathology, inhibition of miR-15/16 family members with LNA-modified oligonucleotides confers protection against cardiomyocyte apoptosis following MI in rodents (Hullinger et al., 2011).
miRNAs have also been documented to perform stress signal modulation functions in the setting of cardiovascular disease, as illustrated by miR-208a (Figure 3B). Numerous forms of cardiac stress, including myocardial infarction, hypertension, and chemotherapy, result in pathological remodeling of the heart and consequent loss of pump function, culminating in heart failure, arrhythmias, and death (Hill and Olson, 2008). A hallmark of heart disease is a switch from the adult alpha-myosin heavy chain (α–MHC) isoform to expression of the fetal β-MHC gene, with concomitant diminution in cardiac function. miR-208a is encoded by an intron of the α-MHC gene and is expressed specifically in the heart along with its host gene (Callis et al., 2009; van Rooij et al., 2007). miR-208a knockout mice are protected from pathologic cardiac remodeling under conditions of chronic stress, indicating that miR-208a is required for the stress response which contributes to many cardiac diseases (Figure 3B). The actions of miR-208a appear to be mediated by a set of transcriptional repressor proteins that govern cardiac gene expression in response to stress. Thrap1/MED13, a component of the Mediator complex, is among the strongest targets of miR-208a (Callis et al., 2009; van Rooij et al., 2007). Upregulation of Thrap1 (and other targets), as occurs in the absence of miR-208a, is believed to impose a blockade to activation of downstream stress-responsive genes, including β-MHC. Systemic delivery of miR-208a inhibitors delays the onset of cardiac dysfunction in hypertensive rats, confirming the key role of this miRNA in pathological cardiac remodeling and suggesting the therapeutic potential of miR-208a inhibitors in heart disease (Montgomery et al., 2011).
Another example of stress signal modulation by a miRNA is provided by miR-126, an endothelial cell-specific miRNA encoded by an intron of the EGFL7 gene (Fish et al., 2008; Kuhnert et al., 2008; Wang et al., 2008), which encodes an endothelial growth factor (Figure 3B). miR-126 knockout mice have fragile, leaky vessels and display impaired angiogenesis in response to injury and angiogenic signaling (Kuhnert et al., 2008; Wang et al., 2008). Deletion of miR-126 impairs angiogenic signaling at least in part due to the consequent upregulation of the miR-126 targets Spred1 and PIK2R2 which function as negative regulators of the pro-angiogenic MAPK/ERK and PI3K/Akt signaling pathways, respectively.
As in cancer, negative feedback loops involving miRNAs play important roles in maintenance of cardiovascular function. A classic example involves the regulation of serum response factor (SRF), a key transcriptional regulator of cardiomyocyte growth and differentiation. SRF activates transcription of miR-133a in cardiomyocytes and miR-133a, in turn, negatively regulates SRF expression (Chen et al., 2006; Liu et al., 2008) (Figure 3C). The importance of this negative feedback loop is demonstrated by the pathologic consequences that result from deletion of the two miR-133a-encoding loci in mice (miR-133a-1 and miR-133a-2). Loss-of-function of these miRNAs results in lethal structural heart defects in approximately half of mutant animals (Liu et al., 2008). Those that survive exhibit aberrant expression of smooth muscle genes in adult cardiomyocytes and ultimately succumb to dilated cardiomyopathy which can be partially attributed to abnormally high SRF activity.
In addition to negative feedback loops, a related mechanism of stress-signal dampening occurs when a stimulus that leads to a specific phenotypic response simultaneously induces an inhibitor of that phenotype. This type of regulation, referred to as incoherent feed-forward, is represented by the regulation of angiogenesis by miR-92a. Ischemia is a potent inducer of angiogenesis which serves to restore blood-flow to poorly perfused tissue sites. Yet ischemia has also been shown to cause the upregulation of miR-92a, which functions as a negative regulator of blood vessel growth. Systemic delivery of antisense oligonucleotides directed against miR-92a in a mouse model of hindlimb ischemia increases blood vessel growth and improves recovery from ischemic damage (Bonauer et al., 2009). The anti-angiogenic actions of miR-92a have been ascribed to its repression of α5 integrin, which exerts pro-angiogenic activity, but undoubtedly other targets also contribute to the effects of miR-92a on angiogenesis. It is noteworthy that miR-92a is encoded by the miR-17-92 cluster, which encodes miR-17, -18a, -19a, -20a, -19b and -92a. miR-18 and -19 have been reported to promote blood vessel growth and tumor angiogenesis through their ability to downregulate the anti-angiogenic factors thrombospondin-1 (Tsp-1) and connective tissue growth factor (CTGF) (Dews et al., 2010). Therefore, the ultimate consequence of expression of this miRNA cluster on angiogenesis is likely dependent on which of its targets plays a dominant role in a given tissue or disease context.
