

Abstract
Although 1,4-cyclohexadienes 2, obtained through the Birch reduction of arenes 1, have found widespread use as masked β-oxo carbonyl synthons 3, the possibility that 2,5-cyclohexadienones 5 might also be employed to the same end has been overlooked despite their ready availability. As part of our ongoing investigation of the synthetic chemistry of nitrenium ions, we have developed a novel and efficient strategy for the stereoselective preparation of di- and trisubstituted azetidinone, pyrrolidinone and piperidinone derivatives, which features the ozonolytic cleavage of azaspirocyclic 2,5-cyclohexadienones 12. For example, ozonolysis of spirodienone 12c in CH2Cl2 and reductive workup with dimethyl sulfide generated unstable β-formyl ester 21 whereas cleavage in MeOH followed by reduction with thiourea led to hemiacetal 22. While both 21 and 22 partially decompose upon exposure to silica gel, they can be trapped in situ, with a variety of weakly basic nucleophiles, to usefully substituted products. The requisite spirodienone substrates are readily accessible through the nitrenium ion cyclization of alkyl ω–arylhydroxamates 10, proceeds with moderate to high diastereoselectivity.

Keywords: nitrenium ions; dearomatization; ozonolysis; 1,3-dicarbonyl synthon; N-heterocycles; hypervalent iodine; alkyl hydroxamates; N-hydroxy lactams; hydroxamic acid; azetidinones; pyrrolidinones and piperidinones
Introduction
The synthetic equivalency of benzenoid systems and carbonyl-based functional groups1 has been widely exploited since Woodward’s biosynthetically inspired use of a veratryl group as a masked hexa-2,4-dienedioic acid during his total synthesis of strychnine.2 Among the numerous reports concerning the oxidative cleavage of aromatic and heteroaromatic rings to form carboxylic acids, lactones and other related functionality,3 the use of disubstituted arenes 1 as latent 1,3-dicarbonyl groups 3 has proven to be particularly fruitful (Scheme 1). Rather than involving a direct oxidation, in this case, Birch reduction of 1 and cleavage of one or both double bonds in the resulting dihydroaromatic compound 2, most commonly through ozonolysis, serves to unmask the latent dicarbonyl system.4 This two-step protocol is tolerant of a wide range of aryl substituents (R1, R2) and has successfully been applied to the synthesis of β-keto esters,5 ω–formyl esters,6 malonate esters,7 β-diketones,8 β-formyl ketones,9 and even higher order polyketides.10
Scheme 1.

1,3-Disubstituted Arenes as Masked β–Oxo Carbonyl Synthons
In contrast to cyclohexadienes 2, there are only a handful of accounts documenting the ozonolytic cleavage of 2,5-cyclohexadienones 5, their electron deficient congeners, to form 1,3-dicarbonyl compounds 7 (Scheme 2).11 Given the complex nature of the reaction between dienones and ozone, the absence of reports concerning this transformation is perhaps understandable. The ozonolysis of cross-conjugated ketones was first studied by Harries, who noted that while treatment of phorone with excess ozone formed mesoxaldialdehyde (6) and acetone,12 exposure of this substrate to one equivalent of ozone, resulted in anomalous ozonolysis13 to produce β,β–dimethyl acrylic acid.
Scheme 2.

2,5-Cyclohexadienones as Potential Masked β–Oxo Carbonyl Synthons
More recently, Caspi11a–c and Rodig11d have independently investigated the ozonolysis of steroidal Δ1,4-3-ketones and found the outcome of this reaction to be similarly complex. Ozonolysis of 1-dehydrotestosterone acetate (8) in ethyl acetate,11c for example, provided keto aldehyde 9 in low yield together with nine other products, which were proposed to arise through anomalous ozonolysis of 8, Baeyer-Villiger oxidation of aldehyde 9,14 and the partial ozonolytic cleavage of the dienone ring.15
 |
(1) |
Notwithstanding these unpromising observations, successful ozonolytic cleavage of systems such as 5 remains synthetically appealing for a number of reasons, not least of which is the prospect that a route to 1,3-dicarbonyl systems, which bear an asymmetric quaternary stereocenter at C-2 might be established through the ozonolysis of differentially 4,4-disubstituted 2,5-cyclohexadienones 5.16 That the requisite dienones precursors 5 are readily available, through with the oxidation of phenols 4 and other electron-rich arenes,17 is an additionally attractive feature of this type of transformation.
Our interest in the ozonolytic cleavage of 2,5-cyclohexadienones was spurred by this laboratory's ongoing study of the synthetic application of acyl nitrenium ions.18,19,20 We have recently reported a stereoselective nitrenium ion spirocyclization involving the treatment of α- and β-substituted methyl 3-(methoxyphenyl)propiohydroxamates 10 (n = 1) with phenyliodine(III) bis(trifluoroacetate) (PIFA) to provide spirolactams 12 with useful levels of diastereoselectivity (Scheme 3).21
Scheme 3.

Spirocyclic Lactams as Masked Heterocyclic β–Oxo Carbonyl Synthons
Given the ease with which spirocycles 12 can be accessed and our ability to control the configuration of the spirocyclic center, we were prompted to consider the possibility that ozonolytic cleavage of the dienone ring would offer convenient access to lactams 13 that possess a nitrogen-bearing quaternary stereogenic center. Since spiroazetidinone, pyrrolidinone and piperidinone systems 12 (n = 0, 1, 2) are all accessible through the oxidative cyclization of alkyl hydroxamates 10,19b and given the fact that this reaction is tolerant of a wide range of aryl and sidechain substituents, this strategy potentially offers a route to a diverse array of target molecules. Among the heterocyclic systems that might potentially be accessed through application of the two-step sequence outlined in Scheme 3, the targets shown in Figure 1 are of particular interest.
Figure 1.

Synthetic targets potentially accessible via nitrenium ion cyclization-dienone cleavage strategy.
The marine natural product (−)-dysibetaine (14) is a neuroexcitotoxin, which is believed to bind to the glutamate receptors present in the central nervous system of mice,22,23 while kaitocephalin (15), an AMPA/NMDA antagonist isolated from the fungus Eupenicillium shearii PF1191, protects hippocampal neurons from kainate toxicity.24 Salinosporamide (17) and lactacystin (18), on the other hand, are both inhibitors of the proteasome, while the latter is additionally a potent cytotoxic agent.25 That the dienone cleavage strategy might also lend itself to the development of an expedient synthetic route to 4,4-disubstituted β-lactams (13, n = 0) is also appealing in view of the pharmacological importance of these systems. Tigemonam (19), for example, is a potent oral antibiotic that is resistant to β-lactamase-mediated hydrolysis.26
Having recently reported the application of the reaction sequence shown in Scheme 3 to the total synthesis of (−)-dysibetaine,27 in this paper, we now disclose a full account of our development of two complementary strategies for the ozonolytic cleavage of spirolactams 2,5-cyclohexadienones 12 and the implementation of this transformation, together with the stereoselective nitrenium ion spirocyclization reaction, as a valuable strategy for the synthesis of di- and trisubstituted lactams and N-hydroxy lactams.
Results and Discussion
1. Substrate Preparation
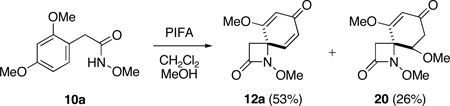
Given Caspie and Rodig’s observations concerning the cleavage of steroidal systems, our initial choice of spirodienone substrate was guided by the recognition that in order to promote efficient ozonolysis, it would be necessary to address the inherent electron-deficiency of these cross-conjugated ketones. Since it is well known that electron-releasing substituents increase the reactivity of alkenes towards ozone,28,29 we opted to examine the cleavage of dienone substrates activated by the presence of a β–methoxy substituent. Our investigation therefore commenced with the preparation of a series of dienones 12, which where accessed through the oxidative spirocyclization of alkyl hydroxamates 10,30 under conditions previously developed in this laboratory (Table 1).21 Thus, upon treatment of a solution of 10 in CH2Cl2 at −78 °C with 1.2 equivalents of phenyliodine(III) bis(trifluoroacetate) (PIFA) in methanol followed by warming to −15 °C, spirocyclization took place smoothly to afford the desired azaspirans 12 in good to excellent yield.31
Table 1.
Preparation of Dienone Substrates 12 through Spirocyclization of Alkyl Hydroxamates 10.a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | substrate | n | R1 | R2 | product | yield (%)b | drc |
| 1 | 10a | 0 | Me | H | 12a | 53d | - |
| 2 | 10a | 0 | Me | H | 12a | 67e | - |
| 3 | 10b | 0 | Bn | H | 12b | 86e | - |
| 4 | 10c | 1 | Me | H | 12c | 81 | - |
| 5 | 10d | 1 | Bn | H | 12d | 98 | - |
| 6 | 10e | 2 | Me | H | 12e | 92 | - |
| 7 | 10f | 2 | Bn | H | 12f | 83 | - |
| 8 | 10g | 1 | Me | Me | 12g | 85f | 92:8 |
| 9 | 10h | 1 | Me | t-Bu | 12h | 82f | 87:13 |
| 10 | 10i | 1 | Me | Bn | 12i | 98g | 92:8 |
| 11 | 10j | 1 | Me | Ph | 12j | 85h | 91:9 |
| 12 | 10k | 1 | Me | OTIPS | 12k | 99i | 90:10 |
| 13 | 10l | 1 | Me | NHBz | 12l | 85f | 91:9 |
| 14 | 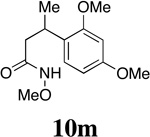 |
- | - | - |  |
90f | 80:20 |
| 15 |  |
- | - | - |  |
94 | - |
Reaction conditions: PIFA (1.2 equiv), CH2Cl2-MeOH (1:1), −78→−15 °C, 1.5 h; H2O, 10 min.
Isolated yield, after purification by flash chromatography.
Diastereomeric ratio (dr) determined by NMR analysis of the appropriate, characteristic proton signals in the unpurified product mixture.
Methanol adduct 20 (26%) was also isolated.31
Reaction performed in the absence of MeOH.
Isolated yield of anti diastereomer, after separation by flash chromatography and recrystallization.
Isolated yield of inseparable 92:8 mixture of anti and syn diastereomers.
Isolated yield of an inseparable 93:7 mixture of anti and syn diastereomers.
Isolated yield of an inseparable 90:10 mixture of anti and syn diastereomers.
As anticipated from our investigation of the stereochemistry of this reaction,21 substrates 10g–m underwent spirocyclization to provide the anti spirolactam diastereomers with reasonable selectivity. Figure 2 shows a possible rationale for this general observation. We believe that spirocyclization of the nitrenium ion generated from 10 preferentially proceeds via conformer B to form anti-12. Conformer A, on the other hand, is destabilized due to nonbonding interactions (benzylic strain)32 between the substituents on the side-chain and the ortho position of the aromatic ring.33
Figure 2.

