

1. Introduction
In 1998, Bikker, Trumpp-Kallmeyer, and Humblet published a Perspective in this journal entitled “G-Protein Coupled Receptors: Models, Mutagenesis and Drug Design” and reviewed the state of the art at that time.1 No high resolution structure of a G protein-coupled receptor (GPCR) had been solved, and researchers were working with models generated with only the structure of bacteriorhodopsin,2 which had been published 8 years earlier and solved using high resolution electron cryomicroscopy and the low resolution electron density footprint of bovine rhodopsin.3 These models, despite greatly improving understanding of GPCR structure and function, posed as many questions as they answered and were not able to clearly rationalize how ligands bound to their target receptor. The authors stated “The principal limitation of the current generation of models when used for rational drug design is that the resolution of the binding cavity is too low to predict specific ligand–receptor interactions. Attempts to dock ligands into various GPCR models are further complicated by difficulty in identifying unique, sensible modes of binding, especially when dealing with molecules of the size of the neurotransmitter ligands.” How things have changed.
Today, there are six GPCRs for which medium to high resolution crystal structures have been solved, in most cases with multiple small molecules ligands. The six receptors are rhodopsin, the β1 and β2 adrenergic receptors, adenosine A2A receptor, chemokine CXCR4 receptor, and dopamine D3 receptor (Table 1 and references therein). In addition, rhodopsin, the β1 and β2 adrenergic receptors (ARs), and the adenosine A2A receptor have been solved with both antagonists and agonists bound (Table 1). Much current research is now engaged in using this new body of structural information for hit identification and drug design purposes, and we will review the state of the art of both structures and the impact they are now having on structure based drug design (SBDD) for GPCR targets in this article.
Table 1. List of Published GPCR Crystal Structures.
| receptor | resolution (Å) | PDB code | date | ref |
|---|---|---|---|---|
| Rhodopsin: bovine rod outer segment | 2.8 | 1F88 | 06/00 | (52) |
| Rhodopsin: bovine rod outer segment | 2.6 | 1L9H | 03/02 | (176) |
| Rhodopsin: bovine rod outer segment | 2.65 | 1GZM | 05/02 | (177) |
| Rhodopsin: bovine rod outer segment | 2.2 | 1U19 | 07/04 | (178) |
| Rhodopsin, photoactivated: bovine rod outer segment | 3.8–4.15 | 2I37 | 08/06 | (179) |
| Rhodopsin: recombinant bovine rhodopsin mutant, N2C/D282C | 3.4 | 2J4Y | 09/06 | (180) |
| Rhodopsin: squid | 3.7 | 2ZIY | 05/07 | (181) |
| Rhodopsin: squid | 2.5 | 2Z73 | 08/07 | (182) |
| Human β2 adrenergic receptor Fab5 complex. Complex with carazalol | 3.4/3.7 | 2R4R | 08/07 | (59) |
| Human β2 adrenergic receptor Fab5 complex. Complex with carazalol | 3.4/3.7 | 2R4S | 08/07 | (59) |
| Human β2 adrenergic receptor: T4 lysozyme replaces ICL3. Complex with carazalol | 2.4 | 2RH1 | 10/07 | (60) |
| Opsin: bovine rod outer segment | 2.9 | 3CAP | 02/08 | (78) |
| Turkey β1 adrenergic receptor: StaR engineered for stability. Complex with cyanopindolol | 2.7 | 2VT4 | 05/08 | (58) |
| Human β2 adrenergic receptor: T4 lysozyme replaces ICL3, E122W stability mutation. Complex with timolol | 2.8 | 3D4S | 05/08 | (183) |
| Opsin in complex with a C-terminal peptide derived from the Gα subunit of transducin | 3.2 | 3DQB | 07/08 | (49) |
| Human adenosine A2A receptor: T4 lysozyme replaces ICL3. In complex with antagonist ZM241385 | 2.6 | 3EML | 09/08 | (65) |
| Methylated β2 adrenergic receptor: Fab complex | 3.4 | 3KJ6 | 11/09 | (184) |
| Human β2 adrenergic receptor: T4 lysozyme replaces ICL3. Complex with the inverse agonist ICI 118,551 | 2.84 | 3NY8 | 07/10 | (117) |
| Human β2 adrenergic receptor: T4 lysozyme replaces ICL3. Complex with a novel inverse agonist | 2.84 | 3NY9 | 07/10 | (117) |
| Human β2 adrenergic receptor: T4 lysozyme replaces ICL3. Complex with alprenolol | 3.16 | 3NYA | 07/10 | (117) |
| CXCR4 chemokine receptor: T4 lysozyme replaces ICL3. Complex with a cyclic peptide antagonist CVX15 | 2.9 | 3OE0 | 08/10 | (70) |
| CXCR4 chemokine receptor: T4 lysozyme replaces ICL3. Complex with a small molecule antagonist IT1t | 3.2 | 3OE6 | 08/10 | (70) |
| CXCR4 chemokine receptor: T4 lysozyme replaces ICL3. Complex with a small molecule antagonist IT1t | 3.1 | 3OE8 | 08/10 | (70) |
| CXCR4 chemokine receptor: T4 lysozyme replaces ICL3. Complex with a small molecule antagonist IT1t | 3.1 | 3OE9 | 08/10 | (70) |
| CXCR4 chemokine receptor: T4 lysozyme replaces ICL3. Complex with a small molecule antagonist IT1t | 2.5 | 3ODU | 08/10 | (70) |
| Crystal structure of bovine rhodopsin with β-ionone | 2.6 | 3OAX | 08/10 | (185) |
| Dopamine D3 receptor: T4 lysozyme replaces ICL3. Complex with D2/D3-selective antagonist | 2.89 | 3PBL | 11/10 | (36) |
| Human β2 adrenergic receptor in active state stabilized with a nanobody: T4 lysozyme replaces ICL3 | 3.5 | 3POG | 01/11 | (80) |
| Human β2 adrenergic receptor with irreversibly bound agonist T4 lysozyme replaces third intracellular loop | 3.5 | 3PDS | 01/11 | (81) |
| Turkey β1 adrenergic receptor: StaR engineered for stability. Complex with dobutamine | 2.5 | 2Y00 | 01/11 | (82) |
| Turkey β1 adrenergic receptor: StaR engineered for stability. Complex with dobutamine | 2.65 | 2Y01 | 01/11 | (82) |
| Turkey β1 adrenergic receptor: StaR engineered for stability. Complex with carmotorol | 2.65 | 2Y02 | 01/11 | (82) |
| Turkey β1 adrenergic receptor: StaR engineered for stability. Complex with isoprenaline | 2.85 | 2Y03 | 01/11 | (82) |
| Turkey β1 adrenergic receptor: StaR engineered for stability. Complex with salbutamol | 3.05 | 2Y04 | 01/11 | (82) |
| Bovine rhodopsin metarhodopsin II | 3.00 | 3PXO | 03/11 | (186) |
| Bovine rhodopsin metarhodopsin II in complex with C-terminal fragment of GR (GRCT2) | 2.85 | 3PQR | 03/11 | (186) |
| Constitutively active rhodopsin mutant with bound Gα (GαCT2) | 3.00 | 2X72 | 03/11 | (46) |
| Human adenosine A2A receptor: T4 lysozyme replaces ICL3. Complex with the agonist UK-432097 | 2.7 | 3QAK | 03/11 | (83) |
| Agonist state human adenosine A2A receptor: StaR engineered for stability. Complex with adenosine | 3.0 | 2YDO | 03/11 | (84) |
| Agonist state human adenosine A2A receptor: StaR engineered for stability. Complex with the agonist NECA | 2.6 | 2YDV | 03/11 | (84) |
| Inverse agonist state human adenosine A2A receptor: StaR engineered for stability. Complex with ZM241385 | 3.29 | 3PWH | 06/11 | (66) |
| Inverse agonist state human adenosine A2A receptor: StaR engineered for stability. Complex with XAC | 3.3 | 3REY | 06/11 | (66) |
| Inverse agonist state human adenosine A2A receptor: StaR engineered for stability. Complex with caffeine | 3.6 | 3RFM | 06/11 | (66) |
While this impressive progress with the structural biology of GPCRs has been coming to fruition, GPCR drug discovery has continued to be a major area of pharmaceutical research. Sixty-three new GPCR drugs have been launched in the past decade, approximately 24% of all drugs reaching the market during this period (Table 2). Figure 1 illustrates the 10 first in class small GPCR molecule drugs (new chemical entities, NCEs) from the past decade (2000–2009). An examination of these molecules and their targets and a consideration of their druglike properties highlight a number of issues associated with recent GPCR drug discovery. First, the molecules are generally at the upper limits of Lipinski’s rules in terms of molecular weight and/or lipophilicity, suggesting that they would have been “high risk” in terms of their ADMET properties during development.4 Indeed, there is a growing body of evidence that molecules with high molecular weight and in particular high lipophilicity have increased risk of both toxicity and cross-reactivity giving a high failure rate in the clinic.5,6 Second, three of the compounds fall into a “lipophilic amine” category that is often associated with promiscuous binders in the GPCR field. This suggests that the discoverers of these drugs would have spent much of their efforts optimizing receptor activity while tuning off target activities, including related receptors, cytochrome P450 enzymes, and ion channels such as the hERG channel. Third, we can see from Figure 1 that usually only one new GPCR target has been drugged per year by an NCE and a consideration of these targets tells us that these drugs are the culmination of very many years of research on these receptors by Pharma. During the same decade four biotherapeutics (NBEs) directed at GPCRs have also been launched, suggesting that a significant number of newer but clinically validated GPCR targets are currently intractable to small molecule drug discovery. Overall, it is certainly not the case that modern GPCR targets could be considered “low hanging fruit”, and the inventors of these drugs are to be congratulated for overcoming the considerable challenges of these first in class targets.
