

Abstract
A common theme in developmental biology is the repeated use of the same gene in diverse spatial and temporal domains, a process that generally involves transcriptional regulation mediated by multiple separate enhancers, each with its own arrangement of transcription factor (TF)-binding sites and associated activities. Here, by contrast, we show that the expression of the Drosophila Nidogen (Ndg) gene at different embryonic stages and in four mesodermal cell types is governed by the binding of multiple cell-specific Forkhead (Fkh) TFs – including Biniou (Bin), Checkpoint suppressor homologue (CHES-1-like) and Jumeau (Jumu) – to three functionally distinguishable Fkh-binding sites in the same enhancer. Whereas Bin activates the Ndg enhancer in the late visceral musculature, CHES-1-like cooperates with Jumu to repress this enhancer in the heart. CHES-1-like also represses the Ndg enhancer in a subset of somatic myoblasts prior to their fusion to form multinucleated myotubes. Moreover, different combinations of Fkh sites, corresponding to two different sequence specificities, mediate the particular functions of each TF. A genome-wide scan for the occurrence of both classes of Fkh domain recognition sites in association with binding sites for known cardiac TFs showed an enrichment of combinations containing the two Fkh motifs in putative enhancers found within the noncoding regions of genes having heart expression. Collectively, our results establish that different cell-specific members of a TF family regulate the activity of a single enhancer in distinct spatiotemporal domains, and demonstrate how individual binding motifs for a TF class can differentially influence gene expression.
Keywords: Transcription factors, Transcription factor binding sites, Forkhead proteins, Transcriptional regulation, Enhancers, Mesoderm
INTRODUCTION
The Drosophila embryonic mesoderm gives rise to multiple tissues and organs in the larva and adult fly, including the heart, the gut musculature, the somatic muscles, fat body and hemocytes. The developmental processes associated with the formation of these derivatives require the coordinated regulation of a diverse array of genes in precise spatial and temporal expression patterns (Frasch, 1999; Tao and Schulz, 2007; Bonn and Furlong, 2008; Busser et al., 2008; Tixier et al., 2010). Previous work has shown that gene expression in subsets of Drosophila embryonic mesodermal cells involves the binding of multiple signal-activated, tissue- and cell-specific transcription factors (TFs) to transcriptional enhancers that integrate these intrinsic and extrinsic combinatorial inputs (Xu et al., 1998; Halfon et al., 2000; Lee and Frasch, 2005; Estrada et al., 2006; Philippakis et al., 2006).
Although the identities of some of the TFs and binding sites that comprise several model mesodermal enhancers have been described (Gajewski et al., 1997; Cripps et al., 1998; Gajewski et al., 1998; Xu et al., 1998; Kremser et al., 1999; Halfon et al., 2000; Gajewski et al., 2001; Knirr and Frasch, 2001; Han et al., 2002; Lee and Frasch, 2005; Wang et al., 2005; Estrada et al., 2006; Philippakis et al., 2006; Tao et al., 2007), the transcriptional codes that are currently known cannot account for the complete specificity of target gene expression at the resolution of single cells. Thus, the challenge is both to expand and refine these transcriptional codes by examining the functions of other TFs that participate in individual regulatory networks. Of particular relevance are those TFs that are expressed in small subsets of cells and thus are candidate cell type-specific regulators. One such class is the Forkhead (Fkh) family of TFs. Although the mesodermal expression patterns and developmental functions of at least 12 mammalian Fkh TFs have been characterized (Carlsson and Mahlapuu, 2002; Wijchers et al., 2006), the functions of only three of the seven Fkh genes expressed in one or more Drosophila mesodermal cell types (Grossniklaus et al., 1992; Zaffran et al., 2001; Lee and Frasch, 2004) are currently known.
In the present study, we identified three Fkh-binding sites in the enhancer of Nidogen (Ndg), a gene that is expressed at different stages and in multiple mesodermal cell types. These binding sites, which correspond to two distinct sequence specificities, were used to examine the roles of Fkh TFs in regulating various temporal and spatial patterns of mesodermal gene expression. Our results show that different tissue-specific Fkh TFs mediate distinct gene expression responses through differential use of the same binding sites in a single enhancer, and support a role for these factors in determining the unique genetic programs that characterize different subtypes of mesodermal cells.
MATERIALS AND METHODS
Identification of Fkh binding sites in the Ndg enhancer
The binding specificities of five mouse Fkh TFs, Foxa2, Foxj1, Foxj3, Foxk1 and Foxl1 (Robasky and Bulyk, 2011) were used to identify three Fkh-binding sites within a previously characterized mesodermal enhancer from the Ndg gene (Philippakis et al., 2006) (Fig. 1; supplementary material Table S1). The ability of these sites to bind relevant Drosophila Fkh TFs was subsequently confirmed by electrophoretic mobility shift assays (EMSAs).
Fig. 1.

Fkh transcription factor binding sites in the Ndg enhancer. (A) Logo representations of the PWMs of the Forkhead primary (FkhP) and secondary (FkhS) binding motifs. (B) The Ndg enhancer is located within the first intron of the gene. The relative locations of three Fkh-binding sites, Fkh1, Fkh2 and Fkh3, are shown, as well as their wild-type sequences (uppercase). The nucleotide substitutions used to create the binding site mutations are shown in bold lowercase. (C) Local alignment among 12 Drosophila genomes showing the Fkh1-binding site. (D) Local alignment showing the Fkh2- and Fkh3-binding sites. (E) Biniou binds to all three wild-type Fkh sites in EMSAs. The binding at each site can be competed with an excess of unlabeled probe corresponding to the relevant wild-type Fkh site, but not the mutated Fkh site or the non-specific competitor. (F) Similarly, CHES-1-like binds to all three wild-type Fkh sites, and the binding at each site can be competed only with an excess of unlabeled probe corresponding to the relevant wild-type Fkh site. (G) By contrast, the binding of Jumu was detected in vitro only at the Fkh2 site, with only an excess of wild-type but not mutated Fkh2 unlabeled probe being capable of competing. (H) The sequences of the probes used in the EMSAs.
Electrophoretic mobility shift assays
EMSAs were performed using biotinylated probes produced by the Biotin 3′ End DNA Labeling Kit (Thermo Fisher Scientific) and Bin, CHES-1-like and Jumu proteins. For Bin and CHES-1-like, GST-FKH domain fusion proteins were synthesized using the PURExpress In Vitro Protein Synthesis Kit (NEB). For Jumu, His-tagged, full-length protein was expressed in E. coli and purified using Ni-NTA agarose resin (Invitrogen). DNA-binding reactions were carried out at room temperature for 30 minutes in a buffer containing biotinylated probes (10 fmol), 10 mM Tris (pH 7.5), 100 mM KCl, 1 mM DTT, 50 ng/μl of Poly (dI-dC), 5% glycerol and proteins. For competitive EMSAs, a 100-fold molar excess of the indicated unlabeled probes was used, which have sequences corresponding to the relevant wild-type Fkh sites, the mutated Fkh sites used in the reporters, and a non-coding sequence from the Drosophila genome lacking specific Fkh-binding sites (the non-specific competitor). Reaction products were resolved by being run on 6% non-denaturing PAGE gels, and signals were produced with the LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific) as described in the manufacturer’s protocol.
DNA constructs and fly transformation
Ndg enhancers with mutant Fkh-binding sites were synthesized by Integrated DNA Technologies, with mutations designed to match nonbinding k-mers from published data (Robasky and Bulyk, 2011). In each case, the mutation was designed to substitute the minimum number of nucleotides and to avoid creating or altering the binding sites of other known TFs. Versions of the Ndg enhancer were cloned into the pWattB-GFP vector (Busser et al., 2012b) and microinjected into Drosophila embryos containing a specific attP site to facilitate site-specific recombination (Groth et al., 2004; Markstein et al., 2008). A P-element construct containing a β-galactosidase reporter driven by the wild-type Ndg enhancer was used as a control (Philippakis et al., 2006).
