

Abstract
Mammalian motor circuits control voluntary movements by transmitting signals from the central nervous system (CNS) to muscle targets. To form these circuits, motor neurons (MNs) must extend their axons out of the CNS. Although exit from the CNS is an indispensable phase of motor axon pathfinding, the underlying molecular mechanisms remain obscure. Here, we present the first identification of a genetic pathway that regulates motor axon exit from the vertebrate spinal cord, utilizing spinal accessory motor neurons (SACMNs) as a model system. SACMNs are a homogeneous population of spinal MNs with axons that leave the CNS through a discrete lateral exit point (LEP) and can be visualized by the expression of the cell surface protein BEN. We show that the homeodomain transcription factor Nkx2.9 is selectively required for SACMN axon exit and identify the Robo2 guidance receptor as a likely downstream effector of Nkx2.9; loss of Nkx2.9 leads to a reduction in Robo2 mRNA and protein within SACMNs and SACMN axons fail to exit the spinal cord in Robo2-deficient mice. Consistent with short-range interactions between Robo2 and Slit ligands regulating SACMN axon exit, Robo2-expressing SACMN axons normally navigate through LEP-associated Slits as they emerge from the spinal cord, and fail to exit in Slit-deficient mice. Our studies support the view that Nkx2.9 controls SACMN axon exit from the mammalian spinal cord by regulating Robo-Slit signaling.
Keywords: Nkx2.9, Robo, SACMN, Slit, Motor axon exit, Mouse
INTRODUCTION
Motor neurons (MNs) project their axons out of the central nervous system (CNS) and make stereotyped connections with peripheral muscle targets to form circuits that control movement (Bonanomi and Pfaff, 2010; Dalla Torre di Sanguinetto et al., 2008; Sharma and Peng, 2001). Motor axons grow in a directed manner to specialized exit points through which they emerge from the CNS (Bravo-Ambrosio and Kaprielian, 2011; Jacob et al., 2001; Lieberam et al., 2005; Schneider and Granato, 2003; Sharma et al., 1998; Shirasaki and Pfaff, 2002). MN subtypes can be distinguished by the positions of their exit points: ventral MNs (vMNs) and dorsal MNs (dMNs) utilize ventral and dorsal exit points, respectively (Chandrasekhar, 2004; Cordes, 2001; Dillon et al., 2005; Guthrie, 2007; Lieberam et al., 2005; Schubert and Kaprielian, 2001; Sharma et al., 1998; Snider and Palavali, 1990). Although Cxcl12-Cxcr4 signaling regulates the growth of vMN axons to their exit points in mice (Lieberam et al., 2005) and myotomal-derived diwanka (plod3 – ZFIN) glycosyltransferase is required for motor axon growth into the periphery in zebrafish (Schneider and Granato, 2006), the molecular mechanisms that control motor axon exit from the vertebrate spinal cord are poorly understood.
Transcription factors (TFs) control axon pathfinding by regulating the expression of cell surface molecules (Broihier et al., 2004; Garcia-Frigola et al., 2008; Labrador et al., 2005; Landgraf et al., 1999; Lee et al., 2008; Wilson et al., 2008). In Drosophila, Zfh1 (Layden et al., 2006) and Nkx6 (HGTX – FlyBase) (Broihier et al., 2004) are required for the exit of vMN axons, whereas Eve (Landgraf et al., 1999) is necessary for dMN axon exit. Nkx6 and Eve are likely to mediate motor axon exit by regulating the expression of Fas3 (Broihier et al., 2004) and Unc5 (Labrador et al., 2005), respectively. In vertebrates, Lhx3/Lhx4 (Sharma et al., 1998) and Phox2b (Hirsch et al., 2007) are required for the directed growth of vMN and dMN axons, respectively, to their exit points. However, these TFs appear to control the specification of vMNs/dMNs rather than motor axon exit per se.
Spinal accessory motor neurons (SACMNs) are branchiomotor dMNs that reside within the cervical spinal cord and project dorsally directed axons to and through a highly localized lateral exit point (LEP) situated midway along the dorsoventral axis of the spinal cord (Dillon et al., 2005; Hirsch et al., 2007; Lieberam et al., 2005). Upon exiting the CNS, SACMN axons execute a rostral turn and assemble into the longitudinally projecting spinal accessory nerve (SAN), which innervates particular neck and back muscles (Dillon et al., 2005; Dillon et al., 2007; Schubert and Kaprielian, 2001; Snider and Palavali, 1990). We identified the immunoglobulin (Ig) domain-containing protein BEN (Alcam or SC1 – Mouse Genome Informatics) (Dillon et al., 2005; Schubert and Kaprielian, 2001) as a selective marker of SACMN cell bodies/axons (Dillon et al., 2005; Schubert and Kaprielian, 2001). Since SACMNs are a molecularly homogenous and readily identifiable population of spinal MNs, which leave the CNS through a circumscribed exit point, they represent an ideal model system for elucidating molecular programs that control motor axon exit.
Our observation that the homeodomain TF Nkx2.9 is likely to be required for SACMN axons to leave the CNS (Dillon et al., 2005) prompted us to further characterize the role of Nkx2.9 in motor axon exit. Here we show that, in mice lacking Nkx2.9, SACMN axons appropriately project to the LEP but assemble into an ectopic longitudinally projecting SAN within the spinal cord. We also identify the axon guidance receptor roundabout 2 (Robo2) (Ypsilanti et al., 2010) as a likely downstream effector of Nkx2.9 by showing that Robo2 expression in SACMNs is downregulated in Nkx2.9 null mice and that SACMN axons fail to exit the spinal cord in Robo2-deficient animals. Furthermore, the Robo2 ligands Slit1-3 are present at the LEP, SACMN axons fail to exit the CNS in Slit null mice, and Slit promotes SACMN axon outgrowth in vitro. Collectively, our findings are consistent with Nkx2.9 controlling SACMN axon exit from the CNS by regulating Robo2-Slit interactions at the LEP.
MATERIALS AND METHODS
Mice
CD-1 wild-type (WT) embryos were used for expression studies (Charles River Laboratories). Nkx2.9 mutant embryos were generated by mating Nkx2.9+/– mice and genotypes determined by PCR (Tian et al., 2006). Nkx2.9 breeding pairs were obtained from J. Locker (Albert Einstein College of Medicine). Pregnant dams were sacrificed as described (Dillon et al., 2005). The morning on which a vaginal plug was detected was considered embryonic day (E) 0.5. Robo and Slit mutant embryos were generated and genotyped as described (Andrews et al., 2008; Farmer et al., 2008; Grieshammer et al., 2004; Long et al., 2004; Lopez-Bendito et al., 2007; Lu et al., 2007; Plump et al., 2002).
RNA isolation, cDNA microarray hybridization, data acquisition and analysis
The Qiagen RNeasy Micro Kit was used to isolate total RNA from E11.5 cervical spinal cords (excluding dorsal spinal cord and floor plate; WT, n=3; Nkx2.9–/–, n=3). RNA was amplified using the WT-Ovation Pico RNA Amplification Kit (NuGEN Technologies), followed by hybridization to GeneChip Mouse Genome 430 2.0 Array (Affymetrix) by the Albert Einstein College of Medicine Affymetrix facility. Results from three independent arrays were averaged. The raw data (CEL files) from each probe set were normalized by RMA methods using GeneSpring GX 10.0 software (Agilent). The log2 transformed signal intensities were averaged for biological replicates and the mean value was used to compute fold change. Differentially expressed genes were identified as those exhibiting a fold change exceeding 1.5, as several previous studies have used this cut-off (Hughes et al., 2000; Schachter et al., 2002; Wang et al., 2003; Yuan et al., 2005) and a fold change of less than 2 can be biologically significant (Hughes et al., 2000).
Immunohistochemistry and in situ hybridization
Antibodies used were: anti-BEN (RIKEN BioResource Center), anti-β-galactosidase (Abcam), anti-NF (DSHB), anti-GFP (Invitrogen), anti-HB9 (S. Pfaff, Salk Institute), anti-Islet1 (DSHB), anti-laminin (Sigma), anti-L1 (Chemicon), anti-myc (Millipore), anti-Phox2b (C. Goridis, INSERM) and anti-Robo2 (R&D Systems). Standard immunohistochemistry, digoxigenin-labeled riboprobe generation and in situ hybridization were carried out as described (Dillon et al., 2005). The mouse Slit1, Slit2, Slit3 (Yuan et al., 1999), Nkx2.9 (Pabst et al., 1998), chick Robo2 (Reeber et al., 2008) and BEN (Dillon et al., 2005) cDNAs were generated as described. Detection of β-galactosidase activity with X-Gal was as described (Corrales et al., 2004). Open-book (OB) spinal cord preparations were generated from E11.5 mouse embryos and subject to whole-mount immunohistochemistry as described (Imondi et al., 2000).