Table 2. GPCR Targeted Drugs Launched in the Past Decade (2000–2009)a.
| family A aminergic, opioid, prostanoid | family A peptidergic, chemokine, other | ||
|---|---|---|---|
| almotriptan | indacaterol | abarelix | mozavaptan |
| alosetron | lafutidine | ambrisentan | olmesartan |
| alvimopan | landiolol | aprepitant | plerixafor |
| arformoterol | levocetirizine | atosiban | prasugrel |
| aripiprazole | methylnaltrexone | bosentan | ramelteon |
| armodafinil | nalfurafine | conivaptan | rimonabant |
| asenapine | paliperidone | degarelix | sitaxsentan |
| bepotastine | perospirone | fosaprepitant | taltirelin |
| betotastine | ramatroban | ganirelix | tolvaptan |
| bimatoprost | rotigotine | icatibant | |
| blonanserin | rupatadine | maraviroc | |
| cevimeline | silodosin | ||
| darifenacin | solifenacin | ||
| desloratadine | tafluprost | ||
| dexmedetomidine | tapentadol | ||
| eletriptan | tegaserod | ||
| fesoterodine | tiotropium | ||
| frovatriptan | travoprost | ||
| Iloperidone | treprostinil | ||
| imidafenacin | ziprasidone | ||
| family B | family C |
|---|---|
| exenatide | cinacalcet |
| liraglutide |
NBEs are shown in italics. First in class drugs are shown in bold.
Figure 1.

First in class GPCR targeted drugs launched in the past decade. The drug, year of launch, protein target, route of administration, MWT, and cLogP are given in each case. The biological drugs are shown next to the GPCR target and year of launch.
2. GPCR Pharmacology
2.1. Classes of GPCRs
There are 390 GPCRs in the human genome (excluding olfactory receptors).7 These fall into three major classes that, although not related by homology, share the same overall structural topology of an extracellular N-terminus, seven-transmembrane spanning domain (TMD), and an intracellular C-terminus. The largest subfamily is the rhodopsin family (also known as family A or class 1). This is the largest and most diverse subfamily with respect to ligand types. Rhodopsin family members can be activated by small molecules including amines, purines, fatty acids, and prostaglandins, as well as peptides to large glycoproteins. Approximately 25% of marketed small molecule drugs act through this subfamily of GPCRs.8 The secretin and adhesion families (also known as family B or class II) have related transmembrane domains but differ in their N-termini. The secretin family is activated by large peptides including glucagon-like peptide (GLP 1), glucagon, and vasoactive intestinal peptide. The secretin family is rich in targets that have been clinically validated through the use of peptides (derived from the natural ligands) such as Byetta9 (exenatide, GLP 1) or Miacalcin10 (calcitonin). However, to date, this family has proved largely intractable to small molecule drug discovery, with one exception being the corticotrophin receptor 1 (CRF 1).11,12 The adhesion family13 has a seven-transmembrane domain linked to a very long and highly glycosylated N-termini. The majority of the adhesion family consists of orphan receptors, and few attempts have been made to drug this class. The final major subclass is the glutamate family (family C, class III), which contains receptors for the amino acids glutamate and γ-aminobutyric acid (GABA) as well as the calcium sensing receptor and a number of taste receptors.14 These receptors also contain a long N-terminus that forms an amino acid binding domain distinct from the TMD. Drugs acting at this class of receptors either bind in the amino acid binding domain (e.g., Lioresal, baclofen15) or are allosteric modulators that bind within the TMD (e.g., Sensipar, cinacalcet16).
Figure 2 shows cartoon views of the three receptor subclasses to illustrate the positions of the natural ligand (orthosteric) binding sites. In family A this is in the top of the TMD bundle. In family B it spans the large N-terminal domain and the TMD, and in family C it exists in the so-called venus fly trap N-terminal domain. In families A and B the ligand directly activates the receptor by at least partly binding within the TMD site, while in family C it is believed that binding of the agonist to the venus fly trap domain causes a conformational change that indirectly causes activation via the TMD site. In families B and C the TMD site is therefore considered allosteric, since the orthosteric agonists interact principally or entirely with the N-terminal domains.
Figure 2.

Overall structures of family A, family B, and family C GPCRs as a cartoon. The seven helices are labeled in each case, and N-terminal ECD is shown. The natural ligand is shown in green. In family B this is the “hot dog in a bun” model where the peptide is shown binding to the ECD and also to the TMD. In family C the ECD is the “Venus fly trap”; conformational change upon binding of ligand is thought to cause receptor signaling.