Benzylic strain in the putative nitrenium ion intermediate leads to selective formation of anti-12.
After separation by chromatography and/or crystallization, the relative stereochemistry of the individual diastereomers was readily assigned on the basis of correlations observed in the 2D-NOESY spectra. In the case of products 12i and 12k, separation of the individual spirodienone diastereomers was not possible and consequently the mixtures were used in the subsequent step.
The successful cyclization of benzyl hydroxamates34 10b, 10d and 10f is noteworthy from a synthetic standpoint since it adds additional flexibility to our methodology: in these cases, hydrogenolysis of the benzyl ether, after dienone cleavage, provides access to N-hydroxy lactams, which are important bioactive targets (vide infra).35
2. Exploratory Dienone Ozonolysis Studies
Proceeding now to examine cleavage of the dienone system, we chose spiropyrrolidinone 12c for the purposes of this exploratory study (Scheme 4).
Scheme 4.

Exploratory Dienone Ozonolysis Studies
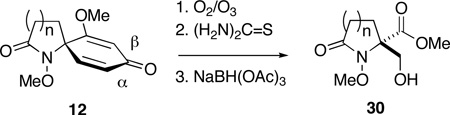
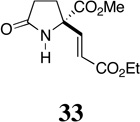
Exposure of a solution of 12c in CH2Cl2 (0.12 M) at −78 °C to a stream of ozonated oxygen for 30 min resulted in complete consumption of starting material and the formation of a less polar product, as indicated by thin-layer chromatography. Subsequent reduction of this ozonide intermediate with dimethyl sulfide (10 equiv) at room temperature for 12 h gave, upon concentration of the reaction mixture, β–formyl ester 21. While the spectroscopic data (1H NMR, 13C NMR, IR, and MS) from this compound were fully consistent with the structure assigned, purification of 21 by flash chromatography resulted in partial decomposition, and the product was isolated in moderate yield. The instability of 21 was further apparent from its rapid decomposition, upon standing at room temperature, to form an intractable mixture of products. That the sterically congested aldehyde group in this compound is highly reactive is also evident from its spontaneous reaction with methanol to generate adduct 22. Ozonolysis of 12c in methanol, followed by reduction of the putative α-methoxy hydroperoxide intermediate36 with thiourea (1.25 equiv),37 proceeded cleanly to yield hemiacetal 22 as a 1:1 mixture of epimers. While this reaction was highly efficient, as evidenced by NMR analysis of the crude product mixture, attempts to purify 22 by flash chromatography resulted in loss of methanol and formation of aldehyde 21, which was also isolated in diminished yield (60%). Fortunately in this case, there was no need for purification since filtration of the reaction mixture through a plug of celite after ozonolysis served to remove the precipitated thiourea S-dioxide and provided material of sufficient purity to be utilized in subsequent manipulations. In view of the instability of both aldehyde 21 and hemiacetal 22, we now examined the possibility that these compounds might be intercepted in situ, either through reduction or reaction with nucleophiles in general, to provide more tractable products (Scheme 5).
Scheme 5.

One-Pot Dienone Ozonolysis and Trapping a
a Reagents and conditions: (a) O3/O2, MeOH, −78 °C, 30 min; thiourea, −78 °C→rt, 30 min; (b) O3/O2, CH2Cl2, −78 °C, 30 min; Me2S, −78 °C→rt, 4 h; (c) H2NOH•HCl, NaOAc, MeOH, rt, 3 h (75%); (d) 2,4-DNPH, HCl (conc), MeOH, 3Å ms, reflux, 3 h (67%); (e) Me2C(CH2OH)2, TsOH, 3 Å m.s., CH2Cl2, reflux, 16 h (decomposition); (f) (CH2SH)2, HCl (conc), rt, 16 h (47%); (g) Ph3P=CHCO2Et, PhCH3, reflux, 2 h (88%); (h) Ph3P=CH2, THF, rt, 14 h (decomposition); (i) LiBH4, THF, rt, 16 h then Ac2O, py, rt 24 h (27%); (j) NaBH(OAc)3, AcOH, rt, 20 h (81%).
Encouragingly, ozonolysis of 12c in methanol, reduction with thiourea and then treatment of the reaction mixture with hydroxylamine hydrochloride (2.4 equiv) and sodium acetate (1.8 equiv) provided stable aldoxime 23 in good yield (75%). Likewise, hemiacetal 22 also underwent condensation with 2,4-dinitrophenylhydrazine to form hydrazone 24, albeit under somewhat more forcing conditions. Although attempts to prepare 4,4-dimethyldioxane acetal 25 under a variety of dehydrating conditions were unsuccessful, treatment of 22 with 1,2-ethanedithiol in the presence of concentrated hydrochloric acid did provide 1,2-dithiolane 26 albeit in modest yield (47%). Homologation of 22, on the other hand, proved to be more successful. Horner-Wittig reaction of 22 with (carboethoxymethylene)triphenylphosphorane (2.2 equiv) proceeded efficiently to provide enoate 27 as a single geometrical isomer.38 In this case, the ozonolysis reaction mixture was concentrated to remove methanol and the carboethoxymethylenation then carried out in toluene. Interestingly, 22 did not undergo methylenation with the ylide generated from methyl triphenylphosphonium bromide and n-butyllithium. In fact, treatment of either 21 or 22 with strongly basic nucleophiles, including Grignard and organozinc reagents, failed to provide the expected addition products. This general observation may be due to decomposition of these substrates through retro-aldol or retro-Claisen processes, which could occur upon deprotonation of the hydroxyl group, in the case of 22, or upon nucleophilic addition to the aldehyde group in 21.39
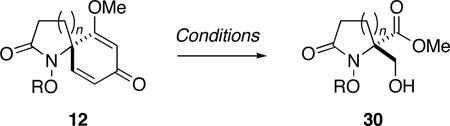
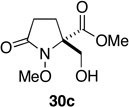
We next turned our attention to the reduction of the initial ozonolysis products and in particular to the transformation of these compounds to β–hydroxy ester 30c via selective reduction of the aldehyde and hemiacetal groups. While attempts to generate 30c through in-situ reduction of 21 with sodium borohydride gave unsatisfactory results, concentration of the ozonolysis reaction mixture and reduction with LiBH4 in THF provided the corresponding bis(hydroxymethyl)pyrrolidinone,40 which because of its polarity, was converted to bis-O-acetate 29 prior to purification. Treatment of hemiacetal 22 with NaBH4 or LiBH4, on the other hand, failed to provide any useful results. Reasoning that a weaker reducing agent might prove selective for the hemiacetal group, without affecting the methyl ester or promoting decomposition, sodium triacetoxyborohydride in acetic acid was evaluated.41 Thus, after ozonolysis of 12c and reductive workup with thiourea, the reaction mixture was concentrated under reduced pressure then treated with NaBH(OAc)3 (4 equiv) in acetic acid. Reduction of latent aldehyde 22 proceeded smoothly at room temperature and was complete within 24 h. Treatment of the reaction mixture with aqueous HCl for 20 min, extractive workup and purification by flash chromatography then provided β-hydroxy ester 30c in 81% overall yield from 12c (Table 2, entry 3).
Table 2.
Ozonolytic Cleavage of Spirodienones 12 and in-situ Reduction.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | substrate | R | product | method Aa | method Bb | ||
| timec | % yieldd | timec | % yieldd | ||||
 |
 |
||||||
| 1 | 12a | Me | 30a | 10 min | 53 | 30 min | 0e |
| 2 | 12b | Bn | 30b | 20 min | 82 | 30 min | 0e |
 |
 |
||||||
| 3 | 12c | Me | 30c | 1 h | 81 | 30 min | 76 |
| 4 | 12d | Bn | 30d | 1 h | 98 | 30 min | 94 |
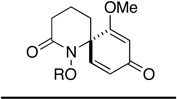 |
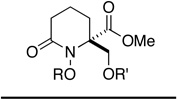 |
||||||
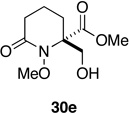
| 5 | 12e | Me | 30e | 3 h | 58 (R' = Ac)f | 30 min | 70 (R' = H) |
| 6 | 12f | Bn | 30f | 2 hf | 56 (R' = Ac)f | 30 min | 65 (R' = H) |
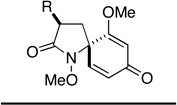 |
 |
||||||
| 7 | 12g | Me | 30g | 1 h | 53 | 30 min | 67 |
| 8 | 12h | t-Bu | 30h | 1 h | 59 | 30 min | 84 |
| 9 | 12i | Bn | 30i | 1 h | 91 | 30 min | 84 |
| 10 | 12j | Ph | 30j | 30 min | 81 | 30 min | 98 |
| 11 | 12k | OTIPS | 30k | 1 h | 91g | 30 min | 89 |
| 12 | 12l | NHBz | 30l | 1 h | 46 | 30 min | 84 |
| 13 | 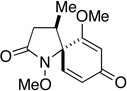 |
- | 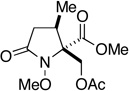 |
3.5 h | 0h | 1 h | 31i |
| 14 | 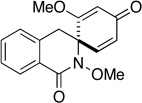 |
- | 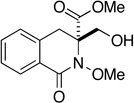 |
30 min | 84 | 30 min | 38 |
Method A: O3/O2, MeOH, −78°C, 30 min; thiourea, −78°C→rt; NaBH(OAc)3, AcOH, rt, 24 h.
Method B: NaBH4, CeCl3•7H2O, MeOH, 0 °C, 1 min; O3/O2, MeOH, −78 °C, 30 min; thiourea, −78 °C→rt, 1 h; NaBH(OAc)3, AcOH, rt, 24 h.
Duration of ozonolysis.
Overall, isolated yield, after purification of the final product by flash chromatography.
A complex, intractable mixture of products was isolated after ozonolysis and reduction.
Isolated yield, after conversion of alcohol to the corresponding O-acetate derivative and purification by flash chromatography.
Isolated as a 91:9 mixture of diastereomers.
A complex, intractable mixture of products was recovered.
Ozonolysis carried out for 1 h and the product converted to acetate 30m derivative before purification.
3. Establishing the Scope and Limitations of Dienone Cleavage
Having successfully established the practical viability of dienone cleavage and in-situ derivatization, we now proceeded to extend our study to include the remaining dienone substrates 12 in order to evaluate the scope and limitations of this chemistry. In view of the synthetic value of β–hydroxy esters of general structure 30, as both masked 2-hydroxymethyl amino acids42 and potential building blocks for the preparation of the natural products dysibetaine (14) and lactacystin (18), we opted to focus our attention on in-situ reduction of the ozonolysis products with sodium triacetoxyborohydride. The results of this study are detailed in Table 2.