Drosophila strains and genetics
The following mutant alleles, deficiencies and transgenes were used: binl1 and UAS-bin (Zaffran et al., 2001); Df(3R)Exel6157, a small deficiency that deletes jumu; Df(1)CHES-1-like1, a null mutation at the CHES-1-like locus (S.M.A., T.R.T., B.W.B., M. T. Nolte, N.J., S. S. Gisselbrecht, N. M. Rusan and A.M.M., unpublished); jumu2.12 and UAS-jumu (Strodicke et al., 2000; Hofmann et al., 2009); TinD-GAL4 (Yin et al., 1997); twi-GAL4 (Greig and Akam, 1993); and Hand-GAL4 (Han and Olson, 2005). Mutant chromosomes were maintained over the TM3, ftz-lacZ balancer.
In situ hybridization, immunohistochemistry and cell counting
Embryo fixation, probe synthesis and histochemical staining were carried out as described previously (Estrada et al., 2006). For all quantitative studies of gene expression, cells in over 100 hemisegments were counted for each genotype examined.
RNA interference assays
RNA interference assays were performed as previously described (Estrada et al., 2006). More than 15 live embryos were analyzed for each injection, and each dsRNA was injected and scored blindly.
Statistical methods
The comparisons of expression changes in cardial cells (CCs) and pericardial cells (PCs) by mutation type are based on comparing the average number of errors (de-repressed cells) per hemisegment in each mutation group. A comparison using t-tests or analysis of variance techniques is not appropriate as the hemisegments for a given embryo are correlated in their likelihood of having errors. Bootstrap and permutation approaches (Good, 1994) were used here to address this correlation and the additional complication that the wild type showed no errors (and hence no variation) for some assays. Permutation tests were used to test for differences in errors per hemisegment between two given mutation types. Bootstrap approaches were used to determine whether non-additive interactions exist among mutation types and if the number of de-repressions per hemisegment differed between morphologically normal and abnormal hemisegments of a given mutation type.
Lever analysis
The Lever algorithm (Warner et al., 2008) was used to derive combinations of motifs that may participate in the regulation of gene sets expressed in different cell types in the Drosophila embryonic mesoderm. An improved method for correcting for the variable lengths of noncoding genomic regions was implemented, as described previously (A.A. and M.L.B., unpublished).
RESULTS
Fkh binding sites in a mesodermal enhancer associated with the Ndg gene
Our previous work identified an enhancer associated with the Ndg gene that recapitulates endogenous expression in multiple mesodermal cell types where Fkh TFs are expressed, including several somatic muscle founder cells (FCs), subsets of both the pericardial cells (PCs) and cardial cells (CCs) of the heart, and the visceral musculature (VM) (Philippakis et al., 2006; see below). Given the relative paucity of information about Fkh protein function in Drosophila, we used the known binding specificities of 5 mouse Fkh TFs (Badis et al., 2009) to detect potential Fkh-binding sites within the Ndg enhancer. The binding preferences of these mammalian Fkh TFs were previously shown to correspond to a canonical (primary) Fkh-binding motif (hereafter referred to as FkhP), the effects of which have been extensively investigated (Zaffran et al., 2001; Zaffran and Frasch, 2002; Popichenko et al., 2007; Zinzen et al., 2009), and a secondary motif (FkhS), which represents an alternate binding preference (Badis et al., 2009). We identified three Fkh-binding sites within the Ndg enhancer: Fkh1, which reflects the FkhP motif; and the adjacent and overlapping sites Fkh2 and Fkh3, which match the FkhS motif (Fig. 1A,B). Of note, sequences corresponding to both the FkhP and FkhS motifs were found at the same sites in other Drosophila species (supplementary material Table S1). Electrophoretic mobility shift assays (EMSAs) confirmed that three Drosophila mesodermal Fkh TFs bind to these sites (Fig. 1E-H). Although the binding to some of these sites is relatively weak, albeit sequence specific, we note that low-affinity binding of some TFs has previously been shown to be developmentally important (Rowan et al., 2010; Parker et al., 2011), and that actual in vivo binding affinities can be modulated by interactions with potential co-factors and collaborating TFs that are not present in in vitro assays.
To test whether Fkh TFs regulate the Ndg enhancer, we used relevant protein binding microarray data to create a series of Fkh-binding site mutations in the wild-type sequence (Fig. 1B; supplementary material Table S1). Competitive EMSAs demonstrated that each of these mutations resulted in significant loss of binding of the relevant Fkh TFs to these sites (Fig. 1E-H). The wild-type and mutant versions of the Ndg enhancer were cloned in GFP reporter vectors and independently inserted into precisely the same location on the third chromosome (Groth et al., 2004; Markstein et al., 2008). Thus, any differences in reporter expression between the wild-type and mutant enhancer constructs is attributable to the Fkh-binding site mutations and not to local positional effects.
Fkh regulation of Ndg expression in the visceral mesoderm
To examine the roles of Fkh TFs in regulating the Ndg enhancer, we examined embryos that contained Ndg-GFP reporters with various Fkh site mutations and in which β-galactosidase expression driven by a NdgWT-lacZ reporter was used as a wild-type reference to assess the activities of the mutant GFP reporters. The wild-type Ndg enhancer-reporter construct is expressed strongly in the midgut VM from stage 15 onwards (Fig. 2A-A″), which is similar to the expression of the endogenous gene (supplementary material Fig. S1D). Mutations in either the Fkh2 or Fkh3 sites did not have a significant effect on GFP reporter expression in the midgut mesoderm (Fig. 2B-C″); however, mutating both Fkh2 and Fkh3 sites simultaneously resulted in a dramatic decrease in VM expression (Fig. 2D-D″). Mutating only the Fkh-binding site also caused a significant decrease in Ndg reporter expression (Fig. 2E-E″). Finally, mutating all three Fkh sites in the same enhancer resulted in complete inactivation of the GFP reporter in the VM (Fig. 2F-F″). Taken together, these results indicate that the wild-type VM expression of Ndg requires binding of a Fkh TF to both the Fkh1 site and either the Fkh2 or the Fkh3 site.
Fig. 2.

Expression driven by the Ndg enhancer in the gut musculature is significantly reduced by mutating either the Fkh1 site or both Fkh2 and Fkh3 sites simultaneously. (A-F″) The expression of GFP reporters for the wild-type (A-A″) and mutated (B-F″) Ndg enhancers was compared at stage 16. Expression of a β-galactosidase reporter from the wild-type Ndg enhancer, NdgWT-lacZ, was used in every embryo as a reference and an internal control. Anterior is towards the left in every panel. Mutating either the Fkh2 (B-B″) or the Fkh3 (C-C″) site alone had no significant effect on reporter expression in the gut musculature, while mutating both these sites simultaneously (D-D″) resulted in a dramatic decrease in visceral muscle expression. Mutating only the Fkh1-binding site (E-E″) also caused a significant decrease in Ndg reporter expression, while mutating all three sites simultaneously (F-F″) resulted in the complete inactivation of GFP reporter expression in the visceral muscle. Note that although Ndg expression in the heart (arrows) is visible in every embryo, it is more readily apparent for the GFP reporters, which is a consequence of: (1) GFP being expressed throughout the entire cytoplasm, as opposed to β-galactosidase, which is confined to the much smaller nuclei; (2) the plane of focus being on the gut muscle and not the heart; and (3) de-repression into additional cardiac cells occurring only for the GFP reporters in which Fkh sites are mutated in B-F″ (also see Fig. 4 and the main text).