Numerous cardiovascular miRNAs establish positive feedback loops that influence phenotypic switching during disease (Figure 3D). Among this class of miRNAs is miR-21, which is upregulated by MAPK/ERK signaling in response to cardiac stress (Patrick et al., 2010; Thum et al., 2008). miR-21 targets Sprouty2 (Spry2), a negative regulator of the MAPK/ERK cascade, thereby further enhancing signaling through this pathway. Systemic delivery of cholesterol-modified miR-21 antagomirs has been reported to diminish MAPK signaling in cardiac fibroblasts and thereby completely prevent and reverse cardiac dysfunction, fibrosis, and hypertrophy in response to pressure overload (Thum et al., 2008). Surprisingly, genetic deletion of miR-21 in mice does not diminish heart disease in response to pressure overload or multiple other stresses (Patrick et al., 2010). Likewise, inhibition of miR-21 with LNA-modified oligonucleotides does not prevent pathological cardiac remodeling in response to stress. These disparate findings may indicate that the therapeutic activity of cholesterol-modified miR-21 inhibitors is due to selective uptake into a key cell type or off-target effects of these molecules. Alternatively, these observations raise the possibility that compensatory mechanisms may overcome the absence of miR-21 in the setting of chronic genetic deletion.
Cardiac stress also induces the expression of the miR-23a/27a/24-2 cluster via activation of the NFAT transcription factor (Lin et al., 2009), which serves as an effector of the calcium-sensitive phosphatase calcineurin (Molkentin et al., 1998). miR-23a, in turn, has been shown to repress expression of the muscle-specific ubiquitin ligase MuRF1, establishing a positive feedback loop that perpetuates stress-dependent cardiac hypertrophy (Figure 3D). Knockdown of miR-23a using miRNA inhibitors confers resistance to cardiac stress and diminishes hypertrophy through dampening of this positive feedback loop. Calcineurin/NFAT signaling also induces expression of miR-199a (da Costa Martins et al., 2010), which negatively regulates the inhibitory NFAT kinase Dyrk1a, thereby further amplifying pathogenic signaling in the heart (Figure 3D). Remarkably, inhibition of miR-199a has been reported not only to prevent cardiac hypertrophy and fibrosis in response to stress, but also to reverse these pathological processes through elevation of Dyrk1a activity (da Costa Martins et al., 2010).
Expression of the miR-23a/27a/24-2 cluster is also induced by VEGF-MAPK signaling, establishing another positive feedback loop involving these miRNAs (Cheng et al., 2009; Zhou et al., 2011). These miRNAs enhance vessel growth by promoting angiogenic signaling through targeting of the MAPK inhibitors Spry2 and Sema6A, which restrain angiogenesis (Figure 3D). These miRNAs have been implicated in pathological angiogenesis of the retina, such that their inhibition represents a potential therapeutic application for this disorder.
Finally, miRNAs have been described that target both positive and negative regulators of a cardiovascular stress response and thereby buffer pathway activity. This is exemplified by the miR-143/145 cluster which regulates phenotypic switching of smooth muscle cells (Boettger et al., 2009; Cheng et al., 2009; Cordes et al., 2009; Xin et al., 2009). Vascular injury and atherosclerosis are accompanied by de-differentiation and excessive proliferation of vascular smooth muscle cells, causing occlusion of the vascular lumen. Expression of miR-143/145 is elevated upon differentiation of smooth muscle cells and is diminished in injured and atherosclerotic vessels. Restoration of miR-145 expression in injured arteries through adenoviral delivery impedes smooth muscle proliferation and neointimal growth. Paradoxically, mice with genetic deletion of miR-143/145 are similarly resistant to pathological vascular remodeling in response to injury (Xin et al., 2009). Thus, both loss and gain of these miRNAs in vivo evoke similar phenotypes. The pro-differentiation and anti-proliferative actions of miR-143/145 have been attributed to negative regulation of genes involved in actin dynamics, cytoskeletal remodeling and actin gene regulation. Among these targets is the transcriptional repressor KLF4, which inhibits smooth muscle cell differentiation and promotes proliferation (Figure 3E). Opposing these actions is the myocardin coactivator MRTF-B, which stimulates smooth muscle differentiation, and is also targeted by miR-145. Although miR-143 and 145 have been reported to be essential for smooth muscle cell differentiation in vitro (Cordes et al., 2009), knockout mice lacking either or both of these miRNAs are born without obvious vascular abnormalities (Xin et al., 2009). However, contractility of the blood vessels of these mice is compromised, suggesting a subtle shift toward a less differentiated phenotype (Boettger et al., 2009). Taken together, the available data support a model whereby miR-143/145 do not play a major role in the initial differentiation of smooth muscle cells. Rather, through the balanced regulation of positive and negative regulators of smooth muscle differentiation and proliferation, this miRNA cluster maintains the phenotypic plasticity of this cell type, facilitating an appropriate (and sometimes pathologic) response to injury.
MicroRNAs in Mendelian and Complex Genetic Disease
The principle that miRNAs function primarily in governing cellular behavior under stress conditions in fully-developed tissues would suggest a limited role for these transcripts in highly-penetrant single-gene genetic disorders since these often manifest as developmental defects in childhood. Moreover, miRNAs represent a limited sequence space in which to accumulate mutations as compared to protein-coding genes and therefore phenotypically-relevant miRNA sequence variants would be expected to occur rarely. Finally, miRNAs and other noncoding RNAs are less sensitive to certain classes of mutations such as insertion/deletions (indels) and nonsense mutations. Whereas these types of variants lead to truncated open-reading frames in mRNAs, upstream indels would have little bearing on downstream critical functional sequences in noncoding RNAs. Thus, it is not surprising that few Mendelian disorders have been linked to mutations in miRNAs. Nevertheless, the small number of human miRNA loss-of-function phenotypes that have been uncovered are revealing, since they point to miRNAs that perform developmental functions.