Encouragingly, the reactivity of spiropyrrolidinone 12c towards ozone appeared to be general: cleavage of the homologous azaspiro[3.5]nonadienone (entries 1 & 2) and azaspiro[5.5]undecadienone (entries 5 & 6) systems proceeded to furnish the respective disubstituted azetidinone and piperidinone products 30. The successful ozonolysis spiroazetidinones 12a and 12b is of particular note since this transformation provides expedient access to usefully functionalized 4,4-disubstituted β–lactams.43 The disparity in yield observed during the formation of 30a and 30b appears to result from the greater polarity of the former product, which hampers its purification by flash chromatography. While the beneficial effect of O-benzyl groups, upon the efficiency of dienone cleavage, was also apparent during the ozonolysis of spiropyrrolidinones 12c and 12d, this effect did not extend to compounds 12e and 12f.
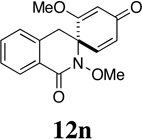
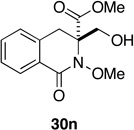
A remarkable structure-reactivity relationship emerged during the cleavage of substrates 12a–e. While the reaction of spiroazetidinones 12a and 12b with ozone was complete within 30 minutes, ozonolysis of the corresponding pyrrolidinones 12c and 12d was significantly slower, requiring 1 h. Even more strikingly, piperidinones 12e and 12f reacted very sluggishly with cleavage not reaching completion even after exposure to ozone for 3 h. Purification of the product mixtures, in the case of this latter pair of substrates, also proved to be problematic due to the presence of several, minor by-products. Fortunately, acetylation of the reaction mixtures, using Ac2O in pyridine, provided the corresponding O-acetate derivatives, which where more amenable to purification by flash chromatography. Rather unexpectedly, spiropiperidinone 12n proved to be considerably more reactive towards ozone than either 12e or 12f and underwent rapid ozonolysis to provide 30n in excellent yield, after in-situ reduction. The successful cleavage of 12n is notable since it opens a possible route to quaternary tetrahydroisoquinoline-3-carboxylic acid (Tic) derivatives,44 which are of interest as conformationally restricted amino acids. A more detailed discussion of the possible origins of this structure-reactivity pattern is presented in Section 5.
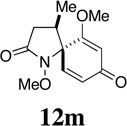
Having established the viability of cleavage in unsubstituted 4, 5 and 6-membered spirolactams, attention now turned to the α and β–substituted spiropyrrolidinones 30g–l. We were encouraged to find, in fact, that the presence of α–substituents in the pyrrolidinone ring was tolerated with cleavage giving the corresponding 2,4-disubstituted pyroglutamate derivatives with moderate to excellent efficiency. That attempts to ozonize 12m, the isomer of compound 12g, failed to provide any trace of compound 30m, is likely a consequence of the increased steric encumbrance imposed upon the dienone ring by the β-methyl substituent in this substrate.
4. An Alternative Strategy for Dienone Cleavage
The extended reaction times and modest yields encountered during the ozonolysis of substrates 12e and 12f, coupled with the failure of β–substituted spiropyrrolidinone 12m to undergo cleavage prompted a search for an alternative, more efficient strategy for cleavage of the dienone ring system. Given the greater reactivity of 1,4-cyclohexadienes towards ozone as compared to their electron-deficient congeners, the logical course of action at this stage seemed to be to reduce the dienone carbonyl group and subject the resulting dienylic alcohol(s) 31 to ozonolysis (Scheme 6).
Scheme 6.

Alternative, Two-Step Protocol for the Cleavage of Spirodienone Lactams 12.
Although the 1,2-reduction of 2,5-cyclohexadienones is well documented,45 a potential caveat with this plan was the propensity of the reduction products 31 to potentially undergo rearrangement during purification.46 Fortunately, reduction of dienone 12c, under Luche conditions (NaBH4, CeCl3, MeOH)47 proceeded rapidly at 0 °C to give a mixture of diastereomers 31, which could be isolated with excellent mass recovery. As these alcohols proved to be highly acid sensitive and decomposed upon exposure to silica gel, they were immediately submitted to ozonolysis without further purification (Table 2).
Gratifyingly, ozonolysis of diene 31 in CH2Cl2 at −78 °C occurred rapidly, with the starting material being consumed within 30 min as opposed to 1 h for dienone 12c. Sequential reduction of the peroxidic intermediates with thiourea (1.25 equiv) and sodium triacetoxyborohydride (5 equiv) in acetic acid then proceeded without incident to furnish β–hydroxy ester 30c in good yield. While this two-step protocol was slightly less efficient than direct dienone ozonolysis in this case (76 vs. 81%), its application to the remaining substrates in Table 2 proved to be considerably more rewarding. With the exception of 12a, 12b and 12n, improvements in yield and a decrease in reaction time were observed in all cases. Indeed, ozonolysis of the intermediate dienylic alcohols was complete within 30 min for all substrates, including spiropiperidinones 12e and 12f. The transformation of hindered 12m to compound 30m,48 albeit in low yield, under these conditions is also of note, since this product could not be prepared through direct ozonolysis of the dienone system. In the case of compounds 12a and 12n, complex mixtures of products where obtained, which we believe arise from decomposition of the intermediate dienylic alcohols prior to ozonolysis.
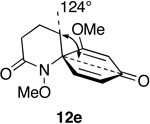
5. Assessment of Spirodienone Structure-Reactivity Relationships
Steric effects are known to play a significant role in determining the rate of 1,3-dipolar cycloaddition between ozone and unsaturated systems, including alkenes and arenes.49 We have rationalized the marked variation in reactivity towards ozone displayed by spirodienones 12 in terms of steric hindrance above (β) and below (α) the plane of the dienone ring system, which varies as a function of the size of the adjoining lactam ring (Table 3).
Table 3.
Relationship between Spirodienone Reactivity and Structure.
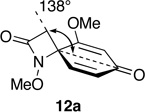

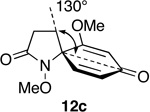
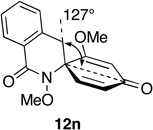
Optimized geometries were obtained through the MMMF force field implemented in Spartan.
Duration of ozonolysis.
Notwithstanding possible dipole and stereoelectronic effects,50 we believe that ozone approaches the spirocyclic cyclohexadienones from the α face, anti to the N-methoxyl group, which in all substrates bisects the dienone ring and thereby disfavors β attack. Our assumption that the N-OMe group is more sterically demanding than the methylene group appears to be born out by the observation that substrate 12m, which possesses a methyl substituent at the β position of the pyrrolidinone ring, is quite inert to ozone. Molecular mechanics minimization (MMMF) of the geometries of dienones 12a, 12c, 12e and 12n using the SPARTAN computational interface51 reveals that methylene group attached to the spirocenter progressively shields the β face of the adjoining dienone ring from ozone cycloaddition as the lactam ring increases in size. The increasing steric influence played by the methylene group in going from azetidinone to piperidinone is apparent from the decrease in the angle (ϕ) formed between the methylene group, spirocenter and the carbonyl carbon of the dienone ring. That the dienone rings in spiroazetidinones 12a and 12b are more accessible to attack is also evident from the fact only these substrates undergo conjugate addition of methanol during azaspirocyclization. While the dramatic increase in reactivity of dienylic alcohols 31, as compared to their corresponding dienone partners can be rationalized in terms of the attenuation of the electron-withdrawing properties of the carbonyl group, it is not immediately apparent why this should result in loss of the structure reactivity relationship.
6. Reductive Cleavage of N-Methoxy and N-Benzyloxy Lactams
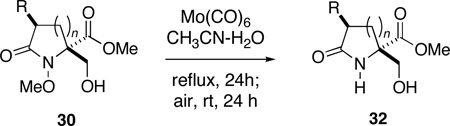
In order to confirm the practical utility of the nitrenium ion cyclization-dienone cleavage strategy, it was necessary now to address the issue of N-O bond scission in the cleavage products 30. Although the reduction of hydroxylamines, hydroxamic acids, and N-hydroxy lactams can readily be accomplished with a variety of reagents, metal ion-mediated reduction of N-alkoxy lactams is often more demanding since these systems lack an acidic chelation site.52 While this transformation has been accomplished with a number of reagents, including: hydrogenolysis over heterogeneous catalysts53 or reduction with Raney nickel;54 SmI2;55 sodium56 and aluminum amalgam;57 LDA;58 tert-butyldimethylsilyl triflate;59 Li/4,4’-di-tert-butylbiphenyl;60 and Birch reduction,61 few are entirely general. From previous studies, we have found the most dependable reducing agent for our relatively hindered substrates to be molybdenum hexacarbonyl.62
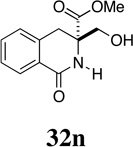
Reductive cleavage of 30 proceeds most efficiently when the substrate is heated with 1.2 equivalents of Mo(CO)6 in a degassed mixture of acetonitrile and water (15:1) for 24 h and the reaction then exposed to air at room temperature for an equivalent period of time (Table 4). Concentration of the black reaction mixture and purification of the residue by flash chromatography then provides the desired lactams 32 in good yield. Exposure of the reaction mixture to air greatly facilitates purification and failure to carry out this step often results in the contamination of the lactam products with unidentified, non-polar molybdenum complexes. As documented in Table 4, reductive cleavage of the N-methoxy lactams proceeded without incident to provide the corresponding pyroglutamate derivatives in good to excellent yield. The successful reduction of N-methoxy-2-azetidinone 30a under these mild conditions is notable since direct cleavage of this class of substrate has previously only been accomplished under more stringent conditions involving alkali metals in ammonia.61
Table 4.
Reductive N-O Bond Cleavage of N-Methoxy Lactams 30.
 | |||||
|---|---|---|---|---|---|
| entrya | substrate | R | n | product | yield (%)b |
| 1 | 30a | H | 0 | 32a | 75 |
| 2 | 30c | H | 1 | 32c | 88 |
| 3 | 30g | Me | 1 | 32g | 70 |
| 4 | 30h | t-Bu | 1 | 32h | 59 |
| 5 | 30i | Bn | 1 | 32i | 73 |
| 6 | 30j | Ph | 1 | 32j | 65 |
| 7 | 30k | OTIPS | 1 | 32k | 90 |
| 8 | 30l | NHBz | 1 | 32l | 77 |
| 9 | 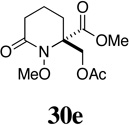 |
- | - | 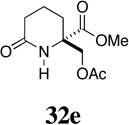 |
76 |
| 10 | 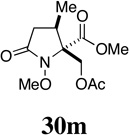 |
- | - | 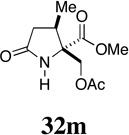 |
86 |
| 11 |  |
- | - |  |
74 |
| 12 |  |
- | - |  |
73 |
Reaction conditions: Mo(CO)6 (1.2 equiv), CH3CN, H2O reflux, 24 h; air, rt, 24 h.