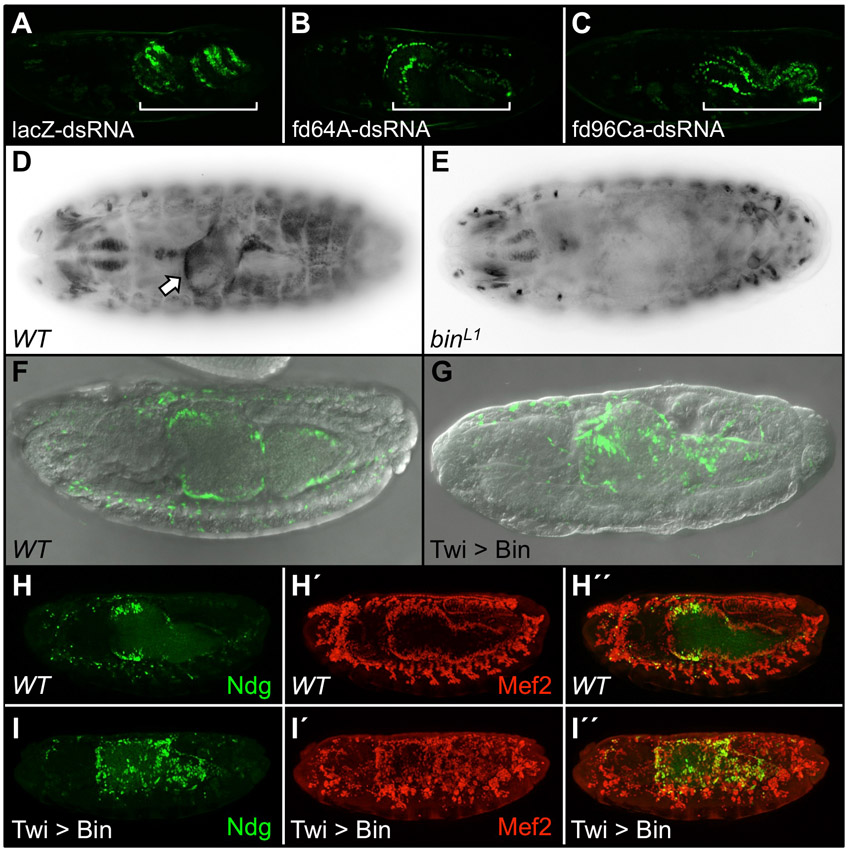
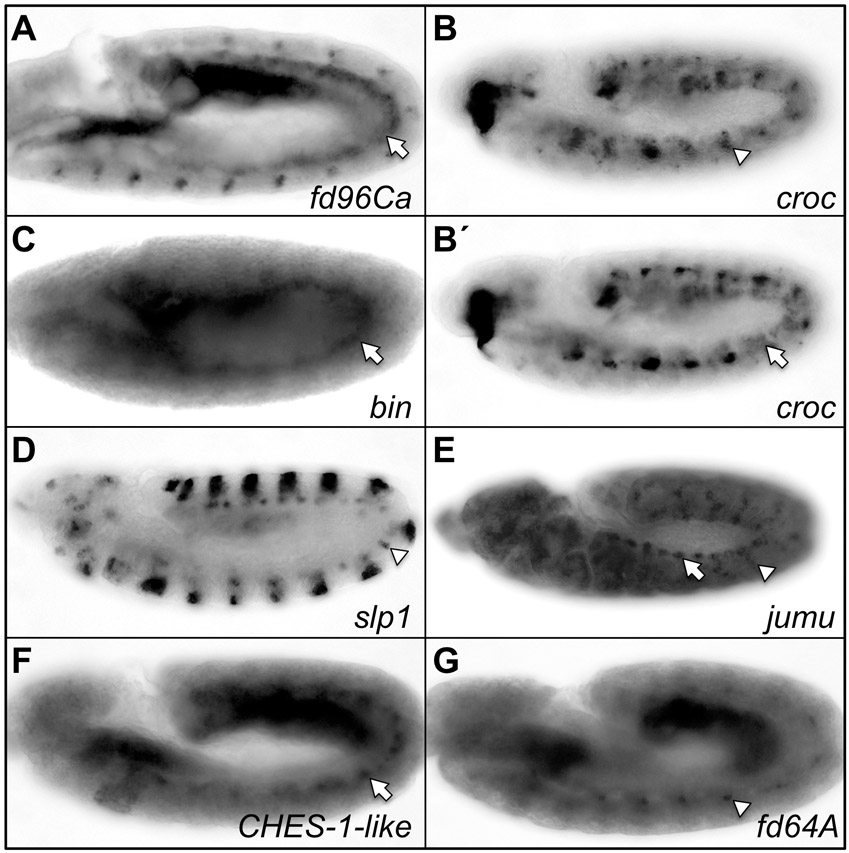
In order to determine which Fkh TF binds to these sites to regulate Ndg expression in the VM, we used both the literature (Lee and Frasch, 2004) and our own whole-embryo in situ hybridizations to identify all Fkh genes that are expressed in this tissue. The Fkh gene fd96Ca is expressed early in the VM, fd64A is expressed late in the gut musculature and biniou (bin) is expressed both early and late in the VM (supplementary material Table S2, Fig. S2). We next assessed whether blocking the function of any of these VM-specific Fkh TFs affects expression of either the wild-type Ndg enhancer-reporter or the endogenous Ndg gene. As no loss-of-function mutations are available for either fd64A or fd96Ca, their effects were assessed by RNA interference. Embryos injected with dsRNAs corresponding to either fd64A or fd96Ca showed no reduction in NdgWT-GFP reporter expression levels compared with the lacZ dsRNA control (supplementary material Fig. S1A-C), indicating that these Fkh genes do not regulate Ndg expression in the VM. Another Fkh TF, crocodile (supplementary material Fig. S2B′), was not considered as a candidate for regulating the late VM expression of Ndg because it is not co-expressed with Ndg in the same visceral muscle layer.
Thus, Bin remained the only candidate Fkh TF for controlling the VM activity of the Ndg enhancer, and in situ hybridization experiments demonstrated that endogenous midgut expression of Ndg is completely eliminated in embryos homozygous for the binL1 allele (supplementary material Fig. S1E). Furthermore, EMSAs showed that Bin bound specifically to all three of the wild-type Fkh motifs in vitro, and that this binding could be competed with the wild type but not with the mutated binding sites (Fig. 1E), results that correlated with the effect of mutated Fkh sites on enhancer activity. However, as loss of bin function results in the transformation of a large proportion of the VM into a somatic body wall muscle fate (Zaffran et al., 2001), it is possible that the absence of Ndg expression in bin mutants could be an indirect effect.
To further assess the potential role of Bin in regulating the Ndg enhancer, twist-GAL4 was used to overexpress Bin throughout the mesoderm. Although this manipulation causes the conversion of somatic and cardiac into visceral mesoderm (Zaffran et al., 2001), an increase in mesodermal Ndg reporter expression was induced by ectopic Bin (supplementary material Fig. S1F-I″). As with loss of bin function, this result is consistent with a direct role of Bin in regulating the Ndg enhancer, but the possibility that it is an indirect consequence of a cell fate transformation cannot be eliminated. Nevertheless, as the Ndg enhancer appears to be inactive in bin mutant embryos but is unaffected by loss of the other VM Fkh TFs, Bin is the most likely candidate Fkh TF regulating Ndg expression in the midgut musculature. Note that this does not preclude other, non-Forkhead, TFs from also playing a role in activating the Ndg enhancer in the VM.