The first human Mendelian disorder to be associated with miRNA loss-of-function was non-syndromic autosomal dominant progressive hearing loss. Linkage analysis of a large family segregating the phenotype mapped the causative mutation to 7q32 (Mencia et al., 2009), which harbors the miR-183/miR-96/miR-182 cluster. In light of the fact that expression of these miRNAs is highly enriched in sensory neurons, including the hair cells of the inner ear (Weston et al., 2006; Xu et al., 2007), this cluster represented an attractive candidate gene. Indeed, in the original family as well as in a second family, heterozygous point mutations in the seed sequence of miR-96 were identified that segregated with the deafness phenotype and are absent in control populations. At the same time, the identification of a mutation in the seed sequence of miR-96 in mice that results in a highly similar phenotype further bolstered the conclusion that altered function of this miRNA leads to adult-onset deafness (Lewis et al., 2009). It is noteworthy that all of the identified mutations occurred at different positions within the miR-96 seed sequence. Thus, while each creates a distinct new seed resulting in the potential acquisition of a unique cadre of novel targets, the similarities in the phenotypes created by each mutation implicate miR-96 haploinsufficiency, rather than gain-of-function, as the cause of the disease. Morpholino-mediated inhibition of miR-96 in zebrafish results in reduced numbers of hair cells, further supporting a loss-of-function pathogenetic mechanism (Li et al., 2010a). Finally, detailed analysis of the phenotype in miR-96 mutant mice demonstrated that the miRNA is not required for hair cell specification and embryonic development. Rather, the miRNA is needed for final postnatal maturation of hair cells, the failure of which leads to their degeneration (Kuhn et al., 2011; Lewis et al., 2009).
Another recently identified Mendelian disorder caused, in some cases, by loss-of-function of a miRNA is Feingold syndrome, an autosomal dominant phenotype characterized by microcephaly, short-stature, specific anomalies of the fingers and toes, developmental delay, and, less frequently, gastrointestinal atresia (Feingold et al., 1997). In over 2/3 of cases, the disease is caused by haploinsufficiency of MYCN (van Bokhoven et al., 2005). One cause of the remaining cases has now been shown to be heterozygous deletion of the miR-17-92 cluster (de Pontual et al., 2011). Comparative genomic hybridization (CGH) revealed the presence of deletions encompassing miR-17-92 and the first exon of the Glypican-5 (GPC5) gene in 3 unrelated Feingold syndrome patients lacking MYCN deletion. The critical contribution of the miR-17-92 cluster to the phenotype as opposed to GPC5 was demonstrated through analysis of mice with germline deletion of these miRNAs. miR-17-92+/− mice exhibit key features of Feingold syndrome including reduced body size, microcephaly, and the characteristic digital abnormalities. Given that the miR-17-92 cluster is known to be transactivated by the Myc family of transcription factors (Bui and Mendell, 2010), these findings support a role for the miR-17-92 cluster as a critical downstream effector of MYCN activity in skeletal development. Furthermore, it is worth noting that among all of the miRNA mutant mice analyzed thus far, miR-17-92 and miR-96 are unique in their manifestation of highly-penetrant developmental phenotypes in heterozygous animals. This is in keeping with the notion that the rare miRNAs with essential roles in development are most likely to be those associated with Mendelian phenotypes. The sensitivity of these phenotypes to miRNA halploinsufficiency likely reflects the existence of key targets whose activity is highly dose-dependent and whose mRNA concentration is limiting such that they are particularly sensitive to changes in miRNA levels.
In addition to rare loss-of-function mutations in miRNAs, single nucleotide polymorphisms (SNPs) that more subtly influence miRNA function have been described. In general, SNPs are underrepresented within miRNA-encoding loci, which is consistent with negative selection acting on these sequences (Saunders et al., 2007). Nevertheless, some polymorphic variants within miRNAs have been associated with human disease risk, most notably cancer (Ryan et al., 2010). While the majority of such studies await rigorous replication in large case-control studies, a promising example is miR-196a-2 which contains a SNP that has been associated with an elevated risk of developing lung, breast, liver, and gastric cancer in independent studies and may influence processing of the miRNA (Hoffman et al., 2009; Li et al., 2010b; Peng et al., 2010; Tian et al., 2009).