Isolated yield, after purification by flash chromatography.
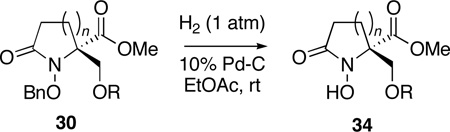
N-Hydroxy lactams are important synthetic targets, imbued with a wide range of biological activities, for which there are relatively few methods of preparation available.63 In this regard, the successful cyclization of the benzyl hydroxamate substrates 12b, 12d and 12f and the subsequent compatibility of the O-benzyl groups with the dienone cleavage protocol now provided an opportunity to access these target molecules, through selective hydrogenolysis of the benzyl ether in N-benzyloxy lactams 30 (Table 5).
Table 5.
Hydrogenolysis of N-Benzyloxy Lactams 30.
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | R | n | product | reaction time (h) |
yield (%)a |
| 1 | 30b | H | 0 | 34b | 0.1 | 99 |
| 2 | 30d | H | 1 | 34d | 0.5 | 90 |
| 3 | 30f | Ac | 2 | 34f | 1.5 | 99 |
Isolated yield, after purification by flash chromatography.
Hydrogenation of 30b, 30d and 30f in ethyl acetate proceeded smoothly at atmospheric pressure, in the presence of 10% Pd-C, to provide the desired N-hydroxy lactams 34 in excellent yield. In all cases, the reaction stopped after debenzylation and no trace of the lactam products, resulting from N-O bond cleavage,64 could be detected.
Conclusions
In conclusion, we have reported a novel and efficient strategy for the stereoselective preparation of di- and trisubstituted azetidinone, pyrrolidinone and piperidinone derivatives, which features the ozonolytic cleavage of azaspirocyclic 2,5-cyclohexadienones. Notable features of this methodology include a) the rapidity with which structural complexity is established; b) flexibility (4, 5, and 6-membered lactams and N-hydroxy lactams can be accessed) and; c) the accessibility of the spirodienone substrates, which can be prepared through the nitrenium ion cyclization of alkyl ω–arylhydroxamates with excellent efficiency and moderate to high diastereoselectivity. The results presented herein demonstrate, for the first time, that the ozonolytic cleavage of 2,5-cyclohexadienones is a synthetically viable and potentially powerful method. Since, in addition to the cyclization of nitrenium ions, spirocyclic 2,5-cyclohexadienones, including lactones, lactams and oxazolines, are readily accessible, through the oxidative spirocyclization of phenol derivatives, we anticipate that the chemistry reported herein may offer a general route to α,α–disubstituted heterocycles. Future directions for this work will include application of the current method to the synthesis of the natural products lactacystin and kaitocephalin as well as an examination of dienone cleavage in the context of other types of spirodienone.
Experimental Section
Reagents
Flash column chromatography was performed according to the method of Still65 using silica gel 60 (mesh 230–400). Phenyliodine(III) bis(trifluoroacetate) (PIFA) was prepared according to the procedure reported by Loudon.66 Solutions of sodium triacetoxyborohydride were prepared freshly by reacting sodium borohydride with acetic acid.
General Procedure A (Preparation of Spirodienones 12 in CH2Cl2). (±)-1,5-Methoxy-1-azaspiro[3.5]nona-5,8-diene-2,7-dione (12a) (Entry 2, Table 1)
To a stirred suspension of phenyliodine(III) bis(trifluoroacetate) (PIFA) (580 mg, 1.35 mmol, 1.2 equiv) in CH2Cl2 (1 mL), under an atmosphere of N2 at −78 °C, was added a cold (−78 °C) solution of alkyl hydroxamate 10a (253 mg, 1.12 mmol) in CH2Cl2 (5 mL) via cannula. The reaction mixture was then allowed to warm to −15 °C (bath temperature) over 1.5 h whereupon H2O (3 mL) was added and the cooling bath removed. After stirring for 10 min, the biphasic mixture was partitioned between CH2Cl2 (10 mL) and saturated aqueous NaHCO3 (5 mL). The aqueous phase was separated, extracted with CH2Cl2 (3 × 10 mL) and the combined organic extracts dried (Na2SO4), filtered, and concentrated under reduced pressure to provide a yellow oil. Purification by flash chromatography (SiO2, EtOAc/hexanes, 1:3) then afforded 12a (158 mg, 67%): white crystals; mp 100–103 °C (EtOAc/hexanes); Rf 0.55 (EtOAc); FTIR (film) υmax 1785, 1676, 1600, 1222 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.72 (d, J = 9.9 Hz, 1 H), 6.36 (dd, J = 1.6, 9.9 Hz, 1 H), 5.78 (d, J = 1.6 Hz, 1 H), 3.84 (s, 3 H), 3.74 (s, 3 H), 3.15 (d, J = 13.7 Hz, 1 H), 2.83 (d, J = 13.7 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 186.7, 169.3, 163.4, 142.2, 132.0, 105.7, 65.4, 61.2, 56.7, 43.8; HRMS-ESI calcd for C10H11NO4Na [M+Na]+ 232.0586, found 232.0591.
General Procedure B (Preparation of Spirodienones 12 in CH2Cl2-MeOH). (±)-1,6-Dimethoxy-1-azaspiro[4.5]deca-6,9-diene-2,8-dione (12c) (Entry 4, Table 1)
To a suspension of phenyliodine(III) bis(trifluoroacetate) (PIFA) (863 mg, 2.01 mmol, 1.2 equiv) in MeOH (2 mL), under an atmosphere of N2 at −78 °C, was added a cold (−78 °C) solution of 10c (400 mg, 1.67 mmol) in CH2Cl2 (8 mL) via cannula. The reaction mixture was then allowed to warm to −20 °C (internal temperature) over 1.5 h whereupon H2O (3 mL) was added and the cooling bath removed. After stirring for 10 min, the biphasic mixture was partitioned between CH2Cl2 (10 mL) and saturated aqueous NaHCO3 (5 mL). After separation, the aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the combined organic extracts dried (MgSO4), filtered and concentrated under reduced pressure to provide a yellow oil. Purification by flash chromatography over silica gel (EtOAc) then afforded 12c (301 mg, 81%): white crystals; mp 133–135 °C (EtOAc/hexanes); Rf 0.24 (EtOAc); FTIR (film) υmax 1728, 1665, 1633, 1602, 1369, 1226 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.61 (d, J = 9.9 Hz, 1 H), 6.28 (dd, J = 1.5, 9.9 Hz, 1 H), 5.61 (d, J = 1.5 Hz, 1 H), 3.76 (s, 3 H), 3.75 (s, 3 H), 2.65-2.55 (m, 1 H), 2.48-2.40 (m, 1 H), 2.26-2.19 (m, 1 H), 2.16-2.08 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 186.4, 173.2, 172.7, 144.3, 130.3, 103.2, 64.7, 62.8, 56.2, 27.3, 26.2; HRMS-ESI calcd for C11H13NO4Na [M+Na]+ 246.0742, found 246.0750.
Methyl (±)-2-Formyl-1-methoxy-5-oxo-pyrrolidine-2-carboxylate (21)
A stream of oxygen and ozone was passed through a solution of 12c (83 mg, 0.37 mmol) in CH2Cl2 (3 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 5 min, Me2S (546 µL, 7.44 mmol) added, the cooling bath removed and the mixture stirred room temperature for 12 h. The reaction mixture was then concentrated under reduced pressure and residual oil purified by flash chromatography (SiO2, EtOAc) to provide a crude sample of unstable 21 (45 mg, 61%): yellow oil; Rf 0.23 (EtOAc); IR (film) υmax 2951, 1734, 1296, 1246, 1059 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.92 (s, 1 H, CHO), 3.94 (s, 3 H), 3.89 (s, 3 H), 2.49-2.26 (m, 4 H); 13C NMR (100 MHz, CDCl3) δ 194.5, 172.6, 168.9, 64.5, 53.5, 42.6, 25.6, 23.3; HRMS-ESI calcd for C8H11NO5Na [M+Na]+ 224.0535, found 224.0538.
Methyl (2S*,6RS*)-2-(Hydroxymethoxymethyl)-1-methoxy-5-oxo-pyrrolidine-2-carboxylate (22)
A stream of oxygen and ozone was passed through a solution of 12c (160 mg, 0.72 mmol) in MeOH (5 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 5 min, thiourea (68 mg, 0.90 mmol, 1.25 equiv) was added, the cooling bath removed and the solution stirred at room temperature for 5 h. The reaction mixture was then filtered through a pad of Celite and the filtrate concentrated under reduced pressure to provide crude 22 (180 mg) as an approximately 1:1 mixture of hemiacetal diastereomers (by 1H NMR): colorless oil; Rf 0.50 (EtOAc); IR (film) υmax 3373 (br), 1741, 1702, 1694, 1440, 1253, 1059 cm−1; 1H NMR (500 MHz, CD3OD) δ (mixture of diastereomers) 5.11 (s, 0.5 H), 5.09 (s, 0.5 H), 3.80 (s, 3.8 H), 3.79 (s, 2.2 H), 3.42 (s, 1.2 H), 3.35 (s 1.5 H), 2.35-2.31 (m, 5 H); 13C NMR (125 MHz, CD3OD) δ (mixture of diastereomers) 174.5, 174.4, 170.8, 170.7, 96.5, 96.3, 71.7, 71.4, 63.4, 63.3, 55.1, 54.8, 52.3, 49.0, 26.2, 26.1, 20.7, 20.1; HRMS-ESI calcd for C9H15NO6Na [M+Na]+ 256.0797, found 256.0791.
(±)-2-(Hydroxyiminomethyl)-1-methoxy-5-oxo-pyrrolidine-2-carboxylic acid methyl ester (23)
A stream of oxygen and ozone was passed through a solution of 12c (200 mg, 0.90 mmol) in MeOH (5 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, thiourea (85 mg, 1.11 mmol) added and the mixture then allowed to warm to room temperature over 40 min. Sodium acetate (132 mg, 1.61 mmol) and NH2OH•HCl (149 mg, 2.15 mmol) were added and the mixture stirred at room temperature for 3 h. The reaction mixture was then concentrated under reduced pressure and the residue purified by flash chromatography (SiO2, EtOAc/hexane, 1:1) to provide 23 (146 mg, 75%) as a 12:1 mixture of geometrical isomers: yellow oil; Rf 0.68 (EtOAc); IR (film) υmax 3293 (br), 1745, 1703, 1439, 1268, 1070, 974 cm−1; 1H NMR (400 MHz, CDCl3) δ (major isomer) 8.96 (br s, 1 H), 7.74 (s, 1 H), 3.89 (s, 3 H), 3.83 (s, 3 H), 2.59-2.51 (m, 1 H), 2.47-2.42 (m, 2 H), 2.26-2.19 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ (major isomer) 173.1, 170.3, 145.9, 68.3, 64.4, 53.5, 26.0, 25.1; HRMS-ESI calcd for C8H12N2O5Na [M+Na]+ 239.0644, found 239.0647.