Fluorescent in situ hybridization followed by immunohistochemistry
Cervical spinal cord-containing cryosections derived from E9.5 mouse embryos were hybridized with Slit1, Slit2 or Slit3 riboprobe for 12 hours at 72°C, and then incubated with anti-digoxigenin POD (Roche) and anti-β-galactosidase (Abcam) overnight at 4°C. Labeled mRNA and protein were visualized using the Tyramide Signal Amplification Plus Cyanine 3 System (Perkin Elmer) and an Alexa Fluor 488-conjugated secondary antibody (Invitrogen), respectively (see Vosshall et al., 2000).
In ovo electroporation
Full-length mouse Nkx2.9 cDNA was subcloned into the pMES expression vector [which contains the chicken β-actin promoter/CMV-IE enhancer and IRES-EGFP (Swartz et al., 2001)]. Purified pMES (2-4 μg/μl), pMESNkx2.9 (2-4 μg/μl) or cytopcig-Slit1-LRR (0.5 μg/μl) vectors (Shiau and Bronner-Fraser, 2009) were microinjected into the central canal of E2 chick embryos, which were then subjected to unilateral electroporation as described (Reeber et al., 2008).
In vitro axon outgrowth assay
Floor plate-containing ventral spinal cord explants were generated from E11.5 WT mouse embryos as described (Imondi and Kaprielian, 2001), placed in 1:1 rat-tail collagen:Matrigel (BD Biosciences) pads and co-cultured with aggregates of mock- or Slit2-transfected (1-2 μg/μl) (Kadison et al., 2006) HEK293 cells for 15-24 hours in 69% OptiMEM with GLUTAMAX (Gibco), 23% F12 (Gibco), 5% FBS (Gemini BioProducts), 2% 2 M glucose and 1% penicillin-streptomycin-glutamine (Gibco). Explants were then fixed in 4% paraformaldehyde for 16 hours at 4°C and subjected to whole-mount immunohistochemistry (Dillon et al., 2005) with anti-BEN (mALCAM antibody, R&D Systems).
Photodocumentation, quantification and statistical analyses
Images were captured and processed as described (Dillon et al., 2005). Confocal images were acquired on a FluoView 500 confocal microscope (Olympus). ImageJ64 software was used to perform measurements. The Mann-Whitney test was performed for all statistical analyses using GraphPad Prism software (version 5.0). Cell body and nerve counts utilized at least five cryosections per animal per genotype. Axon outgrowth was assessed by converting grayscale into binary images using ImageJ64 and outlining areas of equivalent size located between the margins of SACMN axon-containing spinal cord explants and the aggregates of either mock- or Slit2-treated HEK293 cells. The percentage of the delimited area occupied by BEN-positive staining was measured using ImageJ64.
RESULTS
In Nkx2.9–/– mice, SACMN axons fail to exit the spinal cord and inappropriately assemble into an ectopic SAN
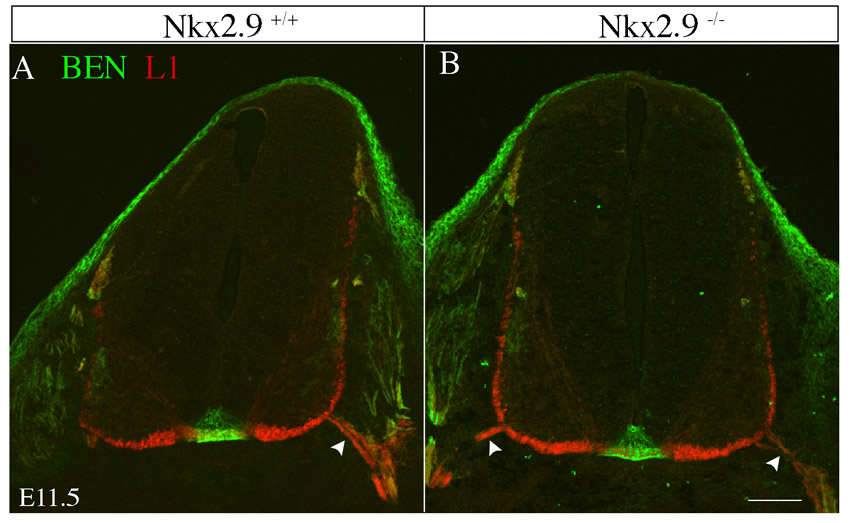
We previously reported that SACMN axons apparently fail to exit the CNS in Nkx2.9 null mice (Dillon et al., 2005). To unambiguously determine whether SACMN axons remain confined to the spinal cord in these mice, we labeled transverse cryosections from the cervical spinal cord of E10.5 Nkx2.9–/– embryos and their WT littermates with anti-BEN, as a selective marker of SACMNs, and anti-laminin, which demarcates the margin of the spinal cord (Fig. 1A-D). BEN-expressing SACMN axons failed to project across the laminin border in Nkx2.9–/– mice (Fig. 1C,D). This does not appear to reflect a developmental delay as SACMN axons do not exit the spinal cord in Nkx2.9–/– mice analyzed as late as E13.5, well after SACMN axons normally assemble into the SAN (Dillon et al., 2005) (data not shown).
Fig. 1.

In Nkx2.9–/– embryos, SACMN axons fail to exit the CNS and assemble into an ectopic SAN within the spinal cord. (A-H) Wild-type (WT) (A,B, E10.5; E,F, E11.5) and Nkx2.9–/– (C,D, E10.5; G,H, E11.5) embryos were labeled with either anti-BEN and anti-laminin (A-D) or anti-BEN and anti-L1 (E-H). (A,B) In E10.5 WT embryos, BEN-labeled spinal accessory motor neuron (SACMN) axons extend through the lateral exit point (LEP) and assemble into a spinal accessory nerve (SAN) located outside the CNS. (C,D) SACMN axons appropriately extend to the LEP in Nkx2.9–/– mice but fail to exit and remain within the anti-laminin-labeled margin of the spinal cord (arrow), where they appear to assemble into an ectopic SAN (arrowhead) (n>5). B and D are magnified views of the boxed areas in A and C, respectively. (E-H) A BEN/L1-expressing SAN (arrowhead) forms outside of the spinal cord and adjacent to the LEP (arrow) in WT embryos, whereas in Nkx2.9–/– mice the SAN inappropriately forms within the spinal cord adjacent to the LEP and BEN-expressing SACMN (H, asterisks). F and H are magnified views of the boxed areas in E and G, respectively. (I) The distance from the base of the floor plate (FP) to the SAN (see upper schematic) was not statistically different in WT (dark gray; SAN) as compared with Nkx2.9–/– (light gray; ectopic SAN) embryos (n=4). Error bars indicate s.d. (J) Schematized open-book (OB) preparation from cervical levels (C1-C4) of the spinal cord. RP, roof plate. (K-N) OB preparations derived from an E11.5 Nkx2.9 null embryo and a WT littermate labeled with anti-BEN. In contrast to WT embryos (K,L), an ectopic SAN is located within the spinal cord of Nkx2.9–/– embryos (M,N; n=3). L and N are magnified views of the boxed areas in K and M, respectively. (O) The trajectory of SACMN axons in WT and Nkx2.9–/– embryos. Scale bars: 50 μm in C for A,C; 25 μm in D for B,D; 100 μm in G for E,G, in K for K,M, in L for L,N; 5 μm in H for F,H.
We next asked whether SACMN axons assemble into an ectopic SAN within the spinal cord of Nkx2.9 null mice by labeling transverse cryosections from E11.5 embryos with anti-BEN and anti-L1 (L1cam – Mouse Genome Informatics), which labels longitudinally projecting spinal axons (Imondi et al., 2000). A BEN/L1-labeled nerve was located adjacent to the LEP but within the spinal cord in Nkx2.9–/– mice (Fig. 1G arrow, 1H arrowhead). To determine whether this ectopic nerve projects along the anterior-posterior (A-P) extent of the cervical spinal cord, we analyzed OB preparations derived from E11.5 Nkx2.9–/– and Nkx2.9+/+ embryos. Consistent with OB preparations lacking an external SAN (Fig. 1J), a BEN-positive SAN was only observed within the spinal cord of Nkx2.9–/– mice (Fig. 1K-O). Together, these findings indicate that Nkx2.9 is selectively required for SACMN axon exit. By contrast, vMN axons appropriately emerge from the spinal cord in Nkx2.9–/– mice (supplementary material Fig. S1).