In contrast to miRNA-encoding loci, target sites encompass a much greater sequence space since each miRNA can potentially target hundreds of transcripts. Therefore functional alterations in these sequences would be expected to be more numerous. Indeed, ~20,000 single nucleotide polymorphisms (SNPs) in predicted miRNA binding sites have been catalogued (Bao et al., 2007; Georges et al., 2006). On the other hand, the gain or loss of a single target within a complex miRNA-regulated network might be expected to have more subtle phenotypic effects. Accordingly, while no Mendelian phenotypes have been linked to target site mutations, there is growing evidence that polymorphic sequence variants in target sites can contribute to complex genetic phenotypes. This concept was initially validated through analysis of quantitative trait loci that influence muscle mass in sheep (Clop et al., 2006). A variant in the 3′ UTR of Myostatin (MSTN) that creates a binding site for the muscle-specific miRNAs miR-1 and miR-206 was found to be highly associated with sheep muscularity. Since MSTN is a potent negative regulator of muscle mass, gain of miRNA targeting and subsequent downregulation of this protein provides a plausible explanation for increased muscle growth in animals harboring this allele. This mechanism was first translated to human populations through analysis of Tourette’s syndrome where a mutation in the 3′ UTR of the SLITRK1 gene that creates a binding site for miR-189 was reported to be associated with the disease (Abelson et al., 2005). However, subsequent studies failed to replicate this association and it has therefore remained controversial (Chou et al., 2007; Deng et al., 2006; Zimprich et al., 2008). Many further studies have linked miRNA target site polymorphisms to a variety of phenotypes including breast cancer, hypertension, asthma, and Parkinson’s disease (Sethupathy and Collins, 2008). For the majority of such studies, it remains to be seen whether the findings will ultimately be reproducible in independent analyses. However, one notable example appears to be a SNP that disrupts a predicted binding site for let-7 in the 3′ UTR of the KRAS oncogene. This variant was first identified by sequencing the KRAS 3′ UTR in non-small cell lung cancer (NSCLC) samples and found to be enriched in patients in two independent NSCLC case-control cohorts (Chin et al., 2008). Further case-control studies showed that the variant was also associated with ovarian cancer risk (Ratner et al., 2010) and reduced survival in oral cancer (Christensen et al., 2009). Despite the reproducibility of this association, questions remain regarding the mechanism through which this allele influences cancer risk. In vitro reporter assays suggest that the risk variant could lead to higher KRAS expression but this has not been confirmed in human tissue samples. Moreover, the SNP is predicted to base-pair with the 3′ end of let-7 which, according to most generally accepted targeting rules, would not be expected to dramatically influence miRNA-mediated regulation. Finally, it remains possible that this allele is in linkage disequilibrium with the true causative variant that is driving the associations. These caveats underscore the difficulty in establishing that miRNA target site variants truly modify disease risk by altering miRNA regulation versus other potential mechanisms. Ideally, animal models could be constructed to test proposed mechanisms directly in vivo, yet given the challenges associated with discerning phenotypes resulting from complete loss-of-function of individual miRNAs, it seems unlikely that effects of these mutations will be easily observable in genetically-engineered mice.
Lastly, genes that influence miRNA biogenesis or function represent an additional source of genetic variation that can potentially influence disease. This concept was convincingly demonstrated in a series of large genome-wide association studies (GWAS) designed to uncover variants associated with the age of onset of menarche, other traits associated with puberty, and height (Hartge, 2009; Lettre et al., 2008). Five independent studies, each analyzing over 15,000 individuals, found strong association signals near the LIN28B gene, a known negative regulator of let-7 biogenesis. Furthermore, transgenic expression of LIN28B in mice increased body size and delayed onset of puberty, strongly supporting the findings in human populations (Zhu et al., 2010). Consistent with a miRNA-dependent mechanism underlying these phenotypes, upregulated genes in tissues from the transgenic animals were enriched for let-7 targets. These studies exemplify the strength of genetic analyses in highly powered case-control studies coupled with functional validation in robust animal models to convincingly pinpoint pathways that regulate human phenotypes. More studies of this nature are needed to further demonstrate a role for miRNAs and their targets in genetically complex human disease.
Extracellular miRNAs as biomarkers of disease
A particularly intriguing, but poorly understood, aspect of the biology of miRNAs is their presence in numerous body fluids, including serum, plasma, saliva, and amniotic fluid (Cortez et al., 2011). miRNAs in serum correlate with the presence of hematologic malignancies and solid tumors and have been reported to be of value for early detection of various types of cancer, preceding diagnosis by conventional methods (Boeri et al., 2011). Circulating miRNAs have also been correlated with a variety of cardiovascular disorders, including myocardial infarction, heart failure, and acute coronary syndrome (Di Stefano et al., 2011; Tijsen et al., 2010). While these studies point to extracellular miRNAs as potential biomarkers for disease, it should be pointed out that most studies to date have involved relatively small numbers of patients, underscoring the need for larger prospective human studies to more accurately assess the clinical value of such measurements.
Despite the uncertainty regarding their clinical utility, the existence of extracellular miRNAs raises interesting questions about their origins and functions. In order to elucidate the biologic functions of circulating miRNAs, it will be important to define their sources to determine whether they are released in an uncontrolled manner from injured or diseased cells or if they are secreted in a regulated fashion as a compensatory response of tissues to stress. Consistent with the latter scenario, in some cases the profile of secreted miRNAs does not directly reflect the miRNA composition of the cell, suggesting regulation of miRNA release or cellular retention (Collino et al., 2010). miRNAs are secreted from cells within lipoprotein complexes and small membranous vesicles known as exosomes. Recently, the majority of miRNAs in plasma were reported to be associated with Ago2 ribonucleo-protein complexes in a non-vesicular fraction, suggesting that miRNAs are released within functional miRNA-induced silencing complexes (Arroyo et al., 2011). The mechanisms that govern selection of miRNAs for incorporation into different extracellular complexes under conditions where miRNA release is regulated will be an important area for future investigation. Once released, it remains to be determined whether circulating miRNAs in mammals act at a distance in inter-tissue communication, perhaps exhibiting hormone-like actions. Given that miRNAs typically act stoichiometrically against mRNA targets, it seems unlikely that the low levels of circulating miRNAs would achieve sufficient concentrations in distal target tissues to participate in target repression. Nevertheless, it remains formally possible that selective uptake or another yet-to-be characterized mechanism might allow sufficient intra-cellular accumulation of a circulating miRNA to result in productive target engagement.