2.2. Pharmacology of Drugs Acting at GPCRs
GPCRs are located in the plasma membrane of all cell types. Here they mediate the action of a diverse set of extracellular messengers through their interaction with membrane bound and intracellular second messenger signaling proteins. Within the membrane, GPCRs are highly flexible and can exist in a number of conformational states ranging from the inactive ground state (commonly known as R) to one or more fully activated states (known as R*) that interact with and activate signaling proteins including G proteins. The ratio of R to R* in the absence of ligand varies from one receptor to another and alters the basal level of receptor activity. Ligands and drugs that bind to GPCRs alter the equilibrium between the different conformational states.17 Until recently little has been known about the molecular nature of the transitions between conformational states; however, the recent development in structural biology, in particular structures of the prototypical GPCR, rhodopsin, is increasing our understanding of this area.18,19 Agonists preferentially bind to and stabilize the R* state of the receptor, resulting in an increase in receptor activity. Inverse agonists preferentially bind to the ground state R and reduce receptor activity. Antagonists act to block GPCR activation by preventing the binding of agonists to the receptor. So-called neutral antagonists in theory would have equal affinity for both R and R*; however, in practice this is rare and most drugs referred to as antagonists are in fact inverse agonists.17 X-ray structures in complex with agonists and antagonist ligands are providing information on the early steps in receptor activation. As discussed above, the binding site of the endogenous ligand for the receptor is referred to as the orthosteric binding site. Many drugs also bind to this site or overlapping sites and are competitive with the natural ligand. Drugs that bind at distinct sites are called allosteric modulators.20
2.3. Allosteric Modulators of G Protein-Coupled Receptors
Much of historical drug discovery focused on GPCRs has targeted the orthosteric binding site. However, the advent of functional screening assays (as opposed to radioligand binding assays) as a screening method of choice has increased the number of allosteric ligands being identified for GPCRs. Allosteric ligands can bind to sites on GPCRs that are topographically distinct from the orthosteric site such that the receptor is able to accommodate two ligands simultaneously. Allosteric binding sites have been identified on many GPCRs including adenosine,21 muscarinic acetylcholine,22 dopamine,23 chemokine,24 calcium sensing,25 and glutamate26 receptors.
The major mechanism of action of allosteric ligands is to either enhance or inhibit the binding of the orthosteric agonist, and ligands are termed positive or negative allosteric modulators (PAM and NAM), respectively. This modulation in agonist affinity is reflected in a change in the resultant potency (i.e., EC50), which is increased or decreased. Allosteric modulators, unlike orthosteric ligands, have an inherent saturability to their effect, i.e., a limit on the maximal degree of inhibition or potentiation of an agonist response. This potential makes their action more subtle and less prone to target related side effects.
The advent of functional assays, as opposed to radioligand binding assays, has had a 2-fold effect. First, it has increased the numbers of allosteric ligands that are being identified, but it has also led to the realization that allosteric modulators can modulate the efficiency of receptor activation as well as directly activate the receptor in their own right (in the absence of orthosteric agonist).27,28 One notable feature of allosteric modulators is that their effects depend on the orthosteric ligand in question; this is referred to as “probe dependence”. For example, the indocarbazole staurosporine is a PAM of N-methylscopolamine binding at the muscarinic M1 receptor but is a NAM of the endogenous ligand acetylcholine.29 This example highlights the importance of using the endogenous agonist for a receptor when engaging in allosteric modulator drug discovery.
Of great interest in more recent years has been the structural basis of allosteric mechanisms. It is thought that by virtue of the fact that allosteric binding sites are not (generally) utilized by endogenous ligands, they would be subject to less evolutionary pressure to be conserved. Thus, they represent an attractive mechanism for developing receptor-subtype-selective ligands within GPCR families. Examples of such selective compounds can be found particularly within the family of muscarinic acetylcholine receptors30 and metabotropic glutamate receptors.26 For family C receptors, the orthosteric ligands bind in the large N-terminal (“Venus fly trap”) domain (Figure 2), whereas most allosteric binding sites have been identified within the transmembrane domain region, such as MPEP and CPPHA at the mGlu5 receptor,31 LY487379 at the mGlu2 receptor,32 and CGP7930 at the GABAB receptor.33 Similarly, allosteric binding sites have been suggested to be present in the transmembrane domain of family B receptors, such as the CRF-1 receptor.11
In family A receptors, where the orthosteric binding site is in the TMD, allosteric binding sites are generally found toward the extracellular region, formed by the top of the helices and the extracellular loops,28 though sites have also been described on the intracellular face of the receptor, most notably for chemokine receptors.34 Unlike in family B and C receptors, allosteric binding sites on family A receptors tend to be much closer to the orthosteric binding site and ligands can have two pharmacophores that engage with both orthosteric and allosteric sites. These have been referred to as “dualsteric”, “multivalent”, or “bitopic” ligands. This property is thought to underlie the selectivity profile of some compounds such as McN-A-343 at the muscarinic M2 receptor35 and R22 (Scheme 4) at the dopamine D3 receptor,36 as they engage with allosteric binding sites that are not conserved between receptor subtypes.
Scheme 4. Selective Dopamine D3 Receptor Antagonist, the Selectivity of Which Can Be Rationalized Using the D3R Crystal Structure.

3. Common Structural Features of GPCRs
Figure 3 illustrates the key features of GPCRs revealed by X-ray crystallography from the family A structures to date, using the β1 adrenergic receptor and dopamine D3 receptor structures as representative. There are a number of recent excellent reviews that describe the similarities and differences between some of the recently published structures.37−40 Throughout this review the Ballesteros–Weinstein residue nomenclature system is used for amino acid residues.41 In this numbering method, which is used for family A, receptors are aligned and the number of the most conserved residue in each helix is assigned 50. Amino acid residues are given two numbers (N1.N2) where N1 refers to the TM number (1–7) and N2 is the number relative to the most conserved (number 50). Numbers decrease toward the N-terminus and increase toward the C-terminus.
Figure 3.

Common structural architecture of family A GPCRs revealed by X-ray crystallography. The structures of β1AR (green, 2VT4) and D3R (red, 3PBL) are overlaid and used as representative. The key features of the structures are illustrated (see main text for details).
3.1. Transmembrane Domain
The TMD consists of seven α-helices approximately perpendicular to the cell membrane. Figure 3 shows the common numbering of the helices and that, in more detail, the α-helices are tilted out of plane. The exact positions and orientations of the helices differ slightly from one structure to the next but can be reasonably predicted using homology modeling methods, especially now that multiple structures are available. Since GPCRs are α-helical, the backbones of these proteins are largely involved in intramolecular interactions and not available to H-bond to ligands. As such, generally only the side chains of binding site residues are involved in binding ligands, which to some extent simplifies the modeling process.
3.2. TMD Ligand Binding Site
The TMD site is a deep hydrophobic cavity containing key H-bonding residues, specific to each receptor family, that engage with ligands. In rhodopsin the ligand retinal is entirely enclosed in its binding site with the transmembrane bundle between TM5 and TM6 and is covalently bound to TM7 via Schiff base linkage to Lys296 (7.43). In the β2 and β1 ARs and the dopamine D3 receptor the binding site is more open but the ligands sit in a similar overall position forming interactions principally with TM3, TM5, and TM7. In the aminergic family A receptors there is always an acidic residue on TM3 at position 3.32. In the adenosine A2A and CXCR4 receptors the antagonist ligands sit higher in the binding site and closer to the extracellular surface and the sites are relatively open. Because of the open nature of the binding sites, the antagonist ligands do not fill the binding cavity, making it challenging in silico to correctly predict their binding modes. Overall it is clear from the structures to date that the position of the orthosteric ligand binding site is conserved for a broad range of family A ligands but that the way in which ligands interact in the site can vary quite considerably. This is dealt with in more detail later, and in particular the differences between binding sites are shown in Table 3 and described in the legends. Where ligands are not small molecules, such as in the chemokine family, it is believed that only part of the natural peptide ligand engages with the TMD site and that a more extended binding site must exist on the extracellular face of these receptors.
Table 3. TMD Binding Sites of Published GPCRs Illustrating Protein–Ligand Interactions for Antagonists (Left-Hand Side)a.