Fkh regulation of Ndg expression in somatic myoblasts
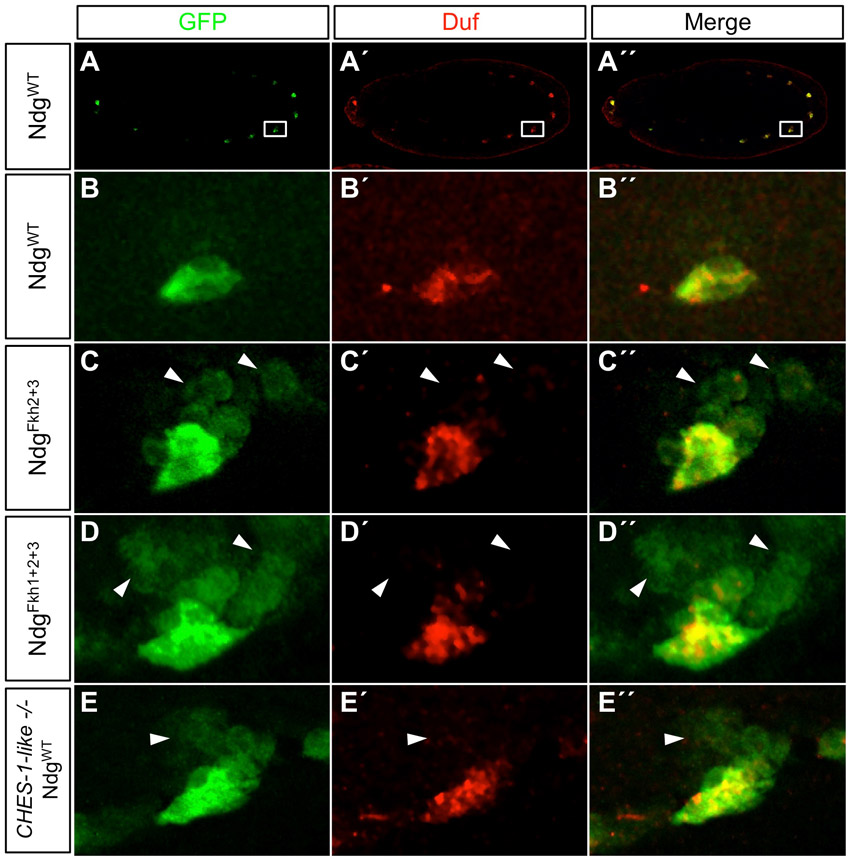
Ndg is expressed at embryonic stage 11 in a group of FCs in the ventral mesoderm of each abdominal hemisegment (Estrada et al., 2006; Philippakis et al., 2006), a pattern that is recapitulated by the wild-type Ndg enhancer-reporter (Fig. 3A-A″′; supplementary material Fig. S3A-B″′, Fig. S4A-B″). Neither the Ndg gene nor this GFP reporter is expressed in FCMs, cells that express the lame duck (lmd) gene and have a different developmental origin from FCs (Duan et al., 2001) (Fig. 3A-A″′). Individual mutations of the Fkh1-, Fkh2- or Fkh3-binding sites in the Ndg enhancer caused no alteration in reporter expression in somatic myoblasts (data not shown). However, mutating both the Fkh2 and Fkh3 sites simultaneously, or all three Fkh sites together, resulted in de-repression of the normal FC-restricted expression of the Ndg reporter into nearby lmd-expressing FCMs without affecting enhancer activity in FCs (Fig. 3B-C″′; supplementary material Figs S3, S4). These findings indicate that one or more TFs repress the Ndg enhancer in FCMs by acting through either the Fkh2 or Fkh3 site.
Fig. 3.

CHES-1-like represses Ndg expression in the FCMs by acting through either the Fkh2 or Fkh3 sites. Lateral views of stage 11 embryos with FCMs marked by the expression of Lmd (red) at low (A-D) and high (A′-D″′) magnification. (A-A″′) GFP expression driven by the wild-type Ndg enhancer (green) is limited to somatic FCs that do not express the FCM marker Lmd. (B-C″′) Mutating either Fkh2 and Fkh3 sites simultaneously (B-B″′) or all three Fkh sites together (C-C″′) results in the de-repression of reporter expression into Lmd-expressing FCMs (arrows) that are adjacent to the normal Ndg-positive FCs. (D-D″′) Similarly, in embryos homozygous for the CHES-1-like null mutation, reporter expression from the wild-type Ndg enhancer is also de-repressed into adjacent FCMs (arrows).
Of the 17 D. melanogaster Fkh genes, only Checkpoint suppressor homologue (CHES-1-like) is expressed in FCMs (Estrada et al., 2006; supplementary material Table S2 and Fig. S2F, Fig. S5A-A″), thus making it the only candidate for the Fkh TF repressor in this subset of mesodermal cells. Moreover, EMSAs showed that CHES-1-like bound specifically to all three of the wild-type Fkh-binding sites in vitro, and that this binding could be competed with the wild type but not with the mutated binding sites (Fig. 1F), results that further strengthen this hypothesis. Therefore, in order to ascertain whether CHES-1-like actually represses Ndg expression in the FCMs, we examined embryos homozygous for the CHES-1-like null mutation Df(1)CHES-1-like1. In the complete absence of CHES-1-like function, activity of the wild-type Ndg enhancer-reporter is de-repressed into Lmd-expressing FCMs (Fig. 3D-D″′; supplementary material Fig. S3G-H″′), demonstrating that CHES-1-like is indeed the Fkh TF responsible for repressing Ndg expression in FCMs. Note, in particular, that this de-repression is a direct consequence of the absence of CHES-1-like on the Ndg enhancer; no supernumerary FCs, which could also explain the additional Ndg-expressing cells, are produced in CHES-1-like mutants (supplementary material Fig. S4E-E″).
Of note, the degree of de-repression of the NdgWT-GFP reporter in CHES-1-like null mutants is lower than that of the NdgFkh1+2+3-GFP or NdgFkh2+3-GFP reporters in wild-type embryos (Fig. 3; supplementary material Figs S3, S4). One plausible explanation for these observations is the existence of another repressor, which binds to at least one region overlapping the Fkh sites in the Ndg enhancer. In CHES-1-like mutants, this additional repressor could still function, resulting in a reduced level of Ndg de-repression. In addition, the de-repression caused by either mutating the Fkh sites in the Ndg enhancer or by eliminating CHES-1-like function is detected in only a small subset of FCMs, further suggesting the existence of another FCM-restricted repressor.
Fkh regulation of Ndg expression in the heart
The Drosophila heart at embryonic stage 16 consists of multiple cell types arranged in a metamerically repeated and stereotyped pattern. Each hemisegment consists of a row of six CCs representing distinct subtypes based on the expression of various marker genes. From anterior to posterior, and named after the TFs they express, there are two Seven-up-CCs (Svp-CCs), two Tinman-Ladybird-CCs (Tin-Lb-CCs) and two CCs expressing only Tin (Tin-CCs). A larger number of PCs surround the cardial cells: four Odd-skipped-PCs (Odd-PCs) are positioned laterally, two Even-skipped-PCs (Eve-PCs) are situated dorsolaterally and a row of Tin-PCs runs immediately ventral to the CCs (Azpiazu and Frasch, 1993; Bodmer, 1993; Jagla et al., 1997; Ward and Skeath, 2000). The wild-type Ndg reporter is expressed in only two out of the six CCs (the Tin-Lb-CCs; Fig. 4A-A″′,G; supplementary material Table S3 and Fig. S6), in all of the Tin-PCs lying immediately ventral to the CCs, and in a small number (less than one cell per hemisegment, on average) of the Odd-PCs (Fig. 4A-A″′,H; supplementary material Table S3).
Fig. 4.

Expression driven by the Ndg enhancer is de-repressed in the heart by mutating any of the Fkh sites. (A-F″′) The posterior-most four cardial cells (the Tin-Lb-CCs and the Tin-CCs) are marked by Tin expression (red), and the Odd-PCs are marked by Zfh1 expression (blue) in abdominal segments A3 and A4. (A-A″′) The GFP reporter driven by the wild-type Ndg enhancer is expressed in only two cardial cells per hemisegment (green): the Tin-Lb-CCs (square bracket). (B-F″′) Mutations in one or more of the Fkh-binding sites result in the de-repression of reporter expression in additional CCs (arrows) and into the Odd-PCs (arrowheads). (G,H) Histograms showing the mean number of cells expressing the reporter and the significance of de-repression into CCs (G) and Odd-PCs (H) as a result of Fkh-binding site mutations.
Mutagenesis of any one of the three Fkh-binding sites in the Ndg enhancer causes significant de-repression of GFP reporter expression from the Tin-Lb-CCs into the neighboring Svp-CCs and Tin-CCs (Fig. 4B-D″′,G; supplementary material Table S3) and in the Odd-PCs (Fig. 4B-D″′,H; supplementary material Table S3). As expected, mutations in multiple Fkh-binding sites also exhibited similar de-repression (Fig. 4E-H; supplementary material Table S3). Activity of each mutant enhancer in the Tin-PCs was entirely unaffected (supplementary material Fig. S7).