Appropriate numbers of SACMNs, which likely arise from Nkx2.9+ progenitors, form in Nkx2.9 null mice
To rule out the possibility that the lack of SACMN axon exit in Nkx2.9–/– embryos reflects a reduction in SACMN number, we first identified a panel of SACMN markers by labeling cryosections from the cervical spinal cord of WT embryos with anti-BEN and antibodies specific for the MN-associated TFs Islet1 (Ericson et al., 1992; Pfaff et al., 1996), HB9 (Mnx1 – Mouse Genome Informatics) (Arber et al., 1999) or Phox2b (Pattyn et al., 2000). At E10.5-11, BEN-positive SACMNs express the generic MN marker Islet1 (Fig. 2A-C) and the branchiomotor neuron marker Phox2b (Hirsch et al., 2007) (Fig. 2A,F,G), but not HB9 (Fig. 2A,D,E). Therefore, we analyzed the numbers of BEN+ Phox2b+ and BEN+ Islet1+ SACMNs in Nkx2.9 null and WT littermates and found that appropriate numbers of properly specified SACMNs are generated in the absence of Nkx2.9 (Fig. 2H-M).
Fig. 2.

Appropriate numbers of properly specified SACMNs are formed in Nkx2.9-deficient mice. (A) Schematic of SACMN and ventral motor neuron (vMN) development in the ventral spinal cord. SACMNs (green circle) and vMNs (red circle) arise from MN progenitors, which migrate away from the ventricular zone to settle in dorsolateral or ventrolateral regions, respectively, of the spinal cord. (B-G) Cryosections derived from WT (B-E, E10.5; F,G, E11) embryos labeled with various SACMN or vMN markers show that SACMNs and their axons (arrow) migrate through vMNs [SACMN (v)] and SACMNs that have settled near the LEP [SACMN (d)] and express BEN, Islet1 and Phox2b, whereas vMNs express Islet1 and HB9. C, E and G are magnified views of the boxed areas in B, D and F, respectively. (H-K) E11.5 WT (H,I) and Nkx2.9–/– (J,K) embryos were labeled with anti-Phox2b (H,J) and colabeled with anti-Phox2b and anti-BEN (I,K). I and K are magnified views of the boxed areas in H and J, respectively. (L,M) The number of Phox2b+ BEN+ (E11.5; n=4) and Islet1+ BEN+ (E10.5; n=3) SACMNs is not statistically different between Nkx2.9–/– and WT/heterozygote littermates. Error bars indicate s.d. Scale bars: 50 μm in F for B,D,F, in J for H,J; 25 μm in G for C,E,G, in K for I,K.
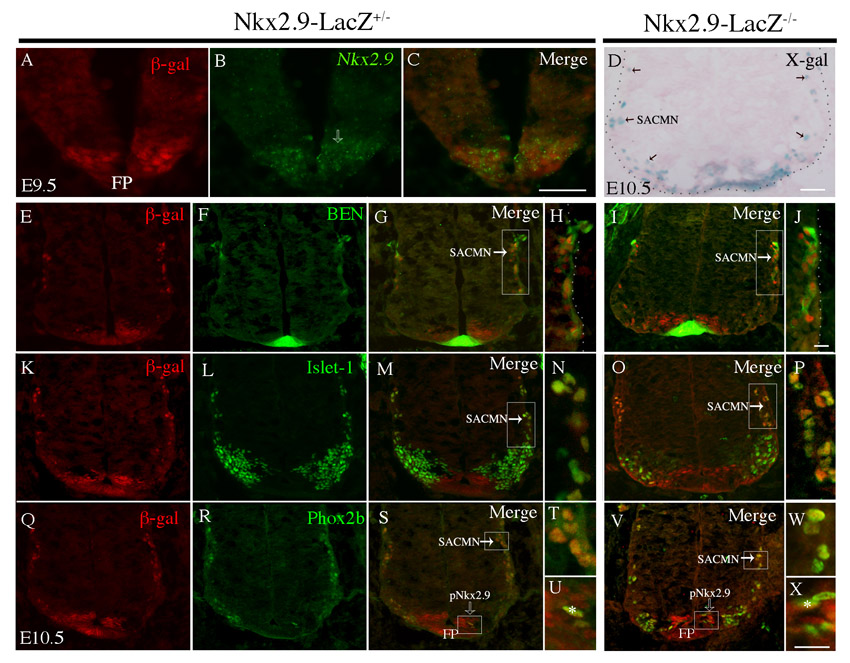
To determine whether Nkx2.9 functions cell-autonomously to facilitate SACMN axon exit we compared the distribution of lacZ-expressing cells and SACMNs within the spinal cord of a Nkx2.9-lacZ knock-in mouse line (Tian et al., 2006). In E9.5 embryos, anti-β-galactosidase staining overlapped with Nkx2.9 mRNA above the floor plate (FP), suggesting that Nkx2.9+ progenitors give rise to lacZ-positive cells (supplementary material Fig. S2A-C). At E10.5, X-Gal staining of sections from Nkx2.9-lacZ–/– embryos identified strong labeling above and adjacent to the FP and within cells (presumably SACMNs) that migrate dorsolaterally towards the LEP (supplementary material Fig. S2D). To investigate the possibility that Nkx2.9 is required to maintain the identity of these lacZ-positive cells, we labeled cryosections derived from E10.5 Nkx2.9-lacZ+/– and Nkx2.9-lacZ–/– embryos with anti-β-galactosidase and antibodies to the SACMN markers BEN, Islet1 or Phox2b. β-galactosidase-expressing neurons were labeled by each of these markers (supplementary material Fig. S2E-X), and a subset of β-galactosidase-positive cells within the ventricular zone, which were likely to be Nkx2.9+ progenitors, expressed Phox2b (supplementary material Fig. S2S,U,V,X, asterisk). These observations are consistent with SACMNs arising from a subset of Nkx2.9+ progenitors and indicate that the specification/identity of SACMNs is unaltered in Nkx2.9–/– mice.
Identifying putative downstream effectors of Nkx2.9
To identify novel downstream effectors of Nkx2.9 that might mediate SACMN axon exit, we used in situ hybridization and immunohistochemistry to examine whether the following known cell surface molecules/axon guidance receptors are expressed by SACMNs: Robo1, Robo2 (Brose et al., 1999; Kidd et al., 1999; Yuan et al., 1999), neuropilin 1 (Npn1) (He and Tessier-Lavigne, 1997; Kolodkin et al., 1997), Npn2 (Chen et al., 1997), NCAM (Ncam1 – Mouse Genome Informatics) (Maness and Schachner, 2007), polysialylated-NCAM (PSA-NCAM) (Boisseau et al., 1991), EphB1-3 (Birgbauer et al., 2001; Holland et al., 1997; Imondi et al., 2000; Jevince et al., 2006; Palmer and Klein, 2003) and VEGFR2 (Kdr – Mouse Genome Informatics) (Ruiz de Almodovar et al., 2009) (data not shown). Robo2 mRNA expression most closely overlapped with the distribution of SACMNs, and Robo2 protein was present on SACMN axons (see below).
We also carried out an unbiased microarray screen to identify genes dysregulated in Nkx2.9–/– embryos (supplementary material Table S1) by comparing total RNA isolated from the ventral half of the cervical spinal cord of E11.5 WT and Nkx2.9–/– embryos. This revealed that Robo2 mRNA levels were ∼2-fold lower in Nkx2.9-deficient mice than in WT mice (supplementary material Table S1).
Given these findings, we focused our subsequent studies on investigating the regulation of Robo2 expression in Nkx2.9–/– embryos and the role of Robo2 in SACMN axon exit.
Robo2 is expressed by SACMNs and is downregulated in Nkx2.9 null mice
To validate the results of our screens, we labeled cryosections derived from E10.5 Nkx2.9–/– embryos and their WT littermates with a Robo2 riboprobe or anti-Robo2. In WT embryos, BEN-expressing SACMNs express Robo2 mRNA (Fig. 3A,C, black arrowheads) and Robo2 protein is expressed on SACMN axons (Fig. 3E, arrows) and on the SAN (Fig. 3E, white arrowheads). In Nkx2.9–/– embryos, SACMN-associated Robo2 mRNA levels are reduced (Fig. 3B,D, black arrowheads) and Robo2 protein is downregulated on SACMN axons (Fig. 3F, arrows) that fail to exit the spinal cord, as well as on the ectopic SAN (Fig. 3F, white arrowheads). These observations are consistent with Robo2 operating as a downstream effector of Nkx2.9.
Fig. 3.

Robo2 expression is reduced in Nkx2.9–/– mice. (A-F) E10.5 cryosections derived from WT (A,C,E) or Nkx2.9–/– (B,D,F) mice were subjected to in vitro hybridization for BEN (Alcam) (A,B) or Robo2 (C,D) mRNA, or labeled with anti-BEN and anti-Robo2 (E,F). Similar levels of BEN mRNA are expressed by SACMNs (black arrowheads) in WT (A) and Nkx2.9–/– (B) embryos (n>3). SACMN-associated Robo2 mRNA is reduced in Nkx2.9–/– embryos (D; n=3) as compared with WT littermates (C). In Nkx2.9–/– embryos, SACMN axons (arrow) and the ectopic SAN (white arrowhead) display reduced levels of Robo2 protein (F; n=3) compared with WT littermates (E). The dotted line indicates the margin of the spinal cord. Scale bars: 100 μm in D for A-D; 25 μm in F for E,F.