The individual ligands are shown keeping an identical view/orientation in the right hand panel (based on a protein structure driven overlay of the different structures using Maestro/Schrödinger). GRID maps to highlight the different shapes and properties of the sites are also shown, with identical energy level contours: Csp3 (C3) at 1 kcal/mol in light gray to define shape, the limit of where a carbon can be. Aromatic C–H probe (C1=) is in yellow at −2.8 kcal/mol for lipophilic/hydrophobic hotspots. Carbonyl group (C=O) is in blue at −4.5 kcal for H-bond acceptor hotspots, and amide NH (N1) is in lilac at −6.6 kcal/mol for H-bond donor hotspots.
3.3. ECL2
The extracellular loop 2 (ECL2) extends from the ends of TM4 and TM5 and sits above the orthosteric binding pocket of family A GPCRs (Figure 3). In rhodopsin this region is very long, having some β-sheet character within it, and it largely encloses the binding site. This feature served to confound many early attempts at homology modeling of GPCRs when only rhodopsin structures were available, and in general, modeling of ECL2 is still challenging because this loop is highly variable and at the same time seems to be generally involved in ligand binding. For example, in β2 and β1 ARs there is a short α-helix within ECL2 that was not predicted by modeling and Phe193 (β2) on the loop is in contact with the bound ligands. The ECL2 often contains one or more disulfide bridges that serve to rigidify the loop. It is thought that for a number of receptors that allosteric binding sites exist at the bridgehead of ECL2 and the top of the orthosteric site and in some cases bitopic ligands span between the orthosteric site and the ECL2 allosteric site.42,43 As discussed above, where the natural ligand for a receptor is a peptide, it is likely that ECL2 is important for ligand binding, and in these cases the orthosteric site may actually extend from the TMD to ECL2 on the extracellular surface.
3.4. Toggle Switch
Immediately below the TMD binding site on TM6 is a highly conserved tryptophan residue W6.48 termed the “toggle switch”44 (Figure 3). This residue is thought to be involved in receptor signaling via a change in its rotational state upon agonist binding that serves to trigger a series of other changes that propagate to the intracellular surface. Other residues involved in this conformational change have been termed “microswitches”.45 Specifically, the toggle switch forms part of the so-called CWxP motif at the bottom of the ligand binding pocket and the rotamer movement is thought to be transmitted through a hydrogen bonding network to the two most conserved residues in TM1 (Asn55 (1.50)), TM2 (Asp83 (2.50)) (Ballesteros–Weinstein nomenclature in parentheses41), and another conserved sequence called the NPxxY motif in TM7. The hydrogen bonding network then extends toward the G protein peptide with water molecules hydrogen bonding to both the receptor and G protein. There is good evidence for this mechanism for rhodopsin, in which the ligand directly interacts with this tryptophan residue,46 but the more recent family A structures have shown no such direct contact with ligands. In the recent agonist bound structures of β2 and β1 ARs the expected conformational change has not occurred (discussed later). However, it remains that this important residue is likely to be involved in the conformational changes that occur on full receptor activation, though perhaps in a more modest way than envisioned in the original rotary toggle switch proposal.
3.5. Ionic Lock
The ionic lock is a salt bridge within the helical bundle on the intracellular face of family A GPCRs.47 In rhodopsin, this salt bridge is between Arg135 (3.50) and Glu247 (6.30) and has been hypothesized to help hold the receptor in an inactive conformation.48 These two residues form the first part of the highly conserved “D(or E)RY” region (Figure 3). Despite this observation, the ionic lock has not been observed in a number of the published structures that bind antagonists, and it has been speculated that this may be due to the presence of antibody or T4 lysozyme fusions (introduced to facilitate crystallization) that have perturbed the intracellular face of the receptor structures or due to the fact that the ligands are not in all cases full inverse agonists.40 The implication is that the ionic lock will only be “closed” in the full ground state (inverse agonist) conformation.
3.6. ICL2 and ICL3
Intracellular loop (ICL) 2 extends between TM3 and TM4 and ICL3 between TM5 and TM6 (Figure 3). These loops are likely to be involved in G protein binding. The structure of bovine opsin bound to the C-terminus of the transducin Gαt protein gives direct support for the role of ICL3 in receptor signaling.49 In this structure the ionic lock is broken and there is outward movement of TM5 and TM6. Comparison of antagonist and agonist structural features is discussed in more detail later.
4. Antagonist Ligand–Receptor Crystal Structures
The individual antagonist structures are briefly described here, focusing on the protein–ligand interactions formed in the TMD binding site and the utility of this information for SBDD approaches. Table 3 also illustrates the protein–ligand interactions for antagonists keeping an identical orientation and is shown with GRID maps to highlight the different shapes and properties of the sites.50,51 The table is intended to allow direct visual comparison of the properties of each of the binding sites to complement the discussion below. Figure 4 shows how each of the ligands overlays in the same consistent binding mode shown in Table 3. The figure indicates how, broadly speaking, all the ligands bind in the TMD site, but we will see that in fine detail they make significantly different interactions to their respective receptors.
Figure 4.
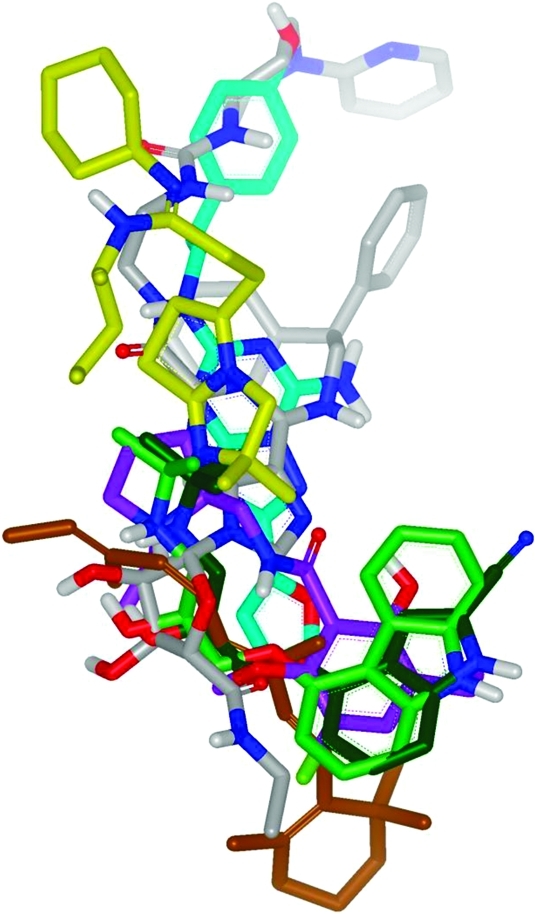
Superposition of ligands from seven different GPCR X-ray structures, based on Cα alignment of the GPCR protein structures: dark green = β1AR antagonist; light green = β2AR antagonist; cyan = A2AR antagonist; lilac = D3R antagonist; yellow = CXCR4 antagonist; brown = rhodopsin (inactive); gray = A2AR agonist.
4.1. Rhodopsin
The first X-ray diffraction structure of a GPCR, bovine rhodopsin, was published in 2000 solved from bovine retinal disk membranes.52 This provided a detailed picture of the ligand binding pocket of a receptor in the full inverse agonist conformation. The 11-cis retinal ligand makes a covalent Schiff base linkage to Lys296 (7.43) in TM7. In addition, residues from TM1, TM2, and TM7 encase the Schiff base, and the β-ionone ring forms interactions with the side chains of Phe208 (5.43) and Trp265 (6.48), from TM5 and TM6 (Table 3 entry 1 and Figure 5B). This and additional structures of the inactive dark-state rhodopsin then provided the basis for GPCR modeling during the following 8 years.53 A great deal of work has been done using bovine rhodopsin as the template for homology modeling of other GPCRs, and there are a number of reviews dealing with these developments.54−56 However, there are several problems associated with rhodopsin as a starting point for GPCR modeling.56,57 First, although rhodopsin shares overall structural features with other family A GPCRs, the actual homology is less than 25% and for other GPCR families such as the secretin, adhesion, and metabotropic receptors, there is no detectable sequence homology at all with rhodopsin. Second, since retinal is covalently bound to the receptor, rhodopsin is likely to have a very different mechanism of activation to other receptors with noncovalent ligands. In rhodopsin, signaling is initially triggered by ligand isomerization via photons of light and the isomerized ligand becomes the agonist. As such, there is no requirement for an entrance to the ligand binding site, and this is actually blocked by the second extracellular loop (ECL2) of the receptor. Despite these concerns, rhodopsin has successfully been used as a starting point for homology modeling, facilitating SBDD efforts, and some examples are given later in this article.