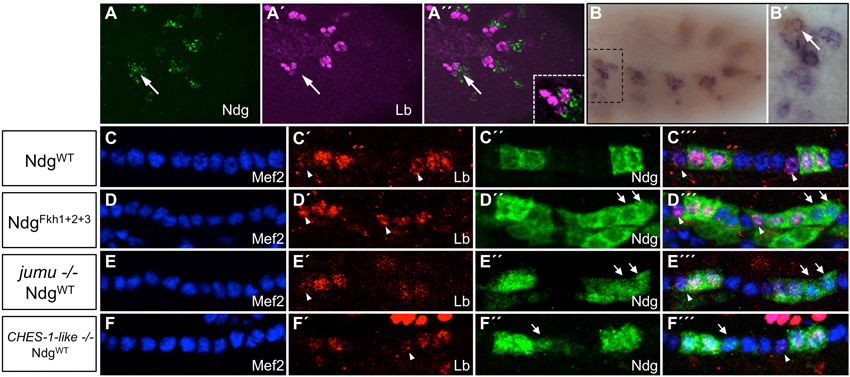
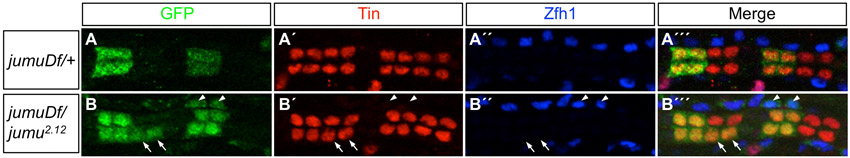
Only two Drosophila Fkh genes, CHES-1-like and jumu, are expressed in the cardiac mesoderm (supplementary material Table S2 and Figs S2, S5), and EMSAs showed that while CHES-1-like bound to all three wild-type Fkh sites in the Ndg enhancer, Jumu bound specifically to only the wild-type Fkh2 site in vitro (Fig. 1F,G). We therefore considered both CHES-1-like and Jumu as candidates for repressing Ndg expression in the heart. Embryos homozygous for the CHES-1-like null mutation, Df(1)CHES-1-like1, homozygous for the jumu null deficiency, Df(3R)Exel6157, or bearing the jumu2.12 hypomorphic allele in trans to Df(3R)Exel6157, all exhibited significant de-repression of the wild-type Ndg enhancer-reporter in both CCs and Odd-PCs (Fig. 5A-D″′; supplementary material Table S3 and Fig. S8), precisely as observed for versions of the Ndg enhancer that contain mutations in Fkh-binding sites. The convergence of results for these cis and trans experiments suggests that CHES-1-like and Jumu both repress the Ndg enhancer in all heart cells, excluding the Tin-Lb-CCs and ventral Tin-PCs.
Fig. 5.

Jumu and CHES-1-like repress activity of the Ndg enhancer in the heart. (A-A″′) A GFP reporter driven by the wild-type Ndg enhancer is expressed in only two cardial cells per hemisegment, the Tin-Lb-CCs (square bracket). (B-B″′) Reporter expression from the wild-type Ndg enhancer is de-repressed into additional CCs (arrows) and Odd-PCs (arrowheads) in embryos homozygous for the CHES-1-like null mutation. The image shows de-repression occurring in both morphologically normal and abnormal (boxed) hemisegments. (C-D″′) Similar de-repression into additional CCs (arrows) and Odd-PCs (arrowheads) is seen for both normal (C-C″′) and abnormal (D-D″′) hemisegments in embryos homozygous for the jumu-null deficiency. The dotted circles in D-D″′ outline the two (instead of four normal) Tin-expressing CCs present in an abnormal hemisegment. (E-E″′) Overexpression of Jumu in the cardiac mesoderm and heart represses reporter expression from the wild-type Ndg enhancer, even in the two Tin-Lb-CCs (square bracket) where Ndg is normally expressed. (F-F″′) Overexpression of Jumu is unable to repress reporter expression from an enhancer with mutations in all three Fkh sites. Rather, these embryos exhibit de-repression into additional CCs (arrows) and Odd-PCs (arrowheads), similar to that seen with wild-type levels of jumu expression (Fig. 4F-F″′). Abdominal segments A3 and A4 are shown in every panel.
Loss-of-function mutations in either jumu or CHES-1-like also result in the generation of morphologically abnormal hemisegments owing to defects in the specification of cardiac cell fates and numbers (S.M.A., T.R.T., B.W.B., M. T. Nolte, N.J., S. S. Gisselbrecht, N. M. Rusan and A.M.M., unpublished; Fig. 5B-B″′,D-D″′). As not all hemisegments in homozygous mutant embryos exhibit this latter phenotype, we assessed whether there is any correlation between the de-repression of a specific gene and the morphological defects associated with mutations in jumu or CHES-1-like. Either no or barely significant differences were observed in the degree of de-repression in either CCs or Odd-PCs between morphologically normal and abnormal hemisegments for mutations in either gene (P=0.02 for CCs and P=0.28 for PCs in jumu mutants; P=0.11 for CCs and P=0.08 for PCs in CHES-1-like mutants). These results suggest that Ndg and the additional targets of these two Fkh TFs that are involved in specifying cardiac cell numbers and fates respond independently to the loss of jumu and CHES-1-like functions. We also determined that the de-repression of Ndg into neighboring CCs was a direct consequence of the absence of either Jumu or CHES-1-like TFs as expression of Ndg in additional CCs was not correlated with an expansion of Tin-Lb-CCs in jumu or CHES-1-like mutants (supplementary material Fig. S6).
We assayed the effects of jumu and CHES-1-like on Ndg enhancer activity at stage 16 owing to the perdurance of GFP in the regular arrangement of CCs and PCs, which afforded ease of quantitation. However, the regulatory effects of these TFs are expected to occur much earlier, that is, in the cardiac mesoderm during stage 11. Thus, we assessed whether the observed cis and trans effects of Fkh regulation of Ndg actually initiate at the earlier stage by double labeling appropriate stage 11 embryos for GFP and endogenous Mef2 expression. These experiments revealed that there is a significant de-repression of the NdgWT reporter in jumu and CHES-1-like mutants, and of the mutated enhancer-reporters in otherwise wild-type embryos, confirming that the repression of Ndg expression in the heart by these two Fkh TFs does occur when they are initially expressed in the cardiac mesoderm (supplementary material Fig. S9).
To confirm that Jumu functions as a transcriptional repressor of Ndg in the heart, we overexpressed Jumu protein by crossing UAS-jumu to both TinD-GAL4 and Hand-GAL4 drivers, which caused a dramatic reduction of Ndg reporter expression in the Tin-Lb-CCs (Fig. 5E-E″′; supplementary material Table S3 and Fig. S10) and in the ventral-most Tin-PCs (data not shown). In addition, if the repressive activity of Jumu is mediated by interaction of this TF with the Fkh sites in the Ndg enhancer, then mutating these binding sites should block the effect of jumu overexpression. We tested this hypothesis by examining the expression of the NdgFkh1+2+3 reporter in embryos in which Jumu was overexpressed under the control of both TinD-GAL4 and Hand-GAL4. NdgFkh1+2+3 was expressed in normal and additional CCs and Odd-PCs in the presence of ectopic Jumu (Fig. 5F-F″′; supplementary material Table S3), indicating that the repressive effect of Jumu is blocked by mutations that prevent Fkh TF binding. These results suggest that Jumu acts through Fkh-binding sites to repress Ndg expression in the heart.
Fkh TF binding sites are significantly overrepresented in combination with known co-regulatory heart TF binding sites in putative cardiac-specific enhancers
The roles of Jumu and CHES-1-like in regulating Ndg expression in both CCs and PCs raise the question of whether these two Fkh TFs also control the expression of other genes involved in cardiac development. To address this issue, we used a computational approach to assess whether Fkh-binding sites are enriched in putative enhancers associated with other heart genes (Philippakis et al., 2006; Warner et al., 2008).
Drosophila cardiac development involves contributions from multiple regulators, including the tissue-specific TFs Twist (Twi) and Tin; and Pointed (Pnt), T-cell factor (Tcf) and Mothers against Dpp (Mad), which are activated by the receptor tyrosine kinase (RTK)/Ras/MAPK, Wingless and Decapentaplegic pathways, respectively (Azpiazu and Frasch, 1993; Bodmer, 1993; Staehling-Hampton et al., 1994; Frasch, 1995; Carmena et al., 1998; Halfon et al., 2000; Halfon et al., 2002). If Jumu and CHES-1-like act in concert with one or more of these TFs to regulate cardiac-specific gene expression, then a significant fraction of heart genes – although not necessarily all – would be expected to have enhancers containing Fkh motifs associated with binding sites for all or subsets of the aforementioned TFs. We tested this possibility by using the Lever algorithm (Warner et al., 2008), which detects significant overrepresentations of given combinations of evolutionarily conserved TF-binding sites within putative cis-regulatory modules in the noncoding regions of defined lists of genes.