MicroRNAs as Therapeutics
As described in detail throughout this review, a predominant paradigm that has emerged from miRNA gain- and loss-of-function studies is that miRNA dysregulation is well-tolerated in normal tissues yet can profoundly influence the behavior of cells and tissues experiencing pathologic stress. It follows from this concept that miRNA inhibition or delivery may provide a highly potent means to modulate a disease process while avoiding unwanted toxic effects in normal tissues. This potential for a wide therapeutic window has stimulated significant effort to develop miRNA-targeted therapeutics.
As discussed above, miRNAs are readily inhibited by antisense oligonucleotides and a variety of modifications have been developed to enhance the specificity, potency, and bioavailability of these antimiRs. Antisense oligonucleotides with the LNA modification, ranging from 7 to ~20 nucleotides in length, can be delivered systemically by intravenous, intraperitoneal, or subcutaneous injection with little or no toxicity (Stenvang et al., 2008). Thus far, methods for oral delivery have not been optimized. Upon systemic delivery, the vast majority of inhibitory LNA oligonucleotide clears through the liver and kidney (Obad et al., 2011). However, sufficient uptake to achieve therapeutic efficacy has been reported in the heart, vascular system, and immune system in rodents.
The most advanced miRNA therapeutic developed to date is an LNA-modified antimiR directed against miR-122 as a treatment for hepatitis C virus (HCV) infection. miR-122 performs an incompletely understood function that is essential for HCV replication and inhibition of this miRNA suppresses viral replication in experimental model systems (Jopling et al., 2005). Likewise, subcutaneous delivery of the LNA miR-122 inhibitor effectively suppresses HCV replication in African green monkeys and is currently being tested in humans (Elmen et al., 2008; Lanford et al., 2010). Results reported thus far for human Phase II studies have shown dose-dependent, prolonged viral reduction of 2 to 3 logs from baseline in HCV RNA after four weeks of treatment, without evidence of toxicity.
Not surprisingly, many miRNAs appear to play beneficial rather than pathologic roles in settings of disease. Thus, the development of miRNA mimics represents an important therapeutic goal. Unlike miRNA inhibitors, the use of injectable, naked miRNA mimics has remained problematic. Challenges to the development of such molecules relate, in part, to the necessity to deliver synthetic RNA duplexes in which one strand, the “guide”, is identical to the miRNA of interest, whereas the complementary strand, or “passenger”, is modified so as to facilitate stability and cellular uptake. Aside from difficulties in cellular uptake of double-stranded mimics, the passenger strand has the potential to counter-productively act as an antimiR. On the other hand, miRNA mimicry has been successfully achieved in rodents by packaging synthetic miRNA duplexes within lipid nanoparticles. Using this approach, systemic delivery of miRNA mimics has been successfully applied in multiple experimental tumor models in mice, resulting in tumor suppression without apparent toxicity in normal tissues (Pramanik et al., 2011; Trang et al., 2011). Adeno-associated virus (AAV) has also been demonstrated to represent an efficacious delivery platform for miRNAs, especially with AAV serotypes that display tropism toward specific tissues such as the liver (Kota et al., 2009).
Despite the importance of miRNAs in disease, and encouraging preliminary studies of antimiRs and miRNA mimics in animal models, many challenges must be overcome prior to the full realization of the promise of miRNA-based therapeutics. Efficient and selective uptake of antimiRs and mimics into different tissues has not been optimized. Moreover, it is conceivable that miRNAs that promote disease in one tissue might play protective roles in another, as illustrated by miR-26a, which suppresses hepatocellular carcinoma (Kota et al., 2009) but may promote gliomagenesis (Huse et al., 2009). The safe application of inhibitors or mimics that target or augment the activity such miRNAs will therefore require methods for tissue-specific delivery. Accordingly, methods for controlled delivery of miRNA inhibitors or mimics through ligand- or antibody-directed targeting are under development. In addition, traditional drug development necessitates a demonstration of correlation between target engagement and therapeutic efficacy. In the case of miRNAs, such correlations are difficult to establish as it is not currently possible to determine the precise concentration of effective antimiR or miRNA mimic in a tissue. Oligonucleotides may be sequestered in various intracellular and extracellular compartments, creating uncertainty with respect to dose-response relationships. In addition, since miRNAs act by modestly repressing myriad high and low affinity targets to evoke their effects, the use of gene expression changes as a surrogate read-out for miRNA inhibition or hyperactivity can provide a qualitative, not quantitative, estimation of miRNA activity. Thus, the development of methods to precisely measure miRNA activity in clinical samples represents an important future goal. Another challenge relates to the intriguing observation that, in some cases, short-term chemical inhibition of a miRNA has been found to result in a beneficial therapeutic response whereas genetic deletion of the miRNA has no influence on the disease process. This scenario is exemplified by miR-21. Administration of inhibitors that target this miRNA prevent pathologic cardiac remodeling yet germ-line deletion of miR-21 has no effect on cardiovascular pathology (Patrick et al., 2010; Thum et al., 2008). While these disparate outcomes may indicate that the beneficial effects of miR-21 inhibitors are due to selective uptake or activity of antimiRs in specific cell types or to off-target effects, another possibility is that chronic loss-of-function of the miRNA results in adaptive responses within the miRNA target network that compensate for its absence. Such theoretical effects raise interesting questions regarding the consequences of acute versus long-term treatments with miRNA inhibitors in the settings of disease. To date, most studies in animals have involved miRNA inhibition over relatively short periods of time. Whether therapeutic efficacy of miRNA inhibitors will decline with time as target networks compensate for diminished miRNA activity remains to be determined.