(±)-2-[(2,4-Dinitrophenyl)-hydrazonomethyl]-1-methoxy-5-oxo-pyrrolidine-2-carboxylic acid methyl ester (24)
A stream of oxygen and ozone was passed through a solution of 12c (178 mg, 0.80 mmol) in MeOH (4 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, thiourea (76 mg, 1.00 mmol) added, the cooling bath removed and the solution allowed to warm to room temperature over 40 min. After filtering the reaction mixture through a plug of Celite, the filtrate was sequentially treated with activated 3Å molecular sieve beads (500 mg), 2,4-dinitrophenylhydrazine (316 mg, 1.59 mmol) and concentrated HCl (300 µL). The reaction mixture was then heated at reflux for 1.5 h, whereupon it was cooled to room temperature, filtered through a plug of Celite and concentrated under reduced pressure. The resulting oil was purified by flash chromatography (SiO2, EtOAc/hexanes, 1:5) to provide hydrazone 24 (204 mg, 67%): orange crystals; mp 174–176 °C (EtOAc/hexanes); Rf 0.67 (EtOAc); FTIR (film) υmax 3296 (br), 1735, 1616, 1593, 1514, 1431, 1335, 751 cm−1; 1H NMR (500 MHz, CDCl3) δ 11.26 (s, 1 H), 9.10 (d, J = 2.5 Hz, 1 H), 8.34 (dd, J = 2.5, 9.5 Hz, 1 H), 7.89 (s, 1H, NH), 7.87 (d, J = 9.5 Hz, 1 H), 3.94 (s, 3 H), 3.90 (s, 3 H), 2.72-2.65 (m, 1 H), 2.61-2.45 (m, 2 H), 2.40-2.33 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 172.7, 170.1, 144.6, 144.3, 138.9, 130.3, 129.8, 123.2, 116.6, 69.3, 64.5, 53.7, 25.9, 25.4; HRMS-ESI calcd for C14H14N5O8 [M−H]− 380.0842, found 380.0844.
(±)-2-[1,3]Dithiolan-2-yl-1-methoxy-5-oxo-pyrrolidine-2-carboxylic acid methyl ester (26)
A stream of oxygen and ozone was passed through a solution of 12c (123 mg, 0.55 mmol) in MeOH (3 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, thiourea (52 mg, 0.69 mmol) added, the cooling bath removed and the solution then allowed to warm to room temperature over 40 min. The reaction mixture was then filtered through a plug of Celite and the filtrate concentrated under reduced pressure. The resulting colorless oil was taken up in CH2Cl2 (6 mL) and ethane-1,2-dithiol (58 µL, 0.69 mmol) and concentrated HCl (300 µL) were added sequentially. The reaction mixture was stirred for 16 h at room temperature, concentrated under reduced pressure and purified by flash chromatography (SiO2, EtOAc/hexanes, 1:5) to provide dithioacetal 26 (72 mg, 47%): colorless oil; Rf 0.66 (EtOAc); FTIR (film) υmax 1735, 1433, 1271, 1057 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.37 (s, 1 H), 3.90 (s, 3 H), 3.81 (s, 3 H), 3.32-3.19 (m, 4 H), 2.51-2.46 (m, 2 H), 2.38-2.29 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 173.6, 171.5, 72.0, 64.2, 55.5, 53.2, 39.4, 39.0, 26.4, 21.8; HRMS-ESI calcd for C10H16NO4S2 [M+H]+ 278.0521, found 278.0526.
Methyl (±)-E-2-(2-Ethoxycarbonylvinyl)-1-methoxy-5-oxo-pyrrolidine-2-carboxylate (27)
A stream of oxygen and ozone was passed through a solution of 12c (200 mg, 0.90 mmol) in MeOH (5 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, thiourea (85 mg, 1.11 mmol) added, the cooling bath removed and the solution then allowed to warm to room temperature over 40 min. The reaction mixture was then concentrated under reduced pressure to provide 22 as an oil, which was dissolved in toluene (5 mL). (Carboethoxymethylene)triphenylphosphorane (687 mg, 1.97 mmol) was then added and the mixture heated at reflux for 2 h. The reaction was then cooled, concentrated under reduced pressure and the resulting oil partitioned between CH2Cl2 (10 mL) and saturated aqueous NH4Cl (7 mL). The organic phase was separated and the aqueous portion extracted with CH2Cl2 (3 × 10 mL), The combined organic extracts were dried (Na2SO4), filtered, concentrated under reduced pressure and the residue purified by flash chromatography (SiO2, EtOAc/hexanes, 1:1) to provide 27 (213 mg, 88%) as a single geometrical isomer: yellow oil; Rf 0.52 (EtOAc); IR (film) υmax 1726, 1659, 1313, 1266, 1184, 1053, 979 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 16.0 Hz, 1 H), 6.10 (d, J = 16.0 Hz, 1 H), 4.20 (q, J = 7.2 Hz, 2 H), 3.93 (s, 3 H), 3.82 (s, 3 H), 2.44-2.15 (m, 4 H), 1.28 (t, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 172.2, 170.3, 165.5, 142.0, 123.5, 69.6, 64.2, 60.9, 53.4, 28.4, 25.7, 14.1; HRMS-ESI calcd for C12H17NO6Na [M+Na]+ 294.0954, found 294.0945.
Acetic acid (±)-2-acetoxymethyl-1-methoxy-5-oxo-pyrrolidin-2-ylmethyl ester (29)
A stream of oxygen and ozone was passed through a solution of 12c (148 mg, 0.66 mmol) in CH2Cl2 (4 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, Me2S (730 µL, 9.94 mmol) added, the cooling bath removed and the mixture stirred at room temperature for 4 h. The reaction mixture was then concentrated under reduced pressure, the residue dissolved in Et2O (5 mL) and this solution then treated with LiBH4 (72 mg). After stirring at room temperature for 16 h, the reaction was quenched with H2O (4 mL) and then stirred for an additional 1 h. The organic layer was then separated and the aqueous layer extracted with EtOAc (5 × 10 mL). The combined organic extracts were dried (Na2SO4), filtered through Celite and concentrated under reduced pressure. The resulting solid was dissolved in pyridine (1 mL) and Ac2O (500 µL) and this mixture then stirred at room temperature for 24 h. The reaction mixture was then concentrated under reduced pressure and the resulting residue purified by flash chromatography (SiO2, EtOAc) to provide 29 (47 mg, 27%): colorless oil; Rf 0.32 (EtOAc); IR (film) υmax 1743, 1724, 1225, 1045 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.26 (d, J = 11.6 Hz, 2 H), 4.12 (d, J = 11.6 Hz, 2 H), 3.84 (s, 3 H), 2.38 (m, 2 H), 2.10 (s, 6 H), 2.03 (m, 2 H); 13C NMR (100 MHz, CDCl3) δ 172.4, 170.3, 64.3, 64.2, 63.8, 26.3, 22.6, 20.7; HRMS-ESI calcd for C11H17NO6Na [M+Na]+ 282.0954, found 282.0958.
Methyl (±)-2-Hydroxymethyl-1-methoxy-5-oxo-pyrrolidine-2-carboxylate (30c)
A stream of oxygen and ozone was passed through a solution of 12c (139 mg, 0.62 mmol) in CH2Cl2 (7 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, Me2S (686 µL, 9.34 mmol) added, the cooling bath removed and the mixture stirred at room temperature for 4 h. The reaction mixture was then filtered through a plug of Celite and the filtrate concentrated under reduced pressure. The resulting residue dissolved in AcOH (2 mL) and this mixture added to a solution of NaBH(OAc)3 in AcOH (0.8 M, 7.8 mL). After stirring for 24 h at room temperature, the reaction was concentrated under reduced pressure, the residual oil diluted with CH2Cl2 (5 mL) and 2 M aqueous HCl (2 mL) added. After standing for 10 min, the organic layer was separated and the aqueous layer extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were concentrated under reduced pressure and the resulting oil purified by flash chromatography (SiO2, EtOAc) to provide 30c (62 mg, 49%): colorless oil; Rf 0.32 (EtOAc); FTIR (film) υmax 3410 (br), 1737, 1706, 1432, 1251, 1056, 969 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.97 (d, J = 11.9 Hz, 1 H), 3.87 (d, J = 11.9 Hz, 1 H), 3.84 (s, 3 H), 3.73 (s, 3 H), 3.35 (br s, 1 H), 2.38-2.34 (m, 2 H), 2.28-2.22 (m, 1 H), 2.08-2.03 (m, 1 H); 13C NMR (125 MHz, CDCl3) δ 173.8, 172.2, 70.0, 64.3, 62.9, 53.2, 26.7, 24.1; HRMS-EI calcd for C8H14NO5 [M+H]+ 204.0872, found 204.0881
General Procedure C (Ozonolysis-Reduction of Spirodienones 12). Methyl (±)-2-Hydroxymethyl-1-methoxy-4-oxo-azetidine-2-carboxylate (30a) (Entry 1, Table 2)
A stream of oxygen and ozone was passed through a solution of 12a (50 mg, 0.24 mmol) in MeOH (3 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, thiourea (23 mg, 0.30 mmol) was added and the solution then allowed to warm to room temperature over 30 min. After stirring for a additional 10 min, the reaction was concentrated under reduced pressure, the residue dissolved in AcOH (500 µL) and this mixture added to a solution of NaBH(OAc)3 in AcOH (0.51 M, 2 mL). After stirring for 24 h at room temperature, the reaction was concentrated under reduced pressure, the residual oil diluted with CH2Cl2 (5 mL) and 2 M aqueous HCl (2 mL) added. After standing for 10 min, the organic layer was separated and the aqueous layer extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were concentrated under reduced pressure and the resulting oil purified by flash chromatography (SiO2, EtOAc) to provide 30a (35 mg, 78%): colorless oil; Rf 0.44 (EtOAc); IR (film) υmax 3445 (br), 1778, 1742, 1072, 1031 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.16 (d, J = 12.4 Hz, 1 H), 4.02 (d, J = 12.4 Hz, 1 H), 3.90 (s, 3 H; OCH3), 3.84 (s, 3 H; OCH3), 2.93 (d, J = 13.8 Hz, 1 H), 2.88 (d, J = 13.8 Hz, 1 H) 2.51 (br s, 1 H); 13C NMR (100 MHz, CDCl3) δ 170.3, 164.4, 67.5, 65.0, 61.4, 53.0, 39.6; HRMS-ESI calcd for C7H11NO5Na [M+Na]+ 212.0535, found 212.0543.