This approach revealed that both Fkh primary and secondary motifs are enriched along with multiple combinations of subsets of Tcf-, Mad-, Ets-, Twi- and Tin-binding sequences within putative enhancers associated with the noncoding regions of CC and PC genes (supplementary material Table S4). Of note, this degree of enrichment is seen only with authentic FkhP and FkhS motifs for genes expressed in the heart, but not with motifs for which the Fkh-binding sequences have been shuffled as a control. By contrast, no enrichment is observed with the actual Fkh site motifs when a collection of random genes is used as the foreground (Fig. 6B). Furthermore, three CC enhancers, including the Ndg enhancer used to test for Fkh motif function in the present study, conform to the 7-way AND combination of motifs that includes those for the five established cardiac TFs and both Fkh primary and secondary sequences (Fig. 6C; supplementary material Table S4 and Fig. S11). Although not all of these binding sites in the Ndg enhancer have been functionally tested, mutating either the Ets site or the Tin site eliminates enhancer activity in the heart (Busser et al., 2012a) (supplementary material Fig. S12).
Fig. 6.

Enrichment of Fkh-binding sites in putative enhancers found within the noncoding regions of genes having heart expression. (A) Logo representations of the PWMs of the TFs used in the Lever analysis. (B) Receiver-operating characteristic (ROC) curves showing the discrimination of CC genes by the indicated AND combinations of Tcf, Mad, Ets, Twi, Tin and the two Fkh-binding motifs FkhP and FkhS. The area under the ROC curve (AUC) for each motif combination is shown. Note that Fkh motifs are over-represented in combination with motifs for all five known regulators of heart gene expression. Two other ROC curves are also shown to demonstrate that using shuffled instead of authentic Fkh motifs, or using a random gene set instead of the CC gene set, both increases the q-value and reduces the AUC. (C) Schematic of two of the three known CC enhancers that contain all seven of the TF-binding motifs used in the Lever analysis, including the Ndg enhancer used in this study (see also supplementary material Table S4).
A similar, although less pronounced result was obtained for PC genes (supplementary material Table S4). Although clearly not all CC and PC genes contain candidate enhancers that conform to the Ndg regulatory model, the present results suggest that Fkh TFs are potentially regulating large numbers of heart genes.
DISCUSSION
In this study, we have characterized the regulatory functions of multiple Fkh TFs and their binding sites in the control of gene expression in different mesodermal cell types in the Drosophila embryo. Specifically, we identified three Fkh-binding sites in a minimal enhancer of the Ndg gene, which mediate the effects of different cell type-specific Fkh TFs and are used in various combinations to regulate enhancer activity in a subset of somatic myoblasts, in differentiated visceral muscle and in progenitors of both the cardial and pericardial cells of the heart (Fig. 7).
Fig. 7.

The different expression patterns of Ndg in various mesodermal cell types involve both different Fkh TFs and distinct modes of use of Fkh-binding sites in the Ndg enhancer. (A) Summary of: (1) the effects of mutating different Fkh binding sites in the Ndg enhancer; and (2) the effects of loss of function of bin, jumu and CHES-1-like on the activity of the wild-type Ndg enhancer in different types of mesodermal cells. (B) In the VM, the convergence of cis, trans and EMSA data suggest that Bin binds either to both Fkh1 and Fkh2 sites or to both Fkh1 and Fkh3 sites to activate Ndg expression. (C) In somatic myoblasts, the cis, trans and EMSA results are consistent with CHES-1-like binding to the Fkh2 or Fkh3 sites to repress Ndg reporter expression in FCMs. The contribution, if any, of CHES-1-like binding at the Fkh1 site to the repression of the Ndg reporter could not be determined by the design of the experiment and is denoted by ‘?’ in the panel. (D,E) CHES-1-like binds to all three sites, whereas Jumu binds to the Fkh2 site to repress Ndg reporter expression in CCs and Odd-PCs.
Differential regulation of mesodermal gene expression by tissue-specific Fkh TFs
To drive expression in the visceral mesoderm, the Fkh1 site in the Ndg enhancer is required in concert with either the Fkh2 or Fkh3 site (Fig. 7A,B). The trans-acting factor responsible for this activity of the Ndg enhancer is likely to be Bin because: (1) Bin bound to all three Fkh sites in vitro; (2) among the three candidate Fkh genes with appropriate VM expression, eliminating the function of only bin resulted in a significant reduction of Ndg expression in this tissue; and (3) Bin overexpression in the mesoderm was associated with Ndg enhancer activity in additional mesodermal cells. There are multiple precedents for Bin activating the expression of other VM genes (Zaffran et al., 2001; Zaffran and Frasch, 2002; Jakobsen et al., 2007; Popichenko et al., 2007). Moreover, chromatin immunoprecipitation assays show that Bin binds in vivo to the Ndg enhancer throughout embryonic stages 14 to 15, precisely when it would be expected to regulate this element in the visceral musculature (Jakobsen et al., 2007; Zinzen et al., 2009).
Somatic muscles in Drosophila are formed by the sequential fusion of individual muscle FCs with multiple FCMs. Both the endogenous Ndg gene and the reporter driven by the minimal enhancer used in this study are expressed in a subset of FCs, but not in any FCMs. Mutating all three Fkh-binding sites had no effect on Ndg expression in FCs, suggesting that Fkh factors do not play a role either in activating Ndg reporter expression in certain FCs, or in repressing it in other FCs. By contrast, binding of the FCM-expressed Fkh TF CHES-1-like to the Fkh2 or Fkh3 sites mediated repression of Ndg expression in FCMs (Fig. 7C). The design of the experiment prevented us from determining unambiguously whether this repression also required CHES-1-like binding to the Fkh1 site.
These results reveal a mechanism for regulating somatic myoblast gene expression that has not been previously recognized. Prior studies have focused on the contributions of signal-activated, tissue-specific and FC identity TFs in specifying the unique genetic programs of this class of myoblast (Halfon et al., 2000; Busser et al., 2008). Similarly, TFs such as Lmd are known to be responsible for activating FCM-specific genes (Duan et al., 2001; Ruiz-Gomez et al., 2002). However, we have now uncovered a novel mode of regulation in which FC genes are excluded from FCMs by an FCM-restricted repressor, in this case in the form of a Fkh domain protein. CHES-1-like is unlikely to be the only repressor playing such a role, as the de-repression in CHES-1-like mutants is limited to only a subset of the FCMs and is weaker than that seen for the Ndg enhancer with mutated Fkh sites. Although not verified functionally, it is possible that Lmd could play a similar repressive role in FCMs as a chromatin immunoprecipitation study found that this TF is bound extensively to FC genes (Cunha et al., 2010). However, given the widespread expression of CHES-1-like in FCMs, we anticipate that many other FC genes will also be repressed by this Fkh domain TF.
Finally, in the heart, we show that CHES-1-like and Jumu repress Ndg expression in Odd-PCs and in all CCs other than Tin-Lb-CCs. Repression in these cardiac cell types is mediated by binding of CHES-1-like to all three of the Fkh sites in the Ndg enhancer, and of Jumu to at least the Fkh2 site (Fig. 7A,D,E).
Use of the same enhancer to respond to different tissue-specific Fkh TFs
A common occurrence in development is the repeated function of the same gene in multiple biological contexts and regulatory processes, which requires that the gene be expressed in distinct spatial and temporal domains. Such expression patterns are often generated by differential transcription mediated by multiple enhancers, each with its own arrangement of TF-binding sites and associated activities (Davidson, 2006). A notable exception is the case of genes regulated by Hox TFs, where different family members exhibit similar binding sequence specificity but exert differential effects on the same target genes (Hueber and Lohmann, 2008; Mann et al., 2009).
The results of the present study identify another class of TFs, the Fkh proteins, which exhibit a similar role in the Drosophila embryonic mesoderm. Specifically, we have shown that various cell-specific members of the Fkh TF family associate with the same binding sites within a single enhancer, thereby regulating the different spatiotemporal expression patterns of the associated target gene. Furthermore, we have shown that the distinct tissue-specific gene expression responses to these Fkh TFs are mediated by the TFs binding to different combinations of Fkh primary and secondary motifs that are represented by these sites. Thus, it was interesting to see that in several Drosophila species, where the Fkh1 site (which is the only site corresponding to the Fkh primary motif in D. melanogaster) is absent, its role may be compensated for by the overlapping Fkh2 and Fkh3 sites (which match only the secondary motif in D. melanogaster), which correspond to both Fkh primary and secondary motifs in these species (supplementary material Table S1). Similar evolutionary shuffling of motifs has been described previously (Ludwig et al., 2000; Hare et al., 2008).