Figure 5.

TMD binding sites of published GPCRs illustrating protein–ligand interactions for agonists (cyan ligands) compared with antagonists (pink ligands): (A) general changes on antagonist to agonist transition exemplified using rhodopsin (red) and opsin (green); (B) rhodopsin agonist structure (green) 2X72 vs antagonist structure (red) 1HZX; (C) β2AR agonist structure (green) 3POG vs antagonist structure (red) 2RH1; (D) A2AR agonist structure (green) 3QAK vs antagonist structure (red) 3EML.
4.2. β1 and β2 Adrenergic Receptors
In 2007 and 2008 the next major breakthroughs in GPCR structural biology were made when the crystal structures of the turkey β158 and human β2 adrenoceptors59,60 were solved. These GPCR structures, in complex with antagonist ligands cyanopindolol (β1) and carazolol (β2), were the first with noncovalently bound small molecules in the binding sites (Table 3, entries 2 and 3). The structure of the human β2AR was first determined at medium resolution (3.5 Å) in complex with an antibody fragment59 and subsequently at higher resolution (2.4 Å) by insertion of the enzyme T4 lysozyme (T4L) into ICL3 of the receptor.60 The fusion proteins were introduced to aid crystallization of the receptors rather than to increase thermal stability (see below). This is the first of two new strategies for the determination of GPCR structures. If the fusion protein approach is used, it has been coupled with a very high potency ligand to aid stability and a highly specialized crystallization method called lipidic cubic phase (LCP) which serves to mimic the environment in the cell membrane. The second strategy, first used in the structure determination of the turkey β1AR to a resolution of 2.7 Å, involved the introduction of a number of point mutations into the receptor construct that increased the thermostability of the protein and enabled crystallization.58 The thermostabilization approach “locks” the receptor in a single homogeneous conformation that greatly facilitates purification and structure determination. The stabilized receptor construct has been termed a “StaR” for stabilized receptor.61 By use of the thermostabilization strategy, crystallization can be achieved using more typical vapor diffusion crystallography methods, albeit in the presence of detergents required to solubilize the receptor. Consistent with the “conformational thermostabilization” hypothesis, the resulting β1AR antagonist StaR displayed high affinity for antagonists and inverse agonists but markedly reduced agonist affinity.58 An important finding is that these two orthogonal crystallization strategies gave highly comparable structure solutions of β1AR and β2AR indicating that (1) none of the point mutations had resulted in perturbations of the β1AR structure and (2) the fusion protein had not significantly perturbed the β2AR structure (Table 3, entries 2 and 3). However, in the case of the β2AR structure and other fusion protein structures, little information can be gained about the intracellular receptor surface because of the presence of the additional protein domain and the fact that the receptor cannot interact with G proteins. In contrast, the β1AR antagonist StaR can still be activated by agonists, albeit at much higher concentrations than required for the wild-type receptor because of the reduced agonist affinity of the StaR compared to the wild-type.58
The ECL2 of both β1AR and β2AR sits above the TMD binding site and contains an α-helical structure that was not predicted computationally using rhodopsin as a template; this is quite different from the β-sheet present in rhodopsin in this region.62,63 The binding pockets for cyanopindolol and carazolol are very similar in the two receptors, which have a high level of sequence conservation in the transmembrane domain regions (Table 3, entries 2 and 3). In fact, in the binding site there are 15 amino acid residues that in β1AR are in contact with cyanopindolol and that are conserved in β2AR.58,60 In both receptors, residues from TM3, TM5, TM6, TM7, and ECL2 make contacts with the ligands, most notably between the amine group of cyanopindolol/carazolol and Asp121/113 (3.32) and Asn329/312 (7.39) and also between Ser211/203 (5.42) (residue numbers in β1AR shown first) and the indole nitrogen of cyanopindolol or the carbazole of carazolol. There are only very small differences between the two structures, such as the rotamer state of Ser211/203 (5.42). However, despite the high degree of conservation of the binding site, there are known ligands that distinguish between the two receptor subtypes pharmacologically (e.g., CGP20712A). Careful inspection of the binding sites reveals that although only two amino acid residues within 8 Å of the β1AR binding site differ in β2AR (Val172 (4.56) and Phe325 (7.35) are Thr164 (4.56) and Tyr308 (7.35), respectively, in β2AR), these changes do result in some subtle differences in the shape and polarity of the binding pocket that may account for, or might now be used in, the design of selective ligands, and this is discussed later. In β2AR, Tyr308 (7.35) has been implicated by site directed mutagenesis (SDM) studies as playing a role in the selectivity of agonists because of its ability to form a hydrogen bond with Asn293 (6.55).64 As in the case of closely related enzyme targets, it seems likely that receptor subtype selectivity could be achieved by SBDD approaches by the exploitation of minor differences either in the primary binding pocket or in the extended region (which includes nonconserved residues) in the ECL2 that contribute to the binding site(s).
4.3. Dopamine D3 Receptor
Very recently the dopamine D3 receptor structure has been solved at a resolution of 3.15 Å in complex with the high affinity antagonist eticlopride using the T4 lysosyme fusion protein strategy.36 As expected, the overall topology is similar to that in the β1AR and β2AR structures and includes an α helix in ICL2 which was also observed for β1AR.58 However, unlike in β1AR and β2AR, the ECL2 was found to be disordered having no apparent secondary structure. This is surprising, since the portion of ECL2 that contributes to ligand binding in the dopamine D3 receptor site is orientated in a similar position relative to the bound ligand compared with the β1 and β2 ARs. Other notable differences from β2AR include an outward tilting of TM6 (by 3 Å) and TM7 (by 2 Å) and an inward tilting of approximately 3.5 Å by TM3 and TM5 at the extracellular face. Significantly, the “ionic lock” between Arg128 (3.50) and Glu324 (6.30) is formed in this structure,36 while it is broken in both of the adrenergic receptor structures (Figure 3). This would suggest that the ligand has induced a full inverse agonist conformation in this structure and/or that the T4 lysozyme fusion has had less impact on the structure than in other receptors. Within the ligand binding pocket it is perhaps of no surprise that there are many similarities with both the β1AR and β2AR structures, given the chemical similarity between the endogenous agonists. Ten of the inward facing residues from a total of 18 amino acids in the primary binding site are conserved. As would be predicted from sequence homology to the β-adrenergic receptors, the tertiary amine of the ligand forms a salt bridge with Asp110 (3.32). The aromatic ring of eticlopride sits in a hydrophobic pocket formed by Phe345 (6.51), Phe346 (6.52), Val189 (5.39), Ser192 (5.42), Ser193 (5.43), Val111 (3.33), and Ile183 in ECL2 (Table 3, entry 4). The pyrrolidine ring sits in a largely aromatic region comprising of Tyr365 (7.35), Phe106 (3.32), Val86 (2.61), and Tyr373 (7.43). Interestingly the ligand forms two intramolecular hydrogen bonds holding the aromatic ring largely in plane with the amide side chain. Overall, as can be seen from Table 3, entry 4, the ligand sits in a very similar position to ligands in β1AR and β2AR.