SACMN axons do not exit the spinal cord in Robo2-deficient mice
To determine whether Robo2 is required for SACMN axon exit, we labeled E10.5 cryosections derived from Robo2–/– mice and their WT littermates with anti-BEN and anti-laminin or anti-neurofilament (NF) (Fig. 4A-J). Phenocopying Nkx2.9 null mice, the majority of SACMN axons appropriately projected to the LEP but failed to exit the CNS in E10.5 Robo2–/– animals (Fig. 4F,G, asterisks), and most SACMN axons remained confined to the spinal cord at E11.5 (Fig. 4H-J). Since a small subset of SACMN axons exited the spinal cord in Robo2–/– mice (Fig. 4H-J, arrowheads), we quantified the presence or absence of an external SAN (SACMN axon-containing nerve bundle) on both sides of the spinal cord in WT and Robo2–/– embryos. Consistent with the failure of most SACMN axons to exit the CNS, there was a significant reduction in the mean number of external SANs in Robo2–/– mice (Fig. 4, top bar chart). Although Robo1 mRNA is expressed by SACMNs and Robo1 and Robo3 protein is present on the SAN (data not shown), SACMN axon pathfinding/exit is not perturbed in Robo1 (Fig. 4K-T, bottom bar chart) and Robo3 (data not shown) null embryos. Accordingly, Robo2 is the sole Robo receptor required for SACMN axon exit from the spinal cord.
Fig. 4.

SACMN axons fail to exit the spinal cord in Robo2-deficient mice. (A-T) Cryosections derived from E10.5 Robo2+/+ (A,B), Robo2–/– (F,G), Robo1+/+ (K,L) and Robo1–/– (P,Q) embryos were labeled with anti-laminin and anti-BEN (first column) or anti-NF alone (second column). E11.5 Robo2+/+ (C-E), Robo2–/– (H-J), Robo1+/+ (M-O) and Robo1–/– (R-T) embryos were colabeled with anti-BEN (third column) and anti-NF (fourth column). BEN/NF-expressing SACMN axons fail to exit the spinal cord and form an ectopic SAN (asterisk) within the CNS in Robo2–/– mice (H-J), whereas in Robo1–/– mice (R-T), just as in Robo2+/+ (C-E) and Robo1+/+ (M-O) embryos, a SAN (A) formed outside the CNS. As in E10.5 Robo2–/– embryos, the majority of BEN/NF-expressing SACMN axons fail to exit the spinal cord at E11.5. At E11.5, several small ectopic SANs (asterisks) form within the spinal cord (H-J), as compared with WT mice (C-E). A small number of SACMN axons emerge from the CNS and form an external SAN at E11.5 (H-J, arrowhead). The bar charts show that the average number of external SANs per section is reduced in E10.5 Robo2–/– mice (top; n=4), but unaltered in Robo1–/– mice (bottom; n=3), as compared with WT mice. Error bars indicate s.d. SAN formation is unperturbed in E11.5 Robo1–/– mice (R-T; n=2), as compared with WT littermates (M-O; n=2). Scale bar: 50 μm.
Slits are expressed at the LEP and SACMN axon exit is perturbed in Slit null mice
Since SACMN axons appropriately project away from the FP and towards the LEP but fail to exit the spinal cord in Robo2–/– mice, short-range interactions between Robo2 and its Slit ligands at the LEP might normally facilitate SACMN axon exit. To test this hypothesis, we labeled serial cryosections derived from E10.5 WT embryos with Slit1, Slit2 or Slit3 riboprobes (Fig. 5A-D). High levels of Slit2 and lower levels of Slit1 and Slit3 are present at the LEP, and each Slit gene is expressed in vMNs and the FP. Based on these observations, we analyzed SACMN axon pathfinding in mice lacking one or more Slits by labeling cryosections from E10.5 Slit1–/–, Slit2–/– or Slit1–/– Slit2–/– embryos with anti-BEN and anti-NF. Whereas SACMN axon pathfinding/exit was not perturbed in the absence of Slit1 (Fig. 6D-F′) or Slit2 (Fig. 6G-I′), SACMN axons failed to exit the spinal cord in mice lacking both Slit1 and Slit2 (Fig. 6J-L′). In the double-mutant mice, SACMN axons formed more than one ectopic SAN within the spinal cord, just as they do in Robo2–/– mice (Fig. 6J-L′, asterisks). The number of ectopic nerve bundles was significantly increased, and there was a corresponding reduction in the number of normally positioned external SANs, in Slit1–/– Slit2–/– embryos as compared with WT mice and mutant mice lacking other combinations of Slits (Fig. 6M,N). These findings identify Slit1 and Slit2 as the LEP-associated Robo2 ligands required for SACMN axon exit, and suggest that the increase in the number of ectopic nerves within Slit1–/– Slit2–/– mice might arise from defasciculation of longitudinally projecting SACMN axons.
Fig. 5.

Distribution of Slit mRNA in the cervical spinal cord. (A-D) Serial cryosections derived from E10.5 WT embryos were labeled with Slit1 (A), Slit2 (B) or Slit3 (C) riboprobes. (D) Pseudo-color image representing the merge of A-C. High levels of Slit2 (B,D, arrowheads) and low levels of Slit1 and Slit3 are expressed at the LEP (A,C,D). High-magnification micrographs depicting LEP-associated expression are included as insets (A-C, arrowheads). Dotted lines demarcate spinal cord margin. Scale bar: 50 μm.
Fig. 6.

SACMN axon exit is perturbed in Slit1–/–Slit2–/– mice. (A-L′) Cryosections derived from E10.5 WT (A-C′) and Slit mutant (D-L′) embryos were labeled with anti-NF and anti-BEN. SACMN axons appropriately exit the spinal cord and assemble into an external SAN (arrow) in WT (A-C′; E10.5, n>5; E11.5, n>5), Slit1–/– (D-F′; E10.5, n=5; E11.5, n=9; data not shown), Slit2–/– (G-I′; E10.5, n=1; E11.5, n=3) or Slit1–/– Slit3–/– (E11.5, n=3; data not shown) mice. (M,N) The average number of SANs located inside or outside of the spinal cord is not statistically different between Slit1–/– mutant, Slit1–/– Slit3–/– mutant and WT mice. Notably, in Slit1–/– Slit2–/– mice, SACMN axons fail to exit the CNS and instead assemble into an ectopic SAN (J-L′, asterisks). The average number of SANs outside the spinal cord in Slit2–/– mice is significantly increased compared with WT embryos (N). *P<0.05; error bars indicate s.e.m. E10.5, n=3; E11.5, n=3; E11.5 data not shown. Scale bars: 50 μm in J for A-C,D-F,G-I,J-L; 25 μm in L′ for C′,F′,I′,L′.
Our expression and genetic data raise the possibility that short-range interactions between Robo2-expressing SACMN axons and LEP-associated Slits normally facilitate SACMN axon exit. Since SACMN cell bodies eventually settle near the LEP (Dillon et al., 2005), it is possible that they themselves might represent the source of Slit mRNA expression. We tested this hypothesis by colabeling cryosections derived from E9.5 Nkx2.9-lacZ+/– embryos with Slit1, Slit2 or Slit3 riboprobes and anti-β-galactosidase to visualize Nkx2.9+ progenitor-derived SACMNs (supplementary material Fig. S2). At E9.5, when the majority of SACMN axons have reached the LEP but have yet to exit the spinal cord, LEP-associated Nkx2.9-lacZ-expressing SACMNs do not express Slit mRNA (Fig. 7A-I, arrowheads). By contrast, SACMNs are likely to express Slit as they begin to migrate away from the FP (Fig. 7A-I). Together, our data suggest that short-range interactions between Slits expressed by LEP-associated (non-SACMN) cells and Robo2-expressing SACMN axons facilitate SACMN axon exit. To determine whether SACMN axons are attracted or repelled by Slits in vitro, we co-cultured SACMN-containing embryonic mouse spinal cord explants and aggregates of Slit2-expressing HEK293 cells. Anti-BEN-labeled SACMN axons exhibited more robust outgrowth towards Slit2-expressing HEK293 cells than towards mock-transfected HEK293 cells (Fig. 8). Therefore, short-range attractive Robo2-Slit interactions at the LEP are likely to promote SACMN axon exit (Fig. 10).
Fig. 7.