Looking to the future
The realization that miRNAs play central roles in disease has provided a new perspective on our understanding of pathophysiologic mechanisms and has offered a new therapeutic modality for disease modification. Moreover, the identification of the mRNA targets that mediate the actions of miRNAs in disease pathways can reveal previously unrecognized components of cellular stress responses that may serve as targets for more traditional drug development. The ability to modulate miRNA activity through the systemic delivery of miRNA inhibitors or mimics without toxicity provides unprecedented opportunities for intervening in disease processes. While challenges remain in this regard, the pace of this field suggests that new discoveries are forthcoming.
Acknowledgments
We thank Kathryn O’Donnell and members of the Mendell and Olson laboratories for helpful comments on the manuscript. J.T.M. is supported by the Cancer Prevention and Research Institute of Texas (CPRIT) and the National Institutes of Health (NIH) (R01CA120185 and P01CA134292). E.N.O. is supported by grants from the NIH (R01HL77439, R01HL093039, and U01HL100401), the Robert A. Welch Foundation, the American Heart Association-Jon Holden DeHaan Foundation, The D.W. Reynolds Center for Clinical Cardiovascular Research, The Leducq Transatlantic Network of Excellence in Cardiovascular Research Program, and CPRIT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- Alexiou P, Maragkakis M, Papadopoulos GL, Reczko M, Hatzigeorgiou AG. Lost in translation: an assessment and perspective for computational microRNA target identification. Bioinformatics. 2009;25:3049–3055. doi: 10.1093/bioinformatics/btp565. [DOI] [PubMed] [Google Scholar]
- Alvarez-Saavedra E, Horvitz HR. Many families of C. elegans microRNAs are not essential for development or viability. Current Biol. 2010;20:367–373. doi: 10.1016/j.cub.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, Zamore PD. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. 2011;108:5003–5008. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Zhou M, Wu L, Lu L, Goldowitz D, Williams RW, Cui Y. PolymiRTS Database: linking polymorphisms in microRNA target sites with complex traits. Nucleic Acids Res. 2007;35:D51–54. doi: 10.1093/nar/gkl797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeri M, Verri C, Conte D, Roz L, Modena P, Facchinetti F, Calabro E, Croce CM, Pastorino U, Sozzi G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc Natl Acad Sci USA. 2011;108:3713–3718. doi: 10.1073/pnas.1100048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Inv. 2009;119:2634–2647. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Current Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, et al. MicroRNA-29 in Aortic Dilation: Implications for Aneurysm Formation. Circ Res. 2011;109:1115–1119. doi: 10.1161/CIRCRESAHA.111.255737. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- Brenner JL, Jasiewicz KL, Fahley AF, Kemp BJ, Abbott AL. Loss of individual microRNAs causes mutant phenotypes in sensitized genetic backgrounds in C. elegans. Current Biol. 2010;20:1321–1325. doi: 10.1016/j.cub.2010.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui TV, Mendell JT. Myc: Maestro of MicroRNAs. Genes and Cancer. 2010;1:568–575. doi: 10.1177/1947601910377491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Inv. 2009;119:2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, Jung J, Gao P, Dang CV, Beer MA, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci USA. 2009;106:3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–233. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, Muller RU, Straka E, Su L, Burki EA, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou IC, Wan L, Liu SC, Tsai CH, Tsai FJ. Association of the Slit and Trk-like 1 gene in Taiwanese patients with Tourette syndrome. Ped Neurol. 2007;37:404–406. doi: 10.1016/j.pediatrneurol.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Christensen BC, Moyer BJ, Avissar M, Ouellet LG, Plaza SL, McClean MD, Marsit CJ, Kelsey KT. A let-7 microRNA-binding site polymorphism in the KRAS 3′ UTR is associated with reduced survival in oral cancers. Carcinogenesis. 2009;30:1003–1007. doi: 10.1093/carcin/bgp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Brennecke J, Stark A. Denoising feedback loops by thresholding--a new role for microRNAs. Genes Dev. 2006;20:2769–2772. doi: 10.1101/gad.1484606. [DOI] [PubMed] [Google Scholar]
- Collino F, Deregibus MC, Bruno S, Sterpone L, Aghemo G, Viltono L, Tetta C, Camussi G. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One. 2010;5:e11803. doi: 10.1371/journal.pone.0011803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Bueso-Ramos C, Ferdin J, Lopez-Berestein G, Sood AK, Calin GA. MicroRNAs in body fluids--the mix of hormones and biomarkers. Nature Rev Clin Oncol. 2011;8:467–477. doi: 10.1038/nrclinonc.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, Croce CM. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci USA. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, Thannickal VJ, Cardoso WV, Lu J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Resp Cell and Mol Biol. 2011;45:287–294. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa Martins PA, Salic K, Gladka MM, Armand AS, Leptidis S, el Azzouzi H, Hansen A, Coenen-de Roo CJ, Bierhuizen MF, van der Nagel R, et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol. 2010;12:1220–1227. doi: 10.1038/ncb2126. [DOI] [PubMed] [Google Scholar]