General Procedure D (Ozonolysis-Reduction-Acetylation of Spirodienones 12). Methyl (±)-2-Acetoxymethyl-1-methoxy-6-oxo-piperidine-2-carboxylate (30e) (Entry 5, Table 2)
A stream of oxygen and ozone was passed through a solution of 12e (50 mg, 0.21 mmol) in MeOH (5 mL) at −78 °C for 3 h. The blue solution was then purged with a stream of argon for 10 min, thiourea (20 mg, 0.26 mmol) was added in one portion and the solution then allowed to warm to room temperature over 30 min. After stirring for an additional 10 min, the reaction was concentrated under reduced pressure, the residue dissolved in AcOH (500 µL). This solution was added to a solution of NaBH(OAc)3 in AcOH (0.17 M, 5 mL). After stirring for 24 h at room temperature, the reaction was concentrated under reduced pressure and the residue dissolved in pyridine (1 mL) and acetic anhydride (500 µL, 5 mmol). The mixture was stirred at room temperature for 8 h, then concentrated under reduced pressure and the residue purified by flash chromatography (SiO2, EtOAc) to provide 30e (32 mg, 58%) and starting material 12e (18 mg): white crystals; mp 72–74 °C (EtOAc/hexanes); Rf 0.37 (EtOAc); FTIR (film) υmax 1745, 1689, 1238, 1051 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.54 (d, J = 11.9 Hz, 1 H), 4.49 (d, J = 11.9 Hz, 1 H), 3.79 (s, 3 H), 3.78 (s, 3 H), 2.54-2.47 (m, 2 H), 2.19-2.16 (m, 1 H), 2.12 (s, 3 H), 2.11-2.05 (m, 1 H), 1.82-1.78 (m 2 H); 13C NMR (125 MHz, CD3OD) δ 171.0 (2 C), 170.7, 70.9, 63.9, 63.7, 53.5, 33.5, 31.3, 21.2, 18.1; HRMS-ESI calcd for C11H17NO6Na [M+Na]+ 282.0954, found 282.0958.
General Procedure E (Luche Reduction-Ozonolysis of Spirodienones 12). Methyl (2S*,4S*)-2-Hydroxymethyl-1-methoxy-5-oxo-4-phenyl-pyrrolidine-2-carboxylate (30j) (Entry 10, Table 2)
To a solution of 12j (117 mg, 0.39 mmol) and CeCl3•7H2O (117 mg, 0.39 mmol) in MeOH (8 mL) at 0 °C was added NaBH4 (16 mg, 0.41 mmol). After stirring for 1 min, the reaction was quenched with water (2 mL), concentrated under reduced pressure and remaining aqueous portion extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried (Na2SO4), filtered and concentrated to provide the unstable dienylic alcohol as a colorless oil, which was immediately subjected to ozonolysis. Thus, a stream of oxygen and ozone was passed through a solution of the dienylic alcohol in MeOH (4 mL) at −78 °C for 30 min. The blue solution was then purged with a stream of argon for 10 min, thiourea (37 mg, 0.49 mmol, 1.25 equiv) was added and the solution then allowed to warm to room temperature over 30 min. After stirring for an additional 30 min, the reaction was concentrated under reduced pressure, the residue dissolved in AcOH (1 mL) and this mixture added to a solution of NaBH(OAc)3 in AcOH (0.65 M, 3 mL, 5 equiv). After stirring for 24 h at room temperature, the reaction was concentrated under reduced pressure, the residual oil diluted with CH2Cl2 (10 mL) and 2 M aqueous HCl (5 mL) added. After standing for 20 min, the organic layer was separated and the aqueous layer extracted with CH2Cl2 (3 × 15 mL). The combined organic extracts were concentrated under reduced pressure and the resulting oil purified by flash chromatography (SiO2, EtOAc) to provide 30j (107 mg, 98%).
General Procedure F (Reductive Cleavage of N-Methoxy Lactams). Methyl (±)-2-Hydroxymethyl-4-oxo-azetidine-2-carboxylate (32a) (Entry 1, Table 4)
A mixture of 30a (210 mg, 1.11 mmol) and Mo(CO)6 (352 mg, 1.33 mmol, 1.2 equiv) in degassed CH3CN-H2O (15:1, 5 mL) was heated at reflux, under N2, for 24 h, whereupon the black reaction mixture was cooled to room temperature, opened to the atmosphere and stirred for 24 h. The reaction was then concentrated under reduced pressure and the resulting residue purified by flash chromatography (SiO2, EtOAc) to yield 32a (133 mg, 75%): colorless oil; Rf 0.37 (EtOAc); FTIR (film) υmax 3312 (br), 1750, 1277, 1228, 1045 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.80 (br s, 1 H), 4.14 (d, J = 11.7 Hz, 1 H), 3.85-3.82 (m, 4 H), 3.12 (d, J = 14.9 Hz, 1 H), 3.04 (d, J = 14.9 Hz, 1 H), 2.71 (br s, 1 H); 13C NMR (100 MHz, CD3OD) δ 172.4, 167.1, 65.1, 59.3, 53.5, 45.2; HRMS-ESI calcd for C6H9NO4 [M+Na]+ 182.0429, found 182.0426.
General Procedure G (Hydrogenolysis of N-Benzyloxy Lactams). Methyl (±)-1-Hydroxy-2-hydroxymethyl-4-oxo-azetidine-2-carboxylate (34b) (Entry 1, Table 5)
A mixture of 30b (40 mg, 0.15 mmol) and 10% Pd/C (2 mg) in EtOAc (2 mL) was stirred under an atmosphere of H2 for 10 min then filtered through a pad of Celite. The filtrate was concentrated under reduced pressure and the residue purified by flash chromatography (SiO2, EtOAc) to yield 34b (26 mg, 99%): colorless oil; Rf 0.25 (EtOAc); FTIR (film) υmax 3390 (br), 1770, 1745, 1247, 1037 cm−1; 1H NMR (400 MHz, CD3OD) δ 4.02 (d, J = 12.4 Hz, 1 H), 3.95 (d, J = 12.4 Hz, 1 H), 3.79 (s, 3 H), 2.89 (d, J = 13.2 Hz, 1 H), 2.82 (d, J = 13.2 Hz, 1 H); 13C NMR (100 MHz, CD3OD) δ 170.0, 164.7, 67.9, 58.4, 51.7, 38.0; HRMS-ESI calcd for C6H9NO5Na [M+Na]+ 198.0378, found 198.0373.
Supplementary Material
Acknowledgement
We thank the National Institutes of Health (GM-67176) for financial support.
Footnotes
Supporting Information Available. Experimental procedures and characterization data for all new compounds. This material is free of charge via the Internet at http://pubs.acs.org.
References and Footnotes
- 1.Ho T-L. Tactics of Organic Synthesis. 1st ed. New York: Wiley; 1994. pp. 50–53. [Google Scholar]
- 2.(a) Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K. J. Am. Chem. Soc. 1954;76:4749. [Google Scholar]; (b) Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K. Tetrahedron. 1963;19:247. [Google Scholar]
- 3.For recent, representative reports concerning the oxidative degradation of benzenoid systems, see: Audouard C, Barsukov I, Fawcett J, Griffith GA, Percy JM, Pintat S, Smith CA. Chem. Commun. 2004:1526. doi: 10.1039/b405067c. Lee YS, Choung WK, Kim KH, Kang TW, Ha DC. Tetrahedron. 2004;60:867. Miranda LSM, Vasconcellos MLAA. Synthesis. 2004:1767. Demir A, Sesenoglu O, Ulku D, Arici C. Helv. Chim. Acta. 2003;86:91. Jaroch S, Holscher P, Rehwinkel H, Sulzle D, Burton G, Hillmann M, McDonald FM. Bioorg. Med. Chem. Lett. 2003;13:1981. doi: 10.1016/s0960-894x(03)00351-2. Mander LN, Williams CM. Tetrahedron. 2003;59:1105. and references therein. Colletti SL, Li C, Fisher MH, Wyvratt MJ, Meinke PT. Tetrahedron Lett. 2000;41:7825. Bringmann G, Munchbach M, Michel M. Tetrahedron: Asymmetry. 2000;11:3167. Zhou W-S, Lu Z-H, Xu Y-M, Liao L-X, Wang Z-M. Tetrahedron. 1999;55:11959.
- 4.Birch AJ, Fitton F, Smith DCC, Steere DE, Stelfox AR. J. Chem. Soc. 1963:2209. [Google Scholar]
- 5.(a) Evans DA, Sjogren EB. Tetrahedron Lett. 1986;27:3119. [Google Scholar]; (b) Evans DA, Gauchet-Prunet JA, Carreira EM, Charette AB. J. Org. Chem. 1991;56:741. [Google Scholar]; (c) Rao AVR, Gurjar MK, Islam A. Tetrahedron Lett. 1993;34:4993. [Google Scholar]
- 6.(a) Corey EJ, Katzenellenbogen JA, Gilman NW, Roman SA, Erickson BWJ. J. Am. Chem. Soc. 1968;90:5618. [Google Scholar]; (b) Moody CJ, Toczek J. Tetrahedron Lett. 1986;27:5253. [Google Scholar]; (c) Moody CJ, Toczek J. J. Chem. Soc.-Perkin Trans. 1. 1988:1397. [Google Scholar]; (d) Kammler R, Polborn K, Wanner KT. Tetrahedron. 2003;59:3359. [Google Scholar]
- 7.(a) Mittelbach M, Poklukar N, Junek H. Liebigs Ann. Chem. 1990:185. [Google Scholar]; (b) Bringmann G, Kuenkel G, Geuder T. Synlett. 1990:253. [Google Scholar]; (c) Bringmann G, Geuder T. Synthesis. 1991:829. [Google Scholar]
- 8.(a) Wipf P, Lim S. J. Am. Chem. Soc. 1995;117:558. [Google Scholar]; (b) Wipf P, Reeves JT. Chem. Commun. 2002:2066. doi: 10.1039/b207383h. [DOI] [PubMed] [Google Scholar]; (c) Jung IC. Eur. J. Org. Chem. 2001:1899. [Google Scholar]
- 9.(a) Zvilichovsky G, Gurvich V. J. Chem. Soc. Perkin Trans. 1. 1995:2509. [Google Scholar]; (b) Zvilichovsky G, Gurvich V. J. Chem. Soc. Perkin Trans. 1. 1997:1069. [Google Scholar]; (c) Zvilichovsky G, Gurvich V. Tetrahedron. 1995;51:5479. [Google Scholar]; (d) Gbara-Haj-Yahia I, Zvilichovsky G, Seri N. J. Org. Chem. 2004;69:4135. doi: 10.1021/jo049731y. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kirkemo CL, White JD. J. Org. Chem. 1985;50:1316. [Google Scholar]; (b) Bringmann G. Liebigs Ann., Chem. 1985:2105. [Google Scholar]
- 11.For reports concerning the ozonolysis of 2,5-cyclohexadienones, see: Caspi E, Schmid W, Khan BT. Tetrahedron. 1962;18:767. Caspi E, Khan BT, Balasubrahmanyam SN. Tetrahedron. 1962;18:1013. Balasubrahmanyam SN, Caspi E, Khan BT. J. Chem. Soc. 1963:761. Rodig OR, Zanati G. J. Org. Chem. 1968;33:914. doi: 10.1021/jo01280a029. Okamoto K, Nitta I, Shingu H. Bull. Chem. Soc. Jpn. 1970;43:1768. Bailey PS, Ferrell TM. J. Org. Chem. 1981;46:5028.