LEP-associated cells express Slits. (A-I) Serial cryosections derived from E9.5 Nkx2.9-lacZ+/– mouse embryos were colabeled with anti-β-galactosidase and Slit1 (A-C), Slit2 (D-F) or Slit3 (G-I) riboprobe. LEP-associated (arrow) cells, but not Nkx2.9-lacZ-expressing SACMNs (arrowheads), express Slits. Scale bar: 50 μm.
Fig. 8.

Slits promote SACMN axon outgrowth in vitro. (A) Schematic depicting the generation of SACMN-containing spinal cord explants and their co-culture with aggregates of Slit2-expressing HEK293 cells. (B,C) Explants were co-cultured with either mock-transfected (B; n=17; limited axon outgrowth) or Slit2-expressing (C; n=10/13 explants showed significant axon outgrowth) HEK293 cells and labeled with anti-BEN (anti-mALCAM; note that this antibody non-specifically labels HEK293 cells). (D,E) Anti-myc labels aggregates of Slit2-expressing (E) but not mock-transfected (D) HEK293 cells. (F) The percentage of the area between SACMN-containing explants and HEK293 aggregates that is anti-BEN-positive is significantly greater for Slit2-expressing than for mock-transfected cells. Error bars indicate s.e.m.; n≥5. Scale bars: 50 μm in C for B,C; 25 μm in E for D,E.
Fig. 10.

A model for the dynamic role of Robo2-Slit interactions in regulating SACMN axon pathfinding towards and through the LEP. (A,B) In WT mice (A, left), Robo2-expressing (Robo2) SACMN axons (green) navigate in close proximity to three sources of Slits (red circles) as they extend away from the FP (1), grow through vMNs (2), and project to and through the LEP (3). SACMNs express Robo2 and Slits as they pathfind away from the FP and traverse vMNs, and through potential cis attenuation of Robo2-Slit signaling their axons become insensitive to FP- and vMN-derived Slits (1, 2). SACMN axons are normally repelled by the FP-derived chemorepellent Netrin (–) and could be guided to the LEP by exit point-derived chemoattractants (+). Once Robo2-positive SACMN axons reach the LEP, they no longer express Slits, become responsive to LEP-associated Slits (3), and attractive Robo2-Slit interactions facilitate their exit from the spinal cord. By contrast, SACMN axons in Nkx2.9–/– (dark gray, Robo2↓, A right), Robo2–/– (light gray, Robo2, B left) and Slit1–/– Slit2–/– (green, Robo2, B right) mice appropriately reach the LEP but fail to exit the spinal cord. (C) Model for Nkx2.9 control of SACMN axon exit via Robo-Slit signaling. (I) Nkx2.9 regulates Robo2 expression in SACMNs. (II) Attractive short-range Robo2-Slit interactions at the LEP facilitate SACMN axon exit from the CNS.
Given that multiple ectopic SANs appear to form in Robo2–/– and Slit1–/– Slit2–/– mice, we asked whether these nerves appropriately project along the A-P axis of the spinal cord by labeling OB preparations derived from the cervical spinal cord of these mice with anti-BEN (Fig. 9A-D). These analyses revealed that SACMN axons form disorganized nerve fascicles along the A-P axis of the spinal cord in mice lacking either Robo2 or Slit1/2 (Fig. 9C,D, arrows), supporting a role for Robo2-Slit interactions in the proper growth of longitudinally projecting SACMN axons.
Fig. 9.

SAN formation is perturbed in Robo2–/– and Slit–/–Slit2–/– mice. (A) Schematic of an OB preparation isolated from the cervical spinal cord. (B-D) E11.5 OB preparations derived from WT (B), Robo2–/– (C) and Slit1–/– Slit2–/– (D) embryos were labeled with anti-BEN. Unlike WT mice (B; n>4), BEN-expressing axons assembled into disorganized nerve fascicles (arrows), within the spinal cord, in Robo2–/– (C; n=6) and Slit1–/– Slit2–/– (D; n=3) mice. Scale bar: 50 μm.
DISCUSSION
The projection of motor axons out of the CNS is a key, but poorly understood, phase of MN development (Bonanomi and Pfaff, 2010; Hirsch et al., 2007; Lieberam et al., 2005; Schneider and Granato, 2003; Shirasaki and Pfaff, 2002; Vermeren et al., 2003). Here we describe the first identification of a genetic program that regulates spinal motor axon exit in vertebrates.
Nkx2.9 controls SACMN axon exit
We show that SACMN axons appropriately project to the LEP but fail to exit the spinal cord in Nkx2.9–/– mice. This indicates that Nkx2.9 is not required for the pathfinding of SACMN axons within the spinal cord and suggests that the FP-derived chemorepellent netrin 1 directs Unc5-expressing SACMN axons away from the FP (Dillon et al., 2005; Dillon et al., 2007) and that LEP-associated chemoattractants (Caton et al., 2000; Guthrie and Lumsden, 1992), possibly secreted by boundary cap cells at motor exit points (Bron et al., 2007; Maro et al., 2004; Mauti et al., 2007; Vermeren et al., 2003), are likely to guide SACMN axons to the LEP. Since normal numbers of properly specified SACMNs are present in Nkx2.9–/– mice (Fig. 2 and supplementary material Fig. S2), and Nkx2.9–/– mice display WT-like ventral spinal cord patterning (Pabst et al., 2003), Nkx2.2 is likely to operate redundantly with Nkx2.9 to control the specification/differentiation of SACMNs (Dillon et al., 2005; Pabst et al., 2003). Although Nkx2.9+ progenitors are capable of differentiating into interneurons (Briscoe et al., 1999; Holz et al., 2010; Pabst et al., 2003), our data indicate that a subset express SACMN markers, suggesting that Nkx2.9 regulates SACMN axon exit in a cell-autonomous manner. Given that Nkx6 is required for vMN axon exit from the Drosophila ventral nerve cord (Broihier et al., 2004), Nkx TFs might have evolutionarily conserved roles in motor axon exit.
Nkx2.9 regulates Robo2 expression
Nkx genes encode homeodomain-containing TFs that regulate cell type specification and organogenesis (Briscoe et al., 2000; Briscoe et al., 1999; Harvey, 1996; Stanfel et al., 2005), and Nkx2.9 is required for FP development and commissural axon guidance (Holz et al., 2010). We show that SACMN-associated Robo2 mRNA and protein are reduced in Nkx2.9–/– embryos, identifying Robo2 as the first candidate CNS-associated downstream effector of any vertebrate Nkx gene. By contrast, we observed no changes in Slit mRNA expression in these animals (data not shown). Although the presence of putative Nkx2 binding sites within the Robo2 promoter (data not shown) is consistent with Robo2 being a direct target of Nkx2.9, forced expression of Nkx2.9 does not modulate Robo2 expression (supplementary material Fig. S3).
Robo2 and Slit1/2 are required for SACMN axon exit
Our findings are the first to show that Robo2-Slit signaling is required to facilitate motor axon exit from the spinal cord. Since Slit2 appears to be the most abundant LEP-associated Slit, it was surprising to find that SACMN axons appropriately exit the spinal cord in mice lacking Slit2. Possible explanations for these observations are that low levels of Slit1 are capable of facilitating SACMN axon exit and/or that Slit1 and Slit2 operate redundantly to regulate exit. The finding that SACMN axons do not exit the spinal cord in Slit1–/– Slit2–/– mice further indicates that LEP-associated Slit3 does not promote SACMN axon exit on its own. Nevertheless, in the absence of published data indicating that Robo2 binds each of the vertebrate Slits with different affinities, it is difficult to reconcile why Slit1, but not Slit3, appears to compensate for the loss of Slit2. In contrast to our loss-of-function data, misexpression of Slits in the chick spinal cord does not perturb SACMN axon pathfinding (supplementary material Fig. S4).
Robo-Slit signaling regulates the guidance of longitudinally projecting axons in the developing mouse brain stem (Dugan et al., 2011; Farmer et al., 2008; Mastick et al., 2010), forebrain (Devine and Key, 2008; Ricano-Cornejo et al., 2011) and spinal cord (Reeber et al., 2008). Consistent with these observations, we show that Robo2 and both Slit1 and Slit2 are required for the proper assembly of SACMN axons into the SAN. Whereas SACMN axons form an organized ectopic nerve bundle in Nkx2.9–/– embryos, disorganized/tangled nerve fascicles are present within the spinal cord of Robo2–/– and Slit1–/– Slit2–/– mice. Ventral spinal cord (including vMN)-associated Slits (Brose et al., 1999; Kadison et al., 2006; Yuan et al., 1999) might play a key role in constraining the orientation of longitudinally projecting SACMN axons as Slit1 and Slit2 position retinal ganglion cell axons at the optic chiasm (Plump et al., 2002).