Finally, we used a computational approach to attempt to generalize the potential involvement of the two classes of Fkh sites in cardiac gene regulation. Specifically, within putative enhancers in the noncoding regions of heart-expressed genes, we observed a statistically significant overrepresentation of combinations of binding sites for known cardiogenic TFs along with primary and secondary Fkh motifs. These observations are in agreement with previous studies that have documented an inability of a single consensus binding site to explain all aspects of in vivo TF binding (Ji et al., 2006; Rabinovich et al., 2008; Cunha et al., 2010). In addition, we have recently shown that the regulatory specificity of a myoblast homeodomain TF is mediated by sequences preferentially bound by that particular homeodomain and not by other related family members (Busser et al., 2012b). In light of these findings, it will be interesting to determine whether other Fkh TFs and members of other TF families mediate differential gene expression responses by acting through distinct sequence motifs.
Supplementary Material
Acknowledgments
We thank H. Jäckle, A. Hofmann, A. Han, H. T. Nguyen, B. Paterson, J. B. Skeath, M. Frasch, and the Bloomington Stock Center for fly lines and reagents, and A. Cohen and C. Sonnenbrot for technical assistance. Stephen Gisselbrecht provided valuable comments throughout the course of these experiments.
Footnotes
Funding
This work was supported by the National Heart, Lung, and Blood Institute (NHLBI) Division of Intramural Research (A.M.M.), by the National Institutes of Health (NIH) [R01 HG005287-01A1 to M.L.B.], by an American Heart Association Predoctoral Fellowship (A.A.) and by an American Heart Association Postdoctoral Fellowship (S.M.A.). Deposited in PMC for immediate release.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.069005/-/DC1
References
- Azpiazu N., Frasch M. (1993). tinman and bagpipe: two homeo box genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes Dev. 7, 1325–1340 [DOI] [PubMed] [Google Scholar]
- Badis G., Berger M. F., Philippakis A. A., Talukder S., Gehrke A. R., Jaeger S. A., Chan E. T., Metzler G., Vedenko A., Chen X., et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R. (1993). The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development 118, 719–729 [DOI] [PubMed] [Google Scholar]
- Bonn S., Furlong E. E. (2008). cis-Regulatory networks during development: a view of Drosophila. Curr. Opin. Genet. Dev. 18, 513–520 [DOI] [PubMed] [Google Scholar]
- Busser B. W., Bulyk M. L., Michelson A. M. (2008). Toward a systems-level understanding of developmental regulatory networks. Curr. Opin. Genet. Dev. 18, 521–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser B. W., Taher L., Kim Y., Tansey T., Bloom M. J., Ovcharenko I., Michelson A. M. (2012a). A machine learning approach for identifying novel cell type-specific transcriptional regulators of myogenesis. PLoS Genet. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser B. W., Shokri L., Jaeger S. A., Gisselbrecht S. S., Singhania A., Berger M. F., Zhou B., Bulyk M. L., Michelson A. M. (2012b). Molecular mechanism underlying the regulatory specificity of a Drosophila homeodomain protein that specifies myoblast identity. Development 139, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson P., Mahlapuu M. (2002). Forkhead transcription factors: key players in development and metabolism. Dev. Biol. 250, 1–23 [DOI] [PubMed] [Google Scholar]
- Carmena A., Gisselbrecht S., Harrison J., Jimenez F., Michelson A. M. (1998). Combinatorial signaling codes for the progressive determination of cell fates in the Drosophila embryonic mesoderm. Genes Dev. 12, 3910–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps R. M., Black B. L., Zhao B., Lien C. L., Schulz R. A., Olson E. N. (1998). The myogenic regulatory gene Mef2 is a direct target for transcriptional activation by Twist during Drosophila myogenesis. Genes Dev. 12, 422–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha P. M., Sandmann T., Gustafson E. H., Ciglar L., Eichenlaub M. P., Furlong E. E. (2010). Combinatorial binding leads to diverse regulatory responses: Lmd is a tissue-specific modulator of Mef2 activity. PLoS Genet. 6, e1001014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E. (2006). The Regulatory Genome: Gene Regulatory Networks in Development and Evolution. London, UK: Academic Press; [Google Scholar]
- Duan H., Skeath J. B., Nguyen H. T. (2001). Drosophila Lame duck, a novel member of the Gli superfamily, acts as a key regulator of myogenesis by controlling fusion-competent myoblast development. Development 128, 4489–4500 [DOI] [PubMed] [Google Scholar]
- Estrada B., Choe S. E., Gisselbrecht S. S., Michaud S., Raj L., Busser B. W., Halfon M. S., Church G. M., Michelson A. M. (2006). An integrated strategy for analyzing the unique developmental programs of different myoblast subtypes. PLoS Genet. 2, e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch M. (1995). Induction of visceral and cardiac mesoderm by ectodermal Dpp in the early Drosophila embryo. Nature 374, 464–467 [DOI] [PubMed] [Google Scholar]
- Frasch M. (1999). Controls in patterning and diversification of somatic muscles during Drosophila embryogenesis. Curr. Opin. Genet. Dev. 9, 522–529 [DOI] [PubMed] [Google Scholar]
- Gajewski K., Kim Y., Lee Y. M., Olson E. N., Schulz R. A. (1997). D-mef2 is a target for Tinman activation during Drosophila heart development. EMBO J. 16, 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski K., Kim Y., Choi C. Y., Schulz R. A. (1998). Combinatorial control of Drosophila mef2 gene expression in cardiac and somatic muscle cell lineages. Dev. Genes Evol. 208, 382–392 [DOI] [PubMed] [Google Scholar]
- Gajewski K., Zhang Q., Choi C. Y., Fossett N., Dang A., Kim Y. H., Kim Y., Schulz R. A. (2001). Pannier is a transcriptional target and partner of Tinman during Drosophila cardiogenesis. Dev. Biol. 233, 425–436 [DOI] [PubMed] [Google Scholar]
- Georgias C., Wasser M., Hinz U. (1997). A basic-helix-loop-helix protein expressed in precursors of Drosophila longitudinal visceral muscles. Mech. Dev. 69, 115–124 [DOI] [PubMed] [Google Scholar]
- Good P. I. (1994). Permutation Tests: A Practical Guide to Resampling Methods for Testing Hypotheses. New York, NY: Springer-Verlag; [Google Scholar]
- Greig S., Akam M. (1993). Homeotic genes autonomously specify one aspect of pattern in the Drosophila mesoderm. Nature 362, 630–632 [DOI] [PubMed] [Google Scholar]
- Grossniklaus U., Pearson R. K., Gehring W. J. (1992). The Drosophila sloppy paired locus encodes two proteins involved in segmentation that show homology to mammalian transcription factors. Genes Dev. 6, 1030–1051 [DOI] [PubMed] [Google Scholar]
- Groth A. C., Fish M., Nusse R., Calos M. P. (2004). Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166, 1775–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker U., Kaufmann E., Hartmann C., Jurgens G., Knochel W., Jackle H. (1995). The Drosophila fork head domain protein crocodile is required for the establishment of head structures. EMBO J. 14, 5306–5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfon M. S., Carmena A., Gisselbrecht S., Sackerson C. M., Jimenez F., Baylies M. K., Michelson A. M. (2000). Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell 103, 63–74 [DOI] [PubMed] [Google Scholar]
- Halfon M. S., Grad Y., Church G. M., Michelson A. M. (2002). Computation-based discovery of related transcriptional regulatory modules and motifs using an experimentally validated combinatorial model. Genome Res. 12, 1019–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z., Olson E. N. (2005). Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Development 132, 3525–3536 [DOI] [PubMed] [Google Scholar]