- 12. Harries C, Turk H. Chem. Ber. 1905;374:1630. Harries C. Ann. 1910;374:288. (c) For an account of Harries' introduction of ozone into organic chemistry, see: Rubin MB. Helv. Chim. Acta. 2003;86:930.
- 13.Bailey PS. In: Ozonation in Organic Chemistry. Trahanovsky W, editor. Vol. 1. New York: Academic Press; 1978. pp. 153–162. Vol. I. [Google Scholar]
- 14.Story PR, Burgess JR. Tetrahedron Lett. 1968:1287. [Google Scholar]
- 15.(a) Shibata K, Takegawa S, Koizumi N, Yamakoshi N, Shimazawa E. Chem. Pharm. Bull. 1992;40:935. doi: 10.1248/cpb.40.935. [DOI] [PubMed] [Google Scholar]; (b) Koizumi N, Takegawa S, Mieda M, Shibata K. Chem. Pharm. Bull. 1996;44:2162. doi: 10.1248/cpb.44.2162. [DOI] [PubMed] [Google Scholar]; (c) Cambie RC, Higgs PI, Read CM, Rutledge PS, Ryan GR, Woodgate PD. Aust. J. Chem. 1990;43:681. [Google Scholar]
- 16.For reviews covering the formation of quaternary stereogenic center, see: Christoffers J, Mann A. Angew. Chem. Int. Ed. 2001;40:4591. doi: 10.1002/1521-3773(20011217)40:24<4591::aid-anie4591>3.0.co;2-v. Corey EJ, Guzman-Perez A. Angew. Chem. Int. Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.
- 17.For recent reviews of phenolic oxidation, see: Wirth T. In: Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis. Wirth T, editor. Vol. 224. Berlin: Springer; 2003. Moriarty RM, Prakash O. Org. React. 2001;57:327. Pelter A, Ward RS. Tetrahedron. 2001;57:273.
- 18.(a) Wardrop DJ, Basak A. Org. Lett. 2001;3:1053. doi: 10.1021/ol015626o. [DOI] [PubMed] [Google Scholar]; (b) Wardrop DJ, Zhang W. Org. Lett. 2001;3:2353. doi: 10.1021/ol0161514. [DOI] [PubMed] [Google Scholar]; (c) Wardrop DJ, Landrie CL, Ortíz JA. Synlett. 2003:1352. [Google Scholar]
- 19.For reviews concerning the chemistry of nitrenium ions, see: Falvey DE. In: Reactive Intermediate Chemistry. Moss RA, Platz MS, Jones M, editors. Chapter 13. Hoboken, NJ: Wiley-Interscience; 2004. Kikugawa Y. Rev. Heteroatom Chem. 1996;15:263. Abramovitch RA, Jeyaraman R. In: Azides and Nitrenes: Reactivity and Utility. Scriven EFV, editor. Chapter 6. Orlando, FL: Academic Press; 1984.
- 20.For the intramolecular reaction acylnitrenium ions and related electron-deficient species with arenes, see: Glover SA, Goosen A, McCleland CW, Schoonraad JL. J. Chem. Soc. Perkin Trans. 1. 1984:2255. Kikugawa Y, Kawase M. J. Am. Chem. Soc. 1984;106:5728. Glover SA, Goosen A, McCleland CW, Schoonraad JL. Tetrahedron. 1987;43:2577. Kawase M, Kitamura T, Kikugawa Y. J. Org. Chem. 1989;54:3394. Cherest M, Lusinchi X. Tetrahedron Lett. 1989;30:715. Glover SA, Rowbottom CA, Scott AP, Schoonraad JL. Tetrahedron. 1990;46:7247. Prata JV, Clemente DTS, Prabhakar S, Lobo AM, Mourato I, Branco PS. J. Chem. Soc. Perkin Trans. 1. 2002:513. Miyazawa E, Sakamoto T, Kikugawa Y. Heterocycles. 2003;59:149. Miyazawa E, Sakamoto T, Kikugawa Y. J. Org. Chem. 2003;68:5429. doi: 10.1021/jo034318w. Kikugawa Y, Nagashima A, Sakamoto T, Miyazawa E, Shiiya M. J. Org. Chem. 2003;68:6739. doi: 10.1021/jo0347009.
- 21.Wardrop DJ, Burge MS, Zhang W, Ortíz JA. Tetrahedron. Lett. 2003;44:2587. [Google Scholar]
- 22.Sakai R, Oiwa C, Takaishi K, Kamiya H, Tagawa M. Tetrahedron Lett. 1999;40:6941. [Google Scholar]
- 23.For syntheses of dysibetaine, see: Langlois N, Le Nguyen BK. J. Org. Chem. 2004;69:7558. doi: 10.1021/jo040216+. Le Nguyen BK, Langlois N. Tetrahedron Lett. 2003;44:5961. Snider BB, Gu Y. Org. Lett. 2001;3:1761. doi: 10.1021/ol015954o.
- 24.(a) Shin-ya K, Kim JS, Furihata K, Hayakawa Y, Seto H. Tetrahedron Lett. 1997;38:7079. [Google Scholar]; (b) Okue M, Kobayashi H, Shin-ya K, Furihata K, Hayakawa Y, Seto H, Watanabe H, Kitahara T. Tetrahedron Lett. 2002;43:857. [Google Scholar]
- 25.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Angew. Chem. Int. Ed. 2003;42:355. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 26.(a) Gordon EM, Ondetti MA, Pluscec J, Cimarusti CM, Bonner DP, Sykes RB. J. Am. Chem. Soc. 1982;104:6053. [Google Scholar]; (b) Koster WH, Bonner DP. In: Frontiers of Antibiotic Research. Umezawa H, editor. New York: Academic Press; 1987. p. 211. [Google Scholar]
- 27.Wardrop DJ, Burge MS. Chem. Commun. 2004:1230. doi: 10.1039/b403081h. [DOI] [PubMed] [Google Scholar]
- 28.(a) Clark RD, Heathcock CH. J. Org. Chem. 1976;41:1396. doi: 10.1021/jo00870a023. [DOI] [PubMed] [Google Scholar]; (b) Keul H, Kuczkowski RL. J. Am. Chem. Soc. 1984;106:5370. [Google Scholar]; (c) Keul H, Kuczkowski RL, Choi HS. J. Org. Chem. 1985;50:3365. [Google Scholar]; (d) Wojciechowski BJ, Pearson WH, Kuczkowski RL. J. Org. Chem. 1989;54:115. [Google Scholar]; (e) Griesbaum K, Kim WS, Nakamura N, Mori M, Nojima M, Kusabayashi S. J. Org. Chem. 1990;55:6153. [Google Scholar]; (f) Bunnelle WH. Chem. Rev. 1991;91:335. [Google Scholar]; (g) Griesbaum K, Kim WS. J. Org. Chem. 1992;57:5574. [Google Scholar]; (h) Hillers S, Niklaus A, Reiser O. J. Org. Chem. 1993;58:3169. [Google Scholar]; (i) Schank K, Beck H, Pistorius S. Helv. Chim. Acta. 2004;87:2025. [Google Scholar]
- 29.For reports of the efficient ozonolysis of dihydropyrones, see: Danishefsky S, Kato N, Askin D, Kerwin JF., Jr J. Am. Chem. Soc. 1982;104:360. Sugiyama T, Yamakoshi H, Nojima M. J. Org. Chem. 1993;58:4212.
- 30.For details of the preparation of 10, see Supporting Information.
-
31.(a) In the case of substrate 10a (entry 1), the formation of β–lactam 12a was also accompanied by compound 20 which arises from the conjugate addition of methanol to the dienone system.31b Although isolated as a single diastereomer, we were unable to unequivocally establish the relative stereochemistry of 20 via spectroscopic means. The formation of this undesired by-product was simply avoided by carrying out the cyclization of 10a and 10b in CH2Cl2, in the absence of methanol (entries 2 and 3).
 Nilsson A, Ronlán A, Parker VD. Tetrahedron Lett. 1975:1107.
Nilsson A, Ronlán A, Parker VD. Tetrahedron Lett. 1975:1107.
- 32.Hoffmann RW. Chem. Rev. 1989;89:1841. [Google Scholar]
- 33.Analogous rationalizations of π-facial diastereoselectivity are proposed in: Wender PA, Ternansky RJ. Tetrahedron Lett. 1985;26:2625. Adam W, Peters EM, Peters K, Prein M, von Schnering HG. J. Am. Chem. Soc. 1995;117:6686. Kamikawa K, Furusyo M, Uno T, Sato Y, Konoo A, Bringmann G, Uemura M. Org. Lett. 2001;3:3667. doi: 10.1021/ol010197f.
- 34.The oxidative Ar2-5 cyclization of 3-substituted 3-aryl-1-benzyloxy ureas has previously been noted: Romero AG, Darlington WH, Jacobsen EJ, Mickelson JW. Tetrahedron Lett. 1996;37:2361. (footnote 7).
- 35.For examples of biologically active N-hydroxy lactams and their derivatives, see: Higashide E, Horii S, Ono H, Mizokami N, Yamazaki T, Shibata M, Yoneda M. J. Antibiot. 1985;38:285. doi: 10.7164/antibiotics.38.285. Schlemminger I, Mole DR, McNeill LA, Dhanda A, Hewitson KS, Tian YM, Ratcliffe PJ, Pugh CW, Schofield CJ. Bioorg. Med. Chem. Lett. 2003;13:1451. doi: 10.1016/s0960-894x(03)00149-5. Wendenbaum S, Demange P, Dell A, Meyer JM, Abdallah MA. Tetrahedron Lett. 1983;24:4877. Swarén P, Massova I, Bellettini JR, Bulychev A, Maveyraud L, Kotra LP, Miller MJ, Mobashery S, Samama JP. J. Am. Chem. Soc. 1999;121:5353. Bulychev A, O'Brien ME, Massova I, Teng M, Gibson TA, Miller MJ, Mobashery S. J. Am. Chem. Soc. 1995;117:5938. Bulychev A, Bellettini JR, O'Brien M, Crocker PJ, Samama JP, Miller MJ, Mobashery S. Tetrahedron. 2000;56:5719.