4.4. Adenosine A2A Receptor
The structure of the A2A receptor was first solved in 2008 using the T4L fusion technology.65 T4L was inserted between Leu209 (5.70) and Ala221 (6.23) in the ICL3 of the receptor. The insertion of the T4L was observed to have somewhat altered the pharmacology of the receptor in that agonists bound to the receptor with a higher affinity than for wild type. A2A-T4L was crystallized using the LCP method with the addition of cholesteryl hemisuccinate (CHS) and in complex with the inverse agonist ligand ZM241385. There is close agreement in the packing of the helices between A2A and other receptor structures; e.g., compared to the adrenergic receptors, the rmsd is 1.8–2.5 Å, despite a relatively low homology (20–40% in the TM regions). In our own laboratories a second structure of the receptor has been solved using the thermostabilization strategy.66 This receptor, known as A2A-StaR2, includes eight thermostabilizing mutations spread throughout the receptor that appear to hold the receptor in an inverse agonist conformation. In contrast to the A2A-T4L construct the A2A-StaR2 has a lower affinity for agonists compared to the wild type receptor but a similar or slightly higher affinity for antagonists.61 As is the case for the β1AR, the mutations in the A2A-StaR2 appear to have very little direct effect on the structure compared to the A2A-T4L but rather seem to facilitate packing between adjacent helices to stabilize the receptor conformation. The two structures are in broad agreement, but there are some significant differences. Most noticeable is a ∼5 Å outward movement of TM5 and TM6 observed in the A2A-T4L compared with A2A-StaR2 most likely due to the presence of the fusion protein displacing these helices. We propose that these are similar to the movements observed during receptor activation as seen in the transition of rhodopsin from the ground state to the active state (opsin) discussed below, and this may account for the observed “agonist-like” pharmacology of A2A-T4L. The differences in TM6 between the structures are particularly noticeable in the region of the ionic lock, which in A2A is formed between Glu228 (6.30) and Arg102 (3.50). In the A2A-StaR2 the ionic lock is present providing further evidence that this receptor is captured in the inverse agonist state, while in the A2A-T4L the movement and rotation of TM6 break the ionic lock.
In the A2A receptor the loop regions are held in place by four disulfide linkages. Cys77-Cys166 links the top of TM3 to ECL2 and is highly conserved in family A GPCRs. Cys71-Cys159 and Cys74-Cys146 link ECL1 and ECL2 and are unique to the A2A receptor. In addition there is an intraloop disulfide bond in ECL3 between Cys259 and Cys262. The disulfide bonds create a rigid structure that produces an open entrance to the ligand binding pocket that may facilitate access of ligands. The inverse agonist ligand ZM241385 sits in quite a different position compared to retinal and to the aminergic ligands within the TMD ligand binding pocket (Table 3, entry 5). In fact, ZM241385 sits almost perpendicular to the membrane plane with its furan ring deep within the binding pocket, interacting with Asn253 (6.55) and Glu169 (ECL2), while at the other end of the ligand the phenol ring projects toward the extracellular region of the receptor. The B factor (or temperature factor, a crystallographic measure of disorder) of the flexible phenol group in the A2A-T4L structure is high (>100 Å2) compared to the rest of the ligand (∼50), suggesting that the position of this substituent should be interpreted with caution.67 As well as the furan, the heterocycle of the ligand also forms a H-bond to the Asn253 (6.55). The Asn253 (6.55) has previously been predicted by SDM experiments to form a key interaction with a broad range of ligands, and its mutation abolishes ligand binding.68 ZM241385 does not bind to the entirety of the binding pocket and the region proposed to interact with the ribose of the natural agonist ligand adenosine, adjacent to the polar residues Thr88 (3.36) and Ser277 (7.42), and the toggle switch Trp246 (6.48) is unoccupied.68,69 However, there is a network of water molecules that interact with the ligand in this region. The involvement of water networks in ligand binding is commonly observed in SBDD but creates an issue for de novo docking to homology models, as it is difficult to predict the exact position and role of waters within a large and open binding cleft in the absence of X-ray data. Crystal structures are normally required to identify water molecules relevant to drug design in a SBDD paradigm.
4.5. Chemokine Receptor CXCR4
CXCR4 is one of the family A chemokine receptor subfamily of which there are 19 members. Chemokines have large peptides as their natural agonists, and in this case the ligand is stromal-derived factor (SDF) 1 (CXCL12). The structure of CXCR4 has recently been obtained in complex with both cyclic peptide and small molecule antagonists.70 This is the first example of a peptide GPCR to be solved and so represents a major breakthrough in understanding the diversity of GPCR structures and for modeling chemokine and other peptide receptors for drug discovery. The receptor structure was obtained using a construct that included the T4L fusion but also contained two thermostabilizing mutations (L125W (3.41) and T240P (6.36)). The antagonists used for crystallization were IT1t (an isothiourea) and CVX15 (a 16-residue cyclic peptide antagonist) (Table 3, entry 6). In all, five structures have been deposited in the Protein Data Bank (PDB) that differ in the truncation of the C-terminus, the presence or absence of the T240P (6.36) mutation, and the ligand (IT1t or CVX15). However, each structure included the L125W (3.41) mutation and the T4L fusion. The N-terminal 26 residues are not visible in the structure and are presumably disordered. Although the overall fold of the TMD is the same, TM1 is shifted toward the core helical bundle compared to the aminergic receptors. This could be interpreted as a feature of peptide receptors in which ligands bind primarily to the N-terminus of the receptor but must still be able to engage the TMD to trigger the conformational changes associated with receptor activation. Other key differences are (1) a rotation in the extracellular end of TM2 resulting from a tighter turn around a conserved proline (2.58), (2) significant differences in the positions of the ends of TM4, and (3) a shift in the extracellular end of TM6 compared to the β-adrenergic and adenosine A2A receptors.
The intracellular C-terminus of CXCR4 is significantly different from the other published GPCR structures. TM7 is shorter by one helical turn, ending in the NPxxY motif, and there is no α-helix in the C-terminal tail, sometimes called helix 8. The structure does not contain the full α-helical motif usually present in this region and does not apparently have a palmitoylation site whose function is to tether the C-terminus to the plasma membrane. It is possible that chemokine receptors do not have the usual helix 8, or perhaps the absence of this feature is an artifact of the crystallization constructs used in these studies. Allosteric modulators have in fact been identified to related chemokine receptors which bind to an intracellular binding site that was believed to be in the region of helix 8. For example, SB265610 behaves as an allosteric inverse agonist of CXCR2. This compound’s binding is affected by mutations K320A, Y314A in the C-terminal tail and D84N (2.40) in TM2.71 A similar binding site has been identified for other CXCR2 antagonists34 and is also thought to exist in CCR4 and CCR5 receptors.72
The CXCR4 structure gives insight into how non-peptide antagonist ligands can block the activity of the much larger peptide agonist CXCL12. Historically, a two-site model of activation or a message–address concept has been suggested to explain the binding of large peptide or hormone ligands. “Site 1” represented the address or ligand recognition site, and in the case of opioids and neurokinins this site was proposed to determine the specificity of ligand/receptor interactions.73,74 In peptide receptors this site was postulated to be located in the extracellular regions of the protein including the N-terminus and/or the extracellular loops such as ECL2. Indeed, NMR structures of the N-terminus of CXCR4 in complex with SDF 1 have been determined, which help to support these ideas.75 Such an extracellular binding site would involve multivalent binding interactions between the receptor and peptide ligand akin to a protein–protein interaction and that would most likely be difficult to inhibit with small molecule antagonists. However, “site 2” or the message region is the binding site that triggers receptor activation and signaling and is believed to involve the N-terminus of the peptide ligand making contact with the TMD binding site, analogous to the orthosteric binding site of aminergic receptors.76 The CXCR4 structure gives us the first view of an example of this “site 2” or “message” binding site for peptide receptors. Acidic residues Asp187 (in ECL2), Glu288 (7.39), and Asp97 (2.63), which are important for the binding of the N-terminus of CXCL12, also interact with the small molecule antagonist IT1t as well as the peptide CVX15 (Table 3, entry 6). IT1t also forms hydrophobic contacts with Trp102 (ECL1), Val112 (3.28), Tyr116 (3.32), Cys186 (ECL2), Arg183 (ECL2), and Ile185 (ECL2). Overall, the small molecule sits higher in the TMD site than other small molecule antagonist ligands in the crystal structures to date, and we might expect that agonist signaling will involve interactions with residues deeper in the binding site and closer to the toggle switch.