- de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, Van Haeringen A, Genevieve D, Goldenberg A, Oufadem M, et al. Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat Genet. 2011;43:1026–1030. doi: 10.1038/ng.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Le WD, Xie WJ, Jankovic J. Examination of the SLITRK1 gene in Caucasian patients with Tourette syndrome. Acta Neurol Scand. 2006;114:400–402. doi: 10.1111/j.1600-0404.2006.00706.x. [DOI] [PubMed] [Google Scholar]
- Dews M, Fox JL, Hultine S, Sundaram P, Wang W, Liu YY, Furth E, Enders GH, El-Deiry W, Schelter JM, et al. The myc-miR-17~92 axis blunts TGF{beta} signaling and production of multiple TGF{beta}-dependent antiangiogenic factors. Cancer Res. 2010;70:8233–8246. doi: 10.1158/0008-5472.CAN-10-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano V, Zaccagnini G, Capogrossi MC, Martelli F. microRNAs as peripheral blood biomarkers of cardiovascular disease. Vasc Pharm. 2011;55:111–118. doi: 10.1016/j.vph.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R. A parsimonious model for gene regulation by miRNAs. Science. 2011;331:550–553. doi: 10.1126/science.1191138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjarn M, Hansen HF, Berger U, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- Farazi TA, Juranek SA, Tuschl T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development. 2008;135:1201–1214. doi: 10.1242/dev.005629. [DOI] [PubMed] [Google Scholar]
- Feingold M, Hall BD, Lacassie Y, Martinez-Frias ML. Syndrome of microcephaly, facial and hand abnormalities, tracheoesophageal fistula, duodenal atresia, and developmental delay. Am J Med Genet. 1997;69:245–249. [PubMed] [Google Scholar]
- Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–284. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges M, Clop A, Marcq F, Takeda H, Pirottin D, Hiard S, Tordoir X, Caiment F, Meish F, Bibe B, et al. Polymorphic microRNA-target interactions: a novel source of phenotypic variation. Cold Spring Harb Symp Quant Biol. 2006;71:343–350. doi: 10.1101/sqb.2006.71.056. [DOI] [PubMed] [Google Scholar]
- Hartge P. Genetics of reproductive lifespan. Nat Genet. 2009;41:637–638. doi: 10.1038/ng0609-637. [DOI] [PubMed] [Google Scholar]
- Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E, Olson EN. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. 2010;18:282–293. doi: 10.1016/j.ccr.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- Hoffman AE, Zheng T, Yi C, Leaderer D, Weidhaas J, Slack F, Zhang Y, Paranjape T, Zhu Y. microRNA miR-196a-2 and breast cancer: a genetic and epigenetic association study and functional analysis. Cancer Res. 2009;69:5970–5977. doi: 10.1158/0008-5472.CAN-09-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. 2011 doi: 10.1161/CIRCRESAHA.111.244442. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, Sohn-Lee C, le Sage C, Agami R, Tuschl T, et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Shi J, Budhu A, Yu Z, Forgues M, Roessler S, Ambs S, Chen Y, Meltzer PS, Croce CM, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361:1437–1447. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Kent OA, Chivukula RR, Mullendore M, Wentzel EA, Feldmann G, Lee KH, Liu S, Leach SD, Maitra A, Mendell JT. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010;24:2754–2759. doi: 10.1101/gad.1950610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Kuhn S, Johnson SL, Furness DN, Chen J, Ingham N, Hilton JM, Steffes G, Lewis MA, Zampini V, Hackney CM, et al. miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells. Proc Natl Acad Sci USA. 2011;108:2355–2360. doi: 10.1073/pnas.1016646108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen CZ, Kuo CJ. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, Eyheramendy S, Voight BF, Butler JL, Guiducci C, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MA, Quint E, Glazier AM, Fuchs H, De Angelis MH, Langford C, van Dongen S, Abreu-Goodger C, Piipari M, Redshaw N, et al. An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet. 2009;41:614–618. doi: 10.1038/ng.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Kloosterman W, Fekete DM. MicroRNA-183 family members regulate sensorineural fates in the inner ear. J Neurosci. 2010a;30:3254–3263. doi: 10.1523/JNEUROSCI.4948-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Li ZG, Song XX, Liu CF. A variant in microRNA-196a2 is associated with susceptibility to hepatocellular carcinoma in Chinese patients with cirrhosis. Pathology. 2010b;42:669–673. doi: 10.3109/00313025.2010.522175. [DOI] [PubMed] [Google Scholar]
- Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol. 2007;27:2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Bezprozvannaya S, Shelton JM, Frisard MI, Hulver MW, McMillan RP, Wu Y, Voelker KA, Grange RW, Richardson JA, et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J Clin Inv. 2011;121:3258–3268. doi: 10.1172/JCI46267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467:86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- Mencia A, Modamio-Hoybjor S, Redshaw N, Morin M, Mayo-Merino F, Olavarrieta L, Aguirre LA, del Castillo I, Steel KP, Dalmay T, et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009;41:609–613. doi: 10.1038/ng.355. [DOI] [PubMed] [Google Scholar]