The dynamic role of Robo-Slit interactions in SACMN axon pathfinding
Given that the Slit-rich FP and ventral spinal cord would be expected to represent repulsive territories for Robo2-expressing SACMN axons, how are these axons capable of growing away from the FP and through the ventral spinal cord (Fig. 10A)? As is the case for vMNs (Bai et al., 2011; Brose et al., 1999), SACMNs are likely to co-express Robo2 and Slits as their axons navigate these initial segments of their trajectory (Figs 7, 10). Thus, Slits produced by SACMN cell bodies might occupy SACMN axon-associated Robo receptors, rendering them insensitive to FP- and ventral spinal cord-associated Slits, analogous to the co-expression of Slit and Robo in vMNs inhibiting the responsiveness of their axons to Netrin (Bai et al., 2011). Similarly, co-expression of Sema3A and Npn1 on vMNs prevents their axons from responding to exogenous Sema3A (Moret et al., 2007), and the attenuation of Eph receptor signaling via ephrins in cis alters the responsiveness of vMN axons to trans ephrins (Kao and Kania, 2011).
Our observations suggest that short-range, presumably attractive/positive (Fig. 8) interactions between Robo2-expressing SACMN axons and LEP-associated Slits promote SACMN axon exit from the spinal cord. Consistent with this possibility, mesodermal cells (Kramer et al., 2001) and tracheal branches (Englund et al., 2002) are attracted to Slit in Drosophila embryos, and human leukocytes are attracted to a Slit source in vitro (Ye et al., 2010). Drosophila mesodermal cells are also capable of responding to Slit as both an attractant and repellent during sequential phases of their migration (Kramer et al., 2001). By analogy, SACMN axons might alter their responsiveness to LEP-associated Slits, first perceiving Slits as attractants and then as repellents, and this would ultimately push SACMN axons out of the spinal cord. We favor the possibility that Slits produced by LEP-associated cells facilitate SACMN axon exit; however, Robo2-expressing SACMN axons could transport ventral midline-associated Slits to the LEP, just as the Frazzled receptor captures and redistributes its Netrin ligand to modulate axon pathfinding within the Drosophila CNS (Hiramoto et al., 2000). Selectively eliminating Slits from LEP-associated cells would clarify which sources of Slits are required for SACMN axon exit.
To ultimately emerge from the spinal cord, motor axons must break through the basement membrane surrounding the neural tube. Although our studies do not directly address this aspect of SACMN axon pathfinding, it seems plausible that Robo2-Slit interactions promote cytoskeletal remodeling, which might alter the shape of SACMN growth cones and facilitate exit. Consistent with this possibility, the complexity of SACMN axon-associated growth cones is dramatically reduced as they exit the LEP (Snider and Palavali, 1990), and this might facilitate their passage through small ‘gaps’ formed by glial end-feet at motor exit points (Fraher et al., 2007).
Alternatively, Slit activation of Robo2 at the LEP might trigger the release of either soluble or membrane-bound proteolytic enzymes such as matrix metalloproteinases (MMPs) (McFarlane, 2003), which are capable of breaking down the basal lamina and potentially promoting SACMN axon exit. Our microarray results indicate that the expression of the ADAM metallopeptidase Adamts3 is reduced in Nkx2.9–/– mice, raising the possibility that MMPs might regulate SACMN axon exit (supplementary material Table S1). In one scenario, SACMN axons may extend small, localized protrusions termed invadopodia that selectively secrete MMPs, which degrade the basement membrane (Bravo-Ambrosio and Kaprielian, 2011). No matter how SACMN axons break through the border between the CNS and the peripheral nervous system, we have provided evidence that short-range Robo2-Slit interactions are likely to regulate this crucial phase of motor axon development.
Supplementary Material
Acknowledgments
We thank Deyou Zheng and Xingyi Guo (Albert Einstein College of Medicine) for assistance with analyses of microarray data; William Andrews (University of London) and Marc Tessier-Lavigne (Genentech) for mutant mice; Minkyung Kim and Brielle Bjorke (University of Nevada) for genotyping; and the following investigators for plasmids: Cathy Krull (University of Michigan; pMES), Hans-Henning Arnold (University of Braunschweig, Germany; mouse Nkx2.9), Marianne Bronner-Fraser (California Institute of Technology; cytopcig-Slit1-LRR), Yi Rao (National Institute of Biological Sciences; Slit2) and Marc Tessier-Lavigne (Robo3 probe). We also thank William Andrews, Jean Hebert, Hannes Buelow, Joseph Locker, Roy Sillitoe and Nozomi Sakai (Albert Einstein College of Medicine) for critical comments on the manuscript. Monoclonal antibodies specific for NF (T. M. Jessell and J. Dodd) and Islet1 (T. M. Jessell and S. Brenner-Morton) were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242, USA.
Footnotes
Funding
This work was supported by National Institutes of Health (NIH) grants [R56NS038505 and R01NS038505 to Z.K., RO1 NS054740, RR024210 and GM103554 to G.M.]. A.B.-A. was supported by fellowships from the Einstein Medical Scientist Training Program and Society for Neuroscience’s Neuroscience Scholars Program, and an Edward A. and Lucille Kimmel Scholarship. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.072256/-/DC1
References
- Andrews W., Barber M., Hernadez-Miranda L. R., Xian J., Rakic S., Sundaresan V., Rabbitts T. H., Pannell R., Rabbitts P., Thompson H., et al. (2008). The role of Slit-Robo signaling in the generation, migration and morphological differentiation of cortical interneurons. Dev. Biol. 313, 648–658 [DOI] [PubMed] [Google Scholar]
- Arber S., Han B., Mendelsohn M., Smith M., Jessell T., Sockanathan S. (1999). Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 23, 659–674 [DOI] [PubMed] [Google Scholar]
- Bai G., Chivatakarn O., Bonanomi D., Lettieri K., Franco L., Xia C., Stein E., Ma L., Lewcock J. W., Pfaff S. L. (2011). Presenilin-dependent receptor processing is required for axon guidance. Cell 144, 106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgbauer E., Oster S. F., Severin C. G., Sretavan D. W. (2001). Retinal axon growth cones respond to EphB extracellular domains as inhibitory axon guidance cues. Development 128, 3041–3048 [DOI] [PubMed] [Google Scholar]
- Boisseau S., Nedelec J., Poirier V., Rougon G., Simonneau M. (1991). Analysis of high PSA-NCAM expression during mammalian spinal cord and peripheral nervous system development. Development 112, 69–82 [DOI] [PubMed] [Google Scholar]
- Bonanomi D., Pfaff S. L. (2010). Motor axon pathfinding. Cold Spring Harb. Perspect. Biol. 2, a001735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Ambrosio A., Kaprielian Z. (2011). Crossing the border: molecular control of motor axon exit. Int. J. Mol. Sci. 12, 8539–8561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J., Sussel L., Serup P., Hartigan-O’Connor D., Jessell T. M., Rubinstein J. L., Ericson J. (1999). Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature 398, 622–627 [DOI] [PubMed] [Google Scholar]
- Briscoe J., Pierani A., Jessell T. M., Ericson J. (2000). A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435–445 [DOI] [PubMed] [Google Scholar]
- Broihier H. T., Kuzin A., Zhu Y., Odenwald W., Skeath J. B. (2004). Drosophila homeodomain protein Nkx6 coordinates motoneuron subtype identity and axonogenesis. Development 131, 5233–5242 [DOI] [PubMed] [Google Scholar]
- Bron R., Vermeren M., Kokot N., Little G. E., Mitchell K. J., Andrews W., Cohen J. (2007). Boundary cap cells constrain spinal motor neuron somal migration at motor exit points by a semaphorin-plexin mechanism. Neural Dev. 2, 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose K., Bland K. S., Wang K. H., Arnott D., Henzel W., Goodman C. S., Tessier-Lavigne M., Kidd T. (1999). Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96, 795–806 [DOI] [PubMed] [Google Scholar]
- Caton A., Hacker A., Naeem A., Livet J., Maina F., Bladt F., Klein R., Birchmeier C., Guthrie S. (2000). The branchial arches and HGF are growth-promoting and chemoattractant for cranial motor axons. Development 127, 1751–1760 [DOI] [PubMed] [Google Scholar]