5. Agonist Ligand–Receptor Crystal Structures
Until very recently, a view of the agonist GPCR conformation and how it varied from the antagonist form was only available from progress in the structural biology of the rhodopsin/opsin system.77 As described above, before 2011 all of the diffusible ligand costructures of family A GPCRs were with antagonist ligands. However, new breakthroughs with structures that bind agonist ligands now start to give a picture of how agonist signaling may occur. The structural features associated with the antagonist to agonist transition will be discussed in this section and are also shown in Figure 5.
5.1. Opsin
Evidence of the structural basis of GPCR activation has come from solutions of the structure of the activated apoprotein form of rhodopsin, called opsin, in complex with a peptide derived from the C-terminal tail of the receptor’s cognate G protein, transducin.49,78 These structures show significant movements of TM5 and TM6 when compared to the ground state of rhodopsin, with the cytoplasmic end of TM6 moving outward by 6–7 Å. Figure 5A indicates the movements of the seven helices; these conformational changes in the transmembrane domain region are thought to underlie the process of receptor activation. Notably, the ionic lock, a salt bridge between Arg135 (3.50) and Glu247 (6.30), is also broken in these structures; this is another element that is thought to contribute to the activation process.79 Changes in other so-called “microswitches” are also observed.45 More recent insights into the stages of receptor activation have come from an agonist-bound structure of rhodopsin containing a constitutively active mutation E113Q (3.28)46 (Figure 5B). The agonist, all-trans-retinal, does not appear to be covalently bound to the receptor. There is also a change in the rotameric state of the toggle switch Trp265 (6.48) that is located at the base of the binding site in the CWxP motif. This conformational change is thought to be caused by a rearrangement of the water-mediated hydrogen bonding network involved in agonist binding and is transmitted through to the two most conserved residues in TM1 (Asn55 (1.50)) and TM2 (Asp83 (2.50)) and the NPxxY motif in TM7. The network of hydrogen bonds continues into the site of receptor–G protein peptide interaction; the rearrangement of interactions from the agonist-binding site to the G-protein-binding site likely represents the mechanism by which the receptor becomes active and able to couple to G proteins. In addition to advancing our knowledge of receptor activation these new structures will be useful for developing models of agonist binding for use in virtual screening and drug discovery.
5.2. β1 and β2 Adrenergic Receptors
A wider level of understanding of the structural basis of receptor activation beyond rhodopsin has come from various recent studies examining agonist-bound structures of the β1 and β2 adrenergic receptors. Initial evidence regarding the differences between agonist and antagonist binding came from the structure of the cyanopindolol-bound β1AR. Docking of adrenaline into the cyanopindolol binding pocket suggested that because of the smaller size of the agonist compared to the antagonist, the distance between the serine residues on TM5 and the catechol hydroxyl groups of the agonist would be too great to form the expected hydrogen bonds. Agonist binding and receptor activation would therefore necessitate a contraction of the binding pocket by 2–3 Å, likely involving movement of TM5.58 However, these proposed changes at the level of the ligand-binding site did not explain how agonist binding would result in the 5–6 Å movement at the base of TM6 which was observed in the rhodopsin/opsin system.77
More recent studies have yielded greater insights into mechanisms of agonist binding and the initial conformational changes involved in receptor activation. These include the description of a crystal structure of the β2AR-T4L fusion in complex with a high affinity agonist BI167107 and a nanobody that acts as a surrogate for the cognate G protein.80 Simultaneously, a crystal structure of an agonist (FAUC50) irreversibly bound to the β2AR has been described81 as well as multiple cocrystal structures of partial and full agonists in complex with the β1AR.82 Figure 6 illustrates the differences between the β1AR agonist and antagonist bound structures in the binding site, while Figures 5C depicts the corresponding β2AR structures.
Figure 6.

Comparison of antagonist and agonist ligands bound to the stabilized β1AR receptor complex, indicating new polar interactions formed upon agonist binding. Antagonist ligand in magenta (2VT4) and agonist in gray (3YO3).
Although the data sets for β1AR have been determined using a receptor construct stabilized by mutagenesis in the antagonist conformation, the receptor can still bind and be activated by agonists,58 albeit at higher concentrations than required for the wild-type receptor. High resolution costructures (all at 3 Å or less) have been determined for both partial agonists (salbutamol and dobutamine) and full agonists (carmeterol and isoprenaline).82 The overall topology of the β1AR is very similar when complexed with antagonists, partial agonists, and full agonists, though there are subtle differences in the binding site. As might be expected from mutagenesis data, the amine moieties of all the agonist ligands (plus the β-hydroxyl for all agonists except dobutamine) form interactions with Asp121 (3.32) and Asn329 (7.39). All of the agonists form a hydrogen bond with Ser211 (5.42); additionally, the full agonists isoprenaline and carmeterol (but not the partial agonists) form a second hydrogen bond with Ser215 (5.46) in conjunction with a change in the rotamer state of Ser212 (5.43) to form a hydrogen bond with Asn310 (6.55) (Figure 6).82
Notably, the number of polar interactions formed by the serine residues on TM5 appears to represent a marker of partial versus full agonism. The formation of these polar interactions causes the binding pocket to contract by approximately 1 Å in comparison with the cyanopindolol costructure (Figure 6). However, it is clear that these conformational changes do not result in the larger scale movements of TM5 and TM6 that might be expected based on the opsin structure. It is likely that, despite binding agonists, because the receptor construct used was one stabilized in an inactive conformation, these changes simply represent the first stage of movements that result in full receptor activation. Similar results have been shown for the β2AR in complex with BI167107 and a nanobody that mimics the actions of the G protein, shifting the receptor into a high-agonist affinity state (Figure 5C). This breakthrough has enabled the agonist-bound structure to be solved at a resolution of 3.5 Å.80 Unlike the β1AR study (where low affinity agonists were profiled), the β2AR-T4L system requires a high affinity ligand, preferably with a slow off-rate, to engender stability to the complex.80 It is this property that enabled the costructure of an irreversibly binding agonist, FAUC50, also to be determined.81
Comparison of the nanobody-bound β2AR structure with the previously solved carazolol-β2AR costructure (lacking the nanobody) shows that there is a clear outward movement at the base of TM6 caused by its rotation, coupled with an inward movement of TM5 and TM7 and an upward shift along the axis of TM3 in the β2AR structure in the presence of the nanobody and BI167107; the 11 Å movement at the base of TM6 is comparable to that seen for the active versus the inactive states of rhodopsin.77 As well as the obvious conformational changes at the cytoplasmic face of the receptor, the changes in the ligand-binding site are larger than those observed in the β1AR80,82 (Figure 5C vs Figure 6). The binding mode of the agonist ligand is very similar to that of the antagonist carazolol with interactions with both Asp113 (3.32) and Asn312 (7.39) (Figure 5C). In addition, polar interactions with serine residues on TM5 Ser203 (5.42) and Ser207 (5.46) are enabled by a ∼2 Å inward movement of TM5 and by smaller movements of TM6 and TM7. Despite the conformational changes throughout the receptor consistent with receptor activation, there was no change in the rotamer state of the Trp286 (6.48), which has been proposed as a “toggle switch” for receptor activation.