- 36.Schreiber SL, Claus RE, Reagan J. Tetrahedron Lett. 1982;23:3867. [Google Scholar]
- 37.Gupta D, Soman R, Dev S. Tetrahedron. 1982;38:3013. [Google Scholar]
- 38.Duhamel P, Kotera M, Monteil T, Marabout B, Davoust D. J. Org. Chem. 1989;54:4419. [Google Scholar]
- 39.The instablity of related systems to basic conditions has been noted: Reddy LR, Saravanan P, Corey EJ. J. Am. Chem. Soc. 2004;126:6230. doi: 10.1021/ja048613p. Trancard D, Tout JB, Giard T, Chichaoui I, Cahard D, Plaquevent JC. Tetrahedron Lett. 2000;41:3843. Duhamel P, Kotera M. J. Org. Chem. 1982;47:1688.
- 40.Bis(hydroxymethyl)pyrrolidinones and their derivatives are of interest as neuraminidase inhibitors: Brouillette WJ, Bajpai SN, Ali SM, Velu SE, Atigadda VR, Lommer BS, Finley JB, Luo M, Air GM. Bioorg. Med. Chem. 2003;11:2739. doi: 10.1016/s0968-0896(03)00271-2. Schieweck F, Altenbach HJ. J. Chem. Soc. Perkin Trans. 1. 2001:3409. Atigadda VR, Brouillette WJ, Duarte F, Ali SM, Babu YS, Bantia S, Chand P, Chu N, Montgomery JA, Walsh DA, Sudbeck EA, Finley J, Luo M, Air GM, Laver GW. J. Med. Chem. 1999;42:2332. doi: 10.1021/jm980707k.
- 41.(a) Ishmuratov GY, Kharisov RY, Yakovleva MP, Botsman OV, Muslukhov RR, Tolstikov GA. Russ. J. Org. Chem. 2001;37:37. [Google Scholar]; (b) Gribble GW. Chem. Soc. Rev. 1998;27:395. [Google Scholar]
- 42.Zhang J, Flippen-Anderson JL, Kozikowski AP. J. Org. Chem. 2001;66:7555. doi: 10.1021/jo010626n. [DOI] [PubMed] [Google Scholar]
- 43.For selected routes to 4,4-disubtituted azetidinones, see: Ageno G, Banfi L, Cascio G, Guanti G, Manghisi E, Riva R, Rocca V. Tetrahedron. 1995;51:8121. DeKimpe N, Tehrani KA, Fonck G. J. Org. Chem. 1996;61:6500. doi: 10.1021/jo960296k. Palomo C, Aizpurua JM, Garc�a JM, Galarza R, Legido M, Urchegui R, Román P, Luque A, Server-Carrio J, Linden A. J. Org. Chem. 1997;62:2070. doi: 10.1021/jo962017z. Gerona-Navarro G, Garcia-López MT, González-Mniz R. J. Org. Chem. 2002;67:3953. doi: 10.1021/jo025571j. Campomanes P, Menendez MI, Sordo TL. J. Org. Chem. 2003;68:6685. doi: 10.1021/jo034803r.
- 44.For the preparation of quaternary tetrahydroisoquinoline-3-carboxylic acid derivatives, see: Vicario JL, Badia D, Carrillo L, Etxebarria J. Curr. Org. Chem. 2003;7:1775. Kawabata T, Ozturk O, Suzuki H, Fuji K. Synthesis. 2003:505. Alezra V, Bonin M, Micouin L, Husson HP. Tetrahedron Lett. 2001;42:2111. Chinchilla R, Galindo N, Najera C. Synthesis. 1999:704. Ma D, Ma Z, Kozikowski AP, Pshenichkin S, Wroblewski JT. Bioorg. Med. Chem. Lett. 1998;8:2447. doi: 10.1016/s0960-894x(98)00409-0. Seebach D, Dziadulewicz E, Behrendt L, Cantoreggi S, Fitzi R. Liebigs Ann. Chem. 1989:1215. Bajgrowicz J, El Achquar A, Roumestant ML, Pigie`re C, Viallefont P. Heterocycles. 1986;24:2165.
- 45.(a) Kondo K, Sodeoka M, Mori M, Shibasaki M. Tetrahedron Lett. 1993;34:4219. [Google Scholar]; (b) Kondo K, Sodeoka M, Mori M, Shibasaki M. Synthesis. 1993:920. [Google Scholar]; (c) Wipf P, Kim Y. J. Am. Chem. Soc. 1994;116:11678. [Google Scholar]; (d) Blades K, Cockerill GS, Easterfield HJ, Lequeux TP, Percy JM. Chem. Commun. 1996:1615. [Google Scholar]
- 46.Banerjee AK, Azocar JA, Vera W. Synth. Commun. 1999;29:2995. [Google Scholar]
- 47.(a) Luche JL. J. Am. Chem. Soc. 1978;100:2226. [Google Scholar]; (b) Abruscato GJ, Tidwell TT. J. Org. Chem. 1972;37:4151. [Google Scholar]
- 48.Acetylation of the crude product was necessary in this case to facilitate chromatographic purification.
- 49.Nakagawa TW, Andrews LJ, Keefer RM. J. Am. Chem. Soc. 1960;82:269. [Google Scholar]
- 50.(a) Wipf P, Jung JK. Chem. Rev. 1999;99:1469. doi: 10.1021/cr9803838. [DOI] [PubMed] [Google Scholar]; (b) Ohkata K, Tamura Y, Shetuni BB, Takagi R, Miyanaga W, Kojima S, Paquette LA. J. Am. Chem. Soc. 2004;126:16783. doi: 10.1021/ja047027t. [DOI] [PubMed] [Google Scholar]
- 51.The calculations were performed with Spartan'04 for Macintosh, Wavefunction Inc. Irvine, CA 4024
- 52.Mattingly PG, Miller MJ. J. Org. Chem. 1980;45:410. [Google Scholar]
- 53.(a) Chang CY, Yang TK. Tetrahedron-Asymmetry. 2003;14:2081. [Google Scholar]; (b) Romero AG, Darlington WH, McMillan MW. J. Org. Chem. 1997;62:6582. [Google Scholar]
- 54.Qabar MN, Kahn M. Tetrahedron Lett. 1996;37:965. [Google Scholar]
- 55.(a) Chiara JL, Destabel C, Gallego P, Marco-Contelles J. J. Org. Chem. 1996;61:359. doi: 10.1021/jo970987w. [DOI] [PubMed] [Google Scholar]; (b) Yang HW, Romo D. J. Org. Chem. 1999;64:7657. [Google Scholar]; (c) Keck GE, Wager TT, McHardy SF. Tetrahedron. 1999;55:11755. [Google Scholar]
- 56.(a) Naruse M, Aoyagi S, Kibayashi C. J. Org. Chem. 1994;59:1358. [Google Scholar]; (b) Blakemore PR, Kim SK, Schulze VK, White JD, Yokochi AFT. J. Chem. Soc. Perkin Trans. 1. 2001:1831. [Google Scholar]; (c) Chow CP, Shea KJ, Sparks SM. Org. Lett. 2002;4:2637. doi: 10.1021/ol026075k. [DOI] [PubMed] [Google Scholar]
- 57.(a) King SB, Ganem B. J. Am. Chem. Soc. 1991;113:5089. [Google Scholar]; (b) Malpass JR, Smith C. Tetrahedron Lett. 1992;33:273. [Google Scholar]
- 58.(a) Graham SL, Scholz TH. Tetrahedron Lett. 1990;31:6269. [Google Scholar]; (b) Anderson JC, Flaherty A, Swarbrick ME. J. Org. Chem. 2000;65:9152. doi: 10.1021/jo0056343. [DOI] [PubMed] [Google Scholar]
- 59.Keck GE, McHardy SF, Murry JA. Tetrahedron Lett. 1994;34:6215. [Google Scholar]
- 60.Yus M, Radivoy G, Alonso F. Synthesis. 2001:914. [Google Scholar]
- 61.(a) Cardani S, Gennari C, Scolastico C, Villa R. Tetrahedron. 1989;45:7397. [Google Scholar]; (b) Williams RM, Lee BH, Miller MM, Anderson OP. J. Am. Chem. Soc. 1989;111:1073. [Google Scholar]
- 62.(a) Cicchi S, Goti A, Brandi A, Guarna A, De Sarlo F. Tetrahedron Lett. 1990;31:3351. [Google Scholar]; (b) Gouverneur V, Ghosez L. Tetrahedron Lett. 1991;32:5349. [Google Scholar]; (c) Ritter AR, Miller MJ. J. Org. Chem. 1994;59:4602. [Google Scholar]
- 63.(a) Black DSC, Brown RFC, Wade AM. Aust. J. Chem. 1972;25:2155. [Google Scholar]; (b) Black DSC, Brown RFC, Wade AM. Aust. J. Chem. 1972;25:2429. [Google Scholar]; (c) Keck GE, Webb RR, Yates JB. Tetrahedron. 1981;37:4007. [Google Scholar]; (d) Crossley MJ, Crumbie RL, Fung YM, Potter JJ, Pegler MA. Tetrahedron Lett. 1987;28:2883. [Google Scholar]; (e) Baldwin JE, Adlington RM, Gollins DW, Schofield CJ. J. Chem. Soc. Chem. Commun. 1990:720. [Google Scholar]; (f) Baldwin JE, Adlington RM, Godfrey CRA, Gollins DW, Schofield CJ. Tetrahedron. 1991;47:5835. [Google Scholar]; (g) Leeson PD, Williams BJ, Rowley M, Moore KW, Baker R, Kemp JA, Priestley T, Foster AC, Donald AE. Bioorg. Med. Chem. Lett. 1993;3:71. [Google Scholar]; (h) Tiecco M, Testaferri L, Tingoli M, Marini F. J. Chem. Soc. Chem. Commun. 1994:221. [Google Scholar]; (i) Thomas A, Rajappa S. Tetrahedron. 1995;51:10571. [Google Scholar]; (j) Forzato C, Nitti P, Pitacco G, Valentin E, Morganti S, Rizzato E, Spinelli D, Dell'Erba C, Petrillo G, Tavani C. Tetrahedron. 2004;60:11011. [Google Scholar]
- 64.Dong L, Miller MJ. J. Org. Chem. 2002;67:4759. doi: 10.1021/jo0256078. [DOI] [PubMed] [Google Scholar]
- 65.Still WC, Kahn M, Mitra A. J. Org. Chem. 1978;43:2923. [Google Scholar]
- 66.Loudon GM, Radhakrishna AS, Almond MR, Blodgett JK, Boutin RH. J. Org. Chem. 1984;49:4272. [Google Scholar]

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.