- Chandrasekhar A. (2004). Turning heads: development of vertebrate branchiomotor neurons. Dev. Dyn. 229, 143–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Chedotal A., He Z., Goodman C. S., Tessier-Lavigne M. (1997). Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron 19, 547–559 [DOI] [PubMed] [Google Scholar]
- Cordes S. P. (2001). Molecular genetics of cranial nerve development in mouse. Nat. Rev. Neurosci. 2, 611–623 [DOI] [PubMed] [Google Scholar]
- Corrales J. D., Rocco G. L., Blaess S., Guo Q., Joyner A. L. (2004). Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development 131, 5581–5590 [DOI] [PubMed] [Google Scholar]
- Dalla Torre di Sanguinetto S. A., Dasen J. S., Arber S. (2008). Transcriptional mechanisms controlling motor neuron diversity and connectivity. Curr. Opin. Neurobiol. 18, 36–43 [DOI] [PubMed] [Google Scholar]
- Devine C. A., Key B. (2008). Robo-Slit interactions regulate longitudinal axon pathfinding in the embryonic vertebrate brain. Dev. Biol. 313, 371–383 [DOI] [PubMed] [Google Scholar]
- Dillon A. K., Fujita S. C., Matise M. P., Jarjour A. A., Kennedy T. E., Kollmus H., Arnold H.-H., Weiner J. A., Sanes J. R., Kaprielian Z. (2005). Molecular control of spinal accessory motor neuron/axon development in the mouse spinal cord. J. Neurosci. 25, 10119–10130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon A. K., Jevince A. R., Hinck L., Ackerman S. L., Lu X., Tessier-Lavigne M., Kaprielian Z. (2007). UNC5C is required for spinal accessory motor neuron development. Mol. Cell. Neurosci. 35, 482–489 [DOI] [PubMed] [Google Scholar]
- Dugan J. P., Stratton A., Riley H. P., Farmer W. T., Mastick G. S. (2011). Midbrain dopaminergic axons are guided longitudinally through the diencephalon by Slit/Robo signals. Mol. Cell. Neurosci. 46, 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C., Steneberg P., Falileeva L., Xylourgidis N., Samakovlis C. (2002). Attractive and repulsive functions of Slit are mediated by different receptors in the Drosophila trachea. Development 129, 4941–4951 [DOI] [PubMed] [Google Scholar]
- Ericson J., Thor S., Edlund T., Jessell T., Yamada T. (1992). Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science 256, 1555–1559 [DOI] [PubMed] [Google Scholar]
- Farmer W. T., Altick A. L., Nural H. F., Dugan J. P., Kidd T., Charron F., Mastick G. S. (2008). Pioneer longitudinal axons navigate using floor plate and Slit/Robo signals. Development 135, 3643–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraher J. P., Dockery P., O’Donoghue O., Riedewald B., O’Leary D. (2007). Initial motor axon outgrowth from the developing central nervous system. J. Anat. 211, 600–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Frigola C., Carreres M. I., Vegar C., Mason C., Herrera E. (2008). Zic2 promotes axonal divergence at the optic chiasm midline by EphB1-dependent and -independent mechanisms. Development 135, 1833–1841 [DOI] [PubMed] [Google Scholar]
- Grieshammer U., Le M., Plump A. S., Wang F., Tessier-Lavigne M., Martin G. R. (2004). SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev. Cell 6, 709–717 [DOI] [PubMed] [Google Scholar]
- Guthrie S. (2007). Patterning and axon guidance of cranial motor neurons. Nat. Rev. Neurosci. 8, 859–871 [DOI] [PubMed] [Google Scholar]
- Guthrie S., Lumsden A. (1992). Motor neuron pathfinding following rhombomere reversals in the chick embryo hindbrain. Development 114, 663–673 [DOI] [PubMed] [Google Scholar]
- Harvey R. P. (1996). NK-2 homeobox genes and heart development. Dev. Biol. 178, 203–216 [DOI] [PubMed] [Google Scholar]
- He Z., Tessier-Lavigne M. (1997). Neuropilin is a receptor for the axonal chemorepellent semaphorin III. Cell 90, 739–751 [DOI] [PubMed] [Google Scholar]
- Hiramoto M., Hiromi Y., Giniger E., Hotta Y. (2000). The Drosophila Netrin receptor Frazzled guides axons by controlling Netrin distribution. Nature 406, 886–889 [DOI] [PubMed] [Google Scholar]
- Hirsch M.-R., Glover J. C., Dufour H. D., Brunet J.-F., Goridis C. (2007). Forced expression of Phox2 homeodomain transcription factors induces a branchio-visceromotor axonal phenotype. Dev. Biol. 303, 687–702 [DOI] [PubMed] [Google Scholar]
- Holland S. J., Gale N. W., Gish G. D., Roth R. A., Songyang Z., Cantley L. C., Henkemeyer M., Yancopoulos G. D., Pawson T. (1997). Juxtamembrane tyrosine residues couple the Eph family receptor EphB2/Nuk to specific SH2 domain proteins in neuronal cells. EMBO J. 16, 3877–3888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz A., Kollmus H., Ryge J., Niederkofler V., Dias J., Ericson J., Stoeckli E. T., Kiehn O., Arnold H. H. (2010). The transcription factors Nkx2.2 and Nkx2.9 play a novel role in floor plate development and commissural axon guidance. Development 137, 4249–4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. R., Marton M. J., Jones A. R., Roberts C. J., Stoughton R., Armour C. D., Bennett H. A., Coffey E., Dai H., He Y. D., et al. (2000). Functional discovery via a compendium of expression profiles. Cell 102, 109–126 [DOI] [PubMed] [Google Scholar]
- Imondi R., Kaprielian Z. (2001). Commissural axon pathfinding on the contralateral side of the floor plate: a role for B-class ephrins in specifying the dorsoventral position of longitudinally-projecting commissural axons. Development 128, 4859–4871 [DOI] [PubMed] [Google Scholar]
- Imondi R., Wideman C., Kaprielian Z. (2000). Complementary expression of transmembrane ephrins and their receptors in the mouse spinal cord: a possible role in constraining the orientation of longitudinally projecting axons. Development 127, 1397–1410 [DOI] [PubMed] [Google Scholar]
- Jacob J., Hacker A., Guthrie S. (2001). Mechanisms and molecules in motor neuron specification and axon pathfinding. BioEssays 23, 582–595 [DOI] [PubMed] [Google Scholar]
- Jevince A. R., Kadison S. R., Pittman A. J., Chien C.-B., Kaprielian Z. (2006). Distribution of EphB receptors and ephrin-B1 in the developing vertebrate spinal cord. J. Comp. Neurol. 497, 734–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadison S. R., Murakami F., Matise M. P., Kaprielian Z. (2006). The role of floor plate contact in the elaboration of contralateral commissural projections within the embryonic mouse spinal cord. Dev. Biol. 296, 499–513 [DOI] [PubMed] [Google Scholar]
- Kao T. J., Kania A. (2011). Ephrin-mediated cis-attenuation of Eph Receptor signaling is essential for spinal motor axon guidance. Neuron 71, 76–91 [DOI] [PubMed] [Google Scholar]
- Kidd T., Bland K. S., Goodman C. S. (1999). Slit is the midline repellent for the Robo receptor in Drosophila. Cell 96, 785–794 [DOI] [PubMed] [Google Scholar]
- Kolodkin A. L., Levengood D. V., Rowe E. G., Tai Y.-T., Ginger R. J., Ginty D. D. (1997). Neuropilin is a semaphorin III receptor. Cell 90, 753–762 [DOI] [PubMed] [Google Scholar]
- Kramer S., Kidd T., Simpson J., Goodman C. (2001). Switching repulsion to attraction: changing responses to slit during transition in mesoderm migration. Science 292, 737–740 [DOI] [PubMed] [Google Scholar]
- Labrador J. P., O’Keefe D., Yoshikawa S., McKinnon R. D., Thomas J. B., Bashaw G. J. (2005). The homeobox transcription factor even-skipped regulates netrin-receptor expression to control dorsal motor-axon projections in Drosophila. Curr. Biol. 15, 1413–1419 [DOI] [PubMed] [Google Scholar]
- Landgraf M., Roy S., Prokop A., VijayRaghavan K., Bate M. (1999). even-skipped determines the dorsal growth of motor axon in Drosophila. Neuron 22, 43–52 [DOI] [PubMed] [Google Scholar]
- Layden M. J., Odden J. P., Schmid A., Garces A., Thor S., Doe C. Q. (2006). Zfh1, a somatic motor neuron transcription factor, regulates axon exit from the CNS. Dev. Biol. 291, 253–263 [DOI] [PubMed] [Google Scholar]
- Lee R., Petros T. J., Mason C. A. (2008). Zic2 regulates retinal ganglion cell axon avoidance of ephrinB2 through inducing expression of the guidance receptor EphB1. J. Neurosci. 28, 5910–5919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberam I., Agalliu D., Nagasawa T., Ericson J., Jessell T. M. (2005). A Cxcl12-Cxcr4 chemokine signaling pathway defines the initial trajectory of mammalian motor axons. Neuron 47, 667–679 [DOI] [PubMed] [Google Scholar]