- Han Z., Fujioka M., Su M., Liu M., Jaynes J. B., Bodmer R. (2002). Transcriptional integration of competence modulated by mutual repression generates cell-type specificity within the cardiogenic mesoderm. Dev. Biol. 252, 225–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison C. T., Gordon D. B., Lee T. I., Rinaldi N. J., Macisaac K. D., Danford T. W., Hannett N. M., Tagne J. B., Reynolds D. B., Yoo J., et al. (2004). Transcriptional regulatory code of a eukaryotic genome. Nature 431, 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare E. E., Peterson B. K., Iyer V. N., Meier R., Eisen M. B. (2008). Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 4, e1000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann A., Brunner M., Korge G. (2009). The winged-helix transcription factor JUMU is a haplo-suppressor/triplo-enhancer of PEV in various tissues but exhibits reverse PEV effects in the brain of Drosophila melanogaster. Chromosome Res. 17, 347–358 [DOI] [PubMed] [Google Scholar]
- Hueber S. D., Lohmann I. (2008). Shaping segments: Hox gene function in the genomic age. BioEssays 30, 965–979 [DOI] [PubMed] [Google Scholar]
- Jagla K., Frasch M., Jagla T., Dretzen G., Bellard F., Bellard M. (1997). ladybird, a new component of the cardiogenic pathway in Drosophila required for diversification of heart precursors. Development 124, 3471–3479 [DOI] [PubMed] [Google Scholar]
- Jakobsen J. S., Braun M., Astorga J., Gustafson E. H., Sandmann T., Karzynski M., Carlsson P., Furlong E. E. (2007). Temporal ChIP-on-chip reveals Biniou as a universal regulator of the visceral muscle transcriptional network. Genes Dev. 21, 2448–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H., Vokes S. A., Wong W. H. (2006). A comparative analysis of genome-wide chromatin immunoprecipitation data for mammalian transcription factors. Nucleic Acids Res. 34, e146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knirr S., Frasch M. (2001). Molecular integration of inductive and mesoderm-intrinsic inputs governs even-skipped enhancer activity in a subset of pericardial and dorsal muscle progenitors. Dev. Biol. 238, 13–26 [DOI] [PubMed] [Google Scholar]
- Kremser T., Gajewski K., Schulz R. A., Renkawitz-Pohl R. (1999). Tinman regulates the transcription of the beta3 tubulin gene (betaTub60D) in the dorsal vessel of Drosophila. Dev. Biol. 216, 327–339 [DOI] [PubMed] [Google Scholar]
- Lee H. H., Frasch M. (2004). Survey of forkhead domain encoding genes in the Drosophila genome: Classification and embryonic expression patterns. Dev. Dyn. 229, 357–366 [DOI] [PubMed] [Google Scholar]
- Lee H. H., Frasch M. (2005). Nuclear integration of positive Dpp signals, antagonistic Wg inputs and mesodermal competence factors during Drosophila visceral mesoderm induction. Development 132, 1429–1442 [DOI] [PubMed] [Google Scholar]
- Ludwig M. Z., Bergman C., Patel N. H., Kreitman M. (2000). Evidence for stabilizing selection in a eukaryotic enhancer element. Nature 403, 564–567 [DOI] [PubMed] [Google Scholar]
- Mann R. S., Lelli K. M., Joshi R. (2009). Hox specificity unique roles for cofactors and collaborators. Curr. Top. Dev. Biol. 88, 63–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M., Pitsouli C., Villalta C., Celniker S. E., Perrimon N. (2008). Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet. 40, 476–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker D. S., White M. A., Ramos A. I., Cohen B. A., Barolo S. (2011). The cis-regulatory logic of Hedgehog gradient responses: key roles for gli binding affinity, competition, and cooperativity. Sci. Signal. 4, ra38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis A. A., Busser B. W., Gisselbrecht S. S., He F. S., Estrada B., Michelson A. M., Bulyk M. L. (2006). Expression-guided in silico evaluation of candidate cis regulatory codes for Drosophila muscle founder cells. PLoS Comput. Biol. 2, e53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popichenko D., Sellin J., Bartkuhn M., Paululat A. (2007). Hand is a direct target of the forkhead transcription factor Biniou during Drosophila visceral mesoderm differentiation. BMC Dev. Biol. 7, 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich A., Jin V. X., Rabinovich R., Xu X., Farnham P. J. (2008). E2F in vivo binding specificity: comparison of consensus versus nonconsensus binding sites. Genome Res. 18, 1763–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robasky K., Bulyk M. L. (2011). UniPROBE, update 2011, expanded content and search tools in the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 39, D124–D128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S., Siggers T., Lachke S. A., Yue Y., Bulyk M. L., Maas R. L. (2010). Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 24, 980–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Gomez M., Coutts N., Suster M. L., Landgraf M., Bate M. (2002). myoblasts incompetent encodes a zinc finger transcription factor required to specify fusion-competent myoblasts in Drosophila. Development 129, 133–141 [DOI] [PubMed] [Google Scholar]
- Sandmann T., Jensen L. J., Jakobsen J. S., Karzynski M. M., Eichenlaub M. P., Bork P., Furlong E. E. (2006). A temporal map of transcription factor activity: mef2 directly regulates target genes at all stages of muscle development. Dev. Cell 10, 797–807 [DOI] [PubMed] [Google Scholar]
- Staehling-Hampton K., Hoffmann F. M., Baylies M. K., Rushton E., Bate M. (1994). dpp induces mesodermal gene expression in Drosophila. Nature 372, 783–786 [DOI] [PubMed] [Google Scholar]
- Stapleton M., Liao G., Brokstein P., Hong L., Carninci P., Shiraki T., Hayashizaki Y., Champe M., Pacleb J., Wan K., et al. (2002). The Drosophila gene collection: identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 12, 1294–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strodicke M., Karberg S., Korge G. (2000). Domina (Dom), a new Drosophila member of the FKH/WH gene family, affects morphogenesis and is a suppressor of position-effect variegation. Mech. Dev. 96, 67–78 [DOI] [PubMed] [Google Scholar]
- Tao Y., Schulz R. A. (2007). Heart development in Drosophila. Semin. Cell Dev. Biol. 18, 3–15 [DOI] [PubMed] [Google Scholar]
- Tao Y., Wang J., Tokusumi T., Gajewski K., Schulz R. A. (2007). Requirement of the LIM homeodomain transcription factor tailup for normal heart and hematopoietic organ formation in Drosophila melanogaster. Mol. Cell. Biol. 27, 3962–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tixier V., Bataille L., Jagla K. (2010). Diversification of muscle types: recent insights from Drosophila. Exp. Cell Res. 316, 3019–3027 [DOI] [PubMed] [Google Scholar]
- Wang J., Tao Y., Reim I., Gajewski K., Frasch M., Schulz R. A. (2005). Expression, regulation, and requirement of the toll transmembrane protein during dorsal vessel formation in Drosophila melanogaster. Mol. Cell. Biol. 25, 4200–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward E. J., Skeath J. B. (2000). Characterization of a novel subset of cardiac cells and their progenitors in the Drosophila embryo. Development 127, 4959–4969 [DOI] [PubMed] [Google Scholar]
- Warner J. B., Philippakis A. A., Jaeger S. A., He F. S., Lin J., Bulyk M. L. (2008). Systematic identification of mammalian regulatory motifs’ target genes and functions. Nat. Methods 5, 347–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijchers P. J., Burbach J. P., Smidt M. P. (2006). In control of biology: of mice, men and Foxes. Biochem. J. 397, 233–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Yin Z., Hudson J. B., Ferguson E. L., Frasch M. (1998). Smad proteins act in combination with synergistic and antagonistic regulators to target Dpp responses to the Drosophila mesoderm. Genes Dev. 12, 2354–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z., Xu X. L., Frasch M. (1997). Regulation of the twist target gene tinman by modular cis-regulatory elements during early mesoderm development. Development 124, 4971–4982 [DOI] [PubMed] [Google Scholar]
- Zaffran S., Frasch M. (2002). The beta 3 tubulin gene is a direct target of bagpipe and biniou in the visceral mesoderm of Drosophila. Mech. Dev. 114, 85–93 [DOI] [PubMed] [Google Scholar]
- Zaffran S., Kuchler A., Lee H. H., Frasch M. (2001). biniou (FoxF), a central component in a regulatory network controlling visceral mesoderm development and midgut morphogenesis in Drosophila. Genes Dev. 15, 2900–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzen R. P., Girardot C., Gagneur J., Braun M., Furlong E. E. (2009). Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature 462, 65–70 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}