- Mendell JT. miRiad roles for the miR-17–92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E. Therapeutic Inhibition of miR-208a Improves Cardiac Function and Survival During Heart Failure. Circulation. 2011;124:1537–1547. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, de Stanchina E, D’Andrea A, Sander C, Ventura A. Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23:2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, van Rooij E, Olson EN. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Inv. 2010;120:3912–3916. doi: 10.1172/JCI43604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Kuang Z, Sheng C, Zhang Y, Xu H, Cheng Q. Association of microRNA-196a-2 gene polymorphism with gastric cancer risk in a Chinese population. Dig Dis Sci. 2010;55:2288–2293. doi: 10.1007/s10620-009-1007-x. [DOI] [PubMed] [Google Scholar]
- Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW, 2nd, van Rooij E, Olson EN. miR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res. 2011;109:670–679. doi: 10.1161/CIRCRESAHA.111.248880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, Zavolan M, Stoffel M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc Natl Acad Sci USA. 2009;106:5813–5818. doi: 10.1073/pnas.0810550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramanik D, Campbell NR, Karikari C, Chivukula R, Kent OA, Mendell JT, Maitra A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol Cancer Ther. 2011;10:1470–1480. doi: 10.1158/1535-7163.MCT-11-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner E, Lu L, Boeke M, Barnett R, Nallur S, Chin LJ, Pelletier C, Blitzblau R, Tassi R, Paranjape T, et al. A KRAS-variant in ovarian cancer acts as a genetic marker of cancer risk. Cancer Res. 2010;70:6509–6515. doi: 10.1158/0008-5472.CAN-10-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders MA, Liang H, Li WH. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci USA. 2007;104:3300–3305. doi: 10.1073/pnas.0611347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethupathy P, Collins FS. MicroRNA target site polymorphisms and human disease. Trends Genet. 2008;24:489–497. doi: 10.1016/j.tig.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenvang J, Silahtaroglu AN, Lindow M, Elmen J, Kauppinen S. The utility of LNA in microRNA-based cancer diagnostics and therapeutics. Semin Cancer Biol. 2008;18:89–102. doi: 10.1016/j.semcancer.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- Tian T, Shu Y, Chen J, Hu Z, Xu L, Jin G, Liang J, Liu P, Zhou X, Miao R, et al. A functional genetic variant in microRNA-196a2 is associated with increased susceptibility of lung cancer in Chinese. Cancer Epidemiol Biomarkers Prev. 2009;18:1183–1187. doi: 10.1158/1055-9965.EPI-08-0814. [DOI] [PubMed] [Google Scholar]
- Tijsen AJ, Creemers EE, Moerland PD, de Windt LJ, van der Wal AC, Kok WE, Pinto YM. MiR423–5p as a circulating biomarker for heart failure. Circ Res. 2010;106:1035–1039. doi: 10.1161/CIRCRESAHA.110.218297. [DOI] [PubMed] [Google Scholar]
- Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, Weidhaas JB, Bader AG, Slack FJ. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Therapy. 2011;19:1116–1122. doi: 10.1038/mt.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, Celli J, van Reeuwijk J, Rinne T, Glaudemans B, van Beusekom E, Rieu P, Newbury-Ecob RA, Chiang C, Brunner HG. MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in Feingold syndrome. Nat Genet. 2005;37:465–467. doi: 10.1038/ng1546. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm RJ, Jr, Olson EN. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell. 2009;17:662–673. doi: 10.1016/j.devcel.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MD, Pierce ML, Rocha-Sanchez S, Beisel KW, Soukup GA. MicroRNA gene expression in the mouse inner ear. Brain Res. 2006;1111:95–104. doi: 10.1016/j.brainres.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Vernooy SY, Guo M, Hay BA. The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Current biol. 2003;13:790–795. doi: 10.1016/s0960-9822(03)00250-1. [DOI] [PubMed] [Google Scholar]
- Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J Biol Chem. 2007;282:25053–25066. doi: 10.1074/jbc.M700501200. [DOI] [PubMed] [Google Scholar]
- Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci USA. 2011;108:9184–9189. doi: 10.1073/pnas.1105398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Gallagher R, Ufret-Vincenty R, Li X, Olson EN, Wang S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23~27~24 clusters. Proc Natl Acad Sci USA. 2011;108:8287–8292. doi: 10.1073/pnas.1105254108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, Viswanathan SR, Takeuchi A, Grasemann C, Rinn JL, Lopez MF, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. 2010;42:626–630. doi: 10.1038/ng.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Hatala K, Riederer F, Stogmann E, Aschauer HN, Stamenkovic M. Sequence analysis of the complete SLITRK1 gene in Austrian patients with Tourette’s disorder. Psych Genet. 2008;18:308–309. doi: 10.1097/YPG.0b013e3283060f6f. [DOI] [PubMed] [Google Scholar]