- Long H., Sabatier C., Ma L., Plump A., Yuan W., Ornitz D. M., Tamada A., Murakami F., Goodman C. S., Tessier-Lavigne M. (2004). Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron 42, 213–223 [DOI] [PubMed] [Google Scholar]
- Lopez-Bendito G., Flames N., Ma L., Fouquet C., Meglio T. D., Chedotal A., Tessier-Lavigne M., Marin O. (2007). Robo1 and Robo2 cooperate to control the guidance of major axonal tracts in the mammalian forebrain. J. Neurosci. 27, 3395–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., van Eerde A. M., Fan X., Quintero-Rivera F., Kulkarni S., Ferguson H., Kim H. G., Fan Y., Xi Q., Li Q. G., et al. (2007). Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am. J. Hum. Genet. 80, 616–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maness P. F., Schachner M. (2007). Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 10, 19–26 [DOI] [PubMed] [Google Scholar]
- Maro G. S., Vermeren M., Voiculescu O., Melton L., Cohen J., Charnay P., Topilko P. (2004). Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat. Neurosci. 7, 930–938 [DOI] [PubMed] [Google Scholar]
- Mastick G. S., Farmer W. T., Altick A. L., Nural H. F., Dugan J. P., Kidd T., Charron F. (2010). Longitudinal axons are guided by Slit/Robo signals from the floor plate. Cell Adh. Migr. 4, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauti O., Domanitskaya E., Andermatt I., Sadhu R., Stoeckli E. T. (2007). Semaphorin6A acts as a gate keeper between the central and the peripheral nervous system. Neural Dev. 2, 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane S. (2003). Metalloproteases: carving out a role in axon guidance. Neuron 37, 559–562 [DOI] [PubMed] [Google Scholar]
- Moret F., Renaudot C., Bozon M., Castellani V. (2007). Semaphorin and neuropilin co-expression in motoneurons sets axon sensitivity to environmental semaphorin sources during motor axon pathfinding. Development 134, 4491–4501 [DOI] [PubMed] [Google Scholar]
- Pabst O., Herbrand H., Arnold H. H. (1998). Nkx2-9 is a novel homeobox transcription factor which demarcates ventral domains in the developing mouse CNS. Mech. Dev. 73, 85–93 [DOI] [PubMed] [Google Scholar]
- Pabst O., Rummelies J., Winter B., Arnold H.-H. (2003). Targeted disruption of the homeobox gene Nkx2. 9 reveals a role in development of the spinal accessory nerve. Development 130, 1193–1202 [DOI] [PubMed] [Google Scholar]
- Palmer A., Klein R. (2003). Multiple roles of ephrins in morphogenesis, neuronal networking, and brain function. Genes Dev. 17, 1429–1450 [DOI] [PubMed] [Google Scholar]
- Pattyn A., Hirsch M.-R., Goridis C., Brunet J.-F. (2000). Control of hindbrain motor neuron differentiation by the homeobox gene Phox2b. Development 127, 1349–1358 [DOI] [PubMed] [Google Scholar]
- Pfaff S. L., Mendelsohn M., Stewart C. L., Edlund T., Jessell T. M. (1996). Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell 84, 309–320 [DOI] [PubMed] [Google Scholar]
- Plump A., Erskine L., Sabatier C., Brose K., Epstein C., Goodman C., Mason C., Tessier-Lavigne M. (2002). Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron 33, 219–232 [DOI] [PubMed] [Google Scholar]
- Reeber S. L., Sakai N., Nakada Y., Dumas J., Dobrenis K., Johnson J. E., Kaprielian Z. (2008). Manipulating Robo expression in vivo perturbs commissural axon pathfinding in the chick spinal cord. J. Neurosci. 28, 8698–8708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricano-Cornejo I., Altick A. L., Garcia-Pena C. M., Nural H. F., Echevarria D., Miquelajauregui A., Mastick G. S., Varela-Echavarria A. (2011). Slit-Robo signals regulate pioneer axon pathfinding of the tract of the postoptic commissure in the mammalian forebrain. J. Neurosci. Res. 89, 1531–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Almodovar C., Lambrechts D., Mazzone M., Carmeliet P. (2009). Role and therapeutic potential of VEGF in the nervous system. Physiol. Rev. 89, 607–648 [DOI] [PubMed] [Google Scholar]
- Schachter P. P., Ayesh S., Schneider T., Laster M., Czerniak A., Hochberg A. (2002). Expression of kinase genes in primary hyperparathyroidism: adenoma versus hyperplastic parathyroid tissue. Surgery 132, 1094–1099 [DOI] [PubMed] [Google Scholar]
- Schneider V. A., Granato M. (2003). Motor axon migration: a long way to go. Dev. Biol. 263, 1–11 [DOI] [PubMed] [Google Scholar]
- Schneider V. A., Granato M. (2006). The myotomal diwanka (lh3) glycosyltransferase and type XVIII collagen are critical for motor growth cone migration. Neuron 50, 683–695 [DOI] [PubMed] [Google Scholar]
- Schubert W., Kaprielian Z. (2001). Identification and characterization of a cell surface marker for embryonic rat spinal accessory motor neurons. J. Comp. Neurol. 439, 368–383 [DOI] [PubMed] [Google Scholar]
- Sharma K., Peng C.-Y. (2001). Spinal motor circuits: Merging development and function. Neuron 29, 321–324 [DOI] [PubMed] [Google Scholar]
- Sharma K., Sheng H. Z., Lettieri K., Li H., Karavanov A., Potter S., Westphal H., Pfaff S. L. (1998). LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell 95, 817–828 [DOI] [PubMed] [Google Scholar]
- Shiau C. E., Bronner-Fraser M. (2009). N-cadherin acts in concert with Slit1-Robo2 signaling in regulating aggregation of placode-derived cranial sensory neurons. Development 136, 4155–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki R., Pfaff S. L. (2002). Transcriptional codes and the control of neuronal identity. Ann. Rev. Neurosci. 25, 251–281 [DOI] [PubMed] [Google Scholar]
- Snider W. D., Palavali V. (1990). Early axon and dendritic outgrowth of spinal accessory motor neurons studied with DiI in fixed tissues. J. Comp. Neurol. 297, 227–238 [DOI] [PubMed] [Google Scholar]
- Stanfel M. N., Moses K. A., Schwartz R. J., Zimmer W. E. (2005). Regulation of organ development by the NKX-homeodomain factors: an NKX code. Cell. Mol. Biol. (Noisy-le-grand) 51 Suppl, OL785–OL799 [PubMed] [Google Scholar]
- Swartz M. E., Eberhart J., Pasquale E. B., Krull C. E. (2001). EphA4/ephrin-A5 interactions in muscle precursor cell migration in the avian forelimb. Development 128, 4669–4680 [DOI] [PubMed] [Google Scholar]
- Tian J., Mahmood R., Hnasko R., Locker J. (2006). Loss of Nkx2.8 deregulates progenitor cells in the large airways and leads to dysplasia. Cancer Res. 66, 10399–10407 [DOI] [PubMed] [Google Scholar]
- Vermeren M., Maro G. S., Bron R., McGonnell I. M., Charnay P., Topilko P., Cohen J. (2003). Integrity of developing spinal motor columns is regulated by neural crest derivatives at motor exit points. Neuron 37, 403–415 [DOI] [PubMed] [Google Scholar]
- Vosshall L. B., Wong A. M., Axel R. (2000). An olfactory sensory map in the fly brain. Cell 102, 147–159 [DOI] [PubMed] [Google Scholar]
- Wang H., Zhu Y. Z., Wong P. T., Farook J. M., Teo A. L., Lee L. K., Moochhala S. (2003). cDNA microarray analysis of gene expression in anxious PVG and SD rats after cat-freezing test. Exp. Brain Res. 149, 413–421 [DOI] [PubMed] [Google Scholar]
- Wilson S. I., Shafer B., Lee K. J., Dodd J. (2008). A molecular program for contralateral trajectory: Rig-1 control by LIM homeodomain transcription factors. Neuron 59, 413–424 [DOI] [PubMed] [Google Scholar]
- Ye B. Q., Geng Z. H., Ma L., Geng J. G. (2010). Slit2 regulates attractive eosinophil and repulsive neutrophil chemotaxis through differential srGAP1 expression during lung inflammation. J. Immunol. 185, 6294–6305 [DOI] [PubMed] [Google Scholar]
- Ypsilanti A. R., Zagar Y., Chedotal A. (2010). Moving away from the midline: new developments for Slit and Robo. Development 137, 1939–1952 [DOI] [PubMed] [Google Scholar]
- Yuan L., Hillman J. D., Progulske-Fox A. (2005). Microarray analysis of quorum-sensing-regulated genes in Porphyromonas gingivalis. Infect. Immun. 73, 4146–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W., Zhou L., Chen J.-h., Wu J., Rao Y., Ornitz D. (1999). The mouse Slit family: secreted ligands for Robo expressed in patterns that suggest a role in morphogenesis and axon guidance. Dev. Biol. 212, 290–306 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}