5.3. Adenosine A2A Receptor
During the preparation of this manuscript the structure of the agonist UK-432097 bound to the adenosine A2A-T4L construct was published.83 This breakthrough allows direct comparison of the structure with the antagonist ligand ZM241385 in the same receptor construct, previously published by the same group.65 The induced fit of this large ligand appears to have trapped the agonist conformation of the receptor, affecting the shape and character of the binding site. Compared with the antagonist-bound structure, there is a small outward tilt and rotation of the cytoplasmic half of TM6, a movement of TM5, and an upward shift of TM3, overall resembling the changes seen between rhodopsin and opsin. The changes within the binding site are more profound than seen for the β adrenergic receptor but are supported by extensive SDM evidence that supports the ligand position and interactions made.68,69 Figure 5D illustrates the binding mode of UK-432097 compared with ZM241385 in the TMD site. The key H-bonding with Asn253 (6.55) is maintained with the purine ring, and the ribose moiety pushes deeply into the bottom of the pocket forming new polar interactions, absent in the antagonist-bound structures, with Thr88 (3.36), His278 (7.43), and Ser277 (7.42). The upward movement of TM3 causes Ile92 (3.40) to move up and create a hydrophobic cavity that is in turn occupied by Pro189 (5.50) on TM5. Very recently a structure of the A2A receptor thermostabilized in the agonist conformation has also been published.84 Thermostabilization has enabled costructures to be obtained with the relatively low affinity agonists adenosine and N-ethylcarboxyadenosine (NECA). This structure shows similar changes to the binding site and helical movements observed in the UK-432097 structure. Overall the changes observed within the binding site are a very useful addition to the tool box of the computational chemist, providing a further template for the homology modeling of the agonist conformation of family A GPCRs from a subfamily different from that of the adrenergic receptors.
6. Extracellular Domains
Family B and family C GPCRs have large extracellular domains (ECD) that are involved in ligand binding (Figure 2). Progress has been made in SBDD with agents that target these binding sites, and examples are given in this section.
6.1. Family B Extracellular Domain Structures
Family B contains the secretin group of 15 receptors that consist of the TMD and an N-terminal ECD that binds peptide hormone ligands including glucagon-like peptide, calcitonin, and parathyroid hormone.7 The proposed mechanism of receptor activation requires binding of the peptide hormone in an α-helical conformation to both the extracellular domain and also into the “message” region within the TMD (Figure 2).85 A number of these receptors represent attractive drug targets, and in some cases modified versions of the native agonist peptides have been used in the clinic. For example, a stabilized version of glucagon-like peptide, liraglutide, was recently approved for the treatment of type II diabetes (Table 2).86 To date, it has been very difficult to discover non-peptide modulators that bind to the TMD allosteric binding site for this class of receptors by using conventional drug discovery approaches such as high throughput screening (HTS). However, structures of the extracellular domains of several members of the family, in some cases in complex with peptide ligands, have been solved by X-ray crystallography87,88 or by NMR spectroscopy.89 These large peptide ligands tend to contain amphipathic helices binding into a central hydrophobic groove formed by a three-layer α–β–β α fold ECD, resembling a “hot dog in a bun” (Figure 2).88 Since this orthosteric site has a large protein–protein interface, it is difficult to block or mimic the peptide agonist with a small molecule inhibitor. This may explain the generally intractable nature of this class of receptors with regard to small molecule drug discovery.
One exception, however, is the progress made against the calcitonin gene related peptide (CGRP) receptor. This receptor is unusual in that it consists of a multimeric complex of the seven-transmembrane protein calcitonin-receptor-like receptor (CLR) and also a single transmembrane protein, receptor activity modifying protein (RAMP) 1.90 CGRP acts as a potent vasodilator and has been implicated in migraine, and several small molecule peptidomimetic CGRP receptor antagonists have entered clinical trials for this indication.91 A recent breakthrough has been the publication of X-ray structures of the CLR/RAMP1 heterodimer ECD in complex with the clinical antagonists olcegepant and telcagepant (Figure 7).92 RAMP1 is a three-helix bundle that interacts with the N-terminus of CLR through hydrophobic and electrostatic interactions (Figure 7A). The antagonist ligands bind to a cleft formed at the interface between the CLR and RAMP1 (Figure 7B). The structures show that there are multiple ligand–protein interactions being formed in this pocket and the ligands need to span the distance between a hydrogen bond donor–acceptor site on CLR and a hydrophobic pocket on RAMP1 for high affinity. Olcegepant forms hydrogen bonds to the backbone of Thr122 and side chain of Asp94 on CLR, the side chain of RAMP1 Asp71, and a water-mediated hydrogen bond at the CLR-RAMP1 interface involving` CLR Arg38 and RAMP1 Arg67. Antagonism via this mode therefore seems to require a high molecular weight compound; both olcegepant and telcagepant are greater than 500 Da in size.
Figure 7.

(A) ECD of the CGRP receptor crystal structure (3N7S). Shown is the N-terminal domain CLR (green) in complex with RAMP 1 (red). (B) Olcegepant (blue) binds at the interface of CLR and RAMP 1.
6.2. Family C Extracellular Domain Structures
The metabotropic glutamate (mGlu) receptors are examples of family C G-protein-coupled receptors, most notable for the presence of a large bilobed N-terminal domain that forms the orthosteric site for glutamate binding. This N-terminal domain is thought to close upon agonist binding (hence the term “Venus fly trap” domain, Figure 2), transmitting receptor activation via a cysteine-rich region to the more familiar seven-transmembrane spanning domain. There are eight mGlu receptor subtypes, which subclassify into three main groups (I–III) based on their pharmacology and G-protein-coupling profile. Types 2 and 3 metabotropic glutamate receptors are members of the group II mGlu receptors and are expressed presynaptically in the CNS where they regulate transmission of the excitatory neurotransmitter glutamate. As schizophrenia is a disease in which there is excessive glutamate release in the cortex, there has been great interest in discovering selective agonists of the mGlu2 and mGlu3 receptors that would act via presynaptic autoreceptors to reduce glutamate levels.93
Despite the high conservation of residues forming the binding site, initial work to discover subtype-selective agonists focused on analogues of the endogenous agonist l-glutamate. Most notable in terms of group II mGlu receptors was the discovery of DCG-IV, a cyclized analogue of glutamate that displayed high affinity and selectivity for mGlu2 and mGlu3 over the other mGlu receptor subtypes.94,95 This has been followed by bicyclic compounds such as LY354740,96 LY379268,97 and LY404039,98 which not only display improved selectivity for mGlu2/3 receptors but had sufficient druglike properties to be considered suitable for clinical development in anxiety and/or schizophrenia. Indeed, recent studies have shown that a prodrug of the active compound LY404039 is effective in a phase IIa trial in schizophrenia, prompting further interest in this approach.99
It was during the development of these agents that the crystal structures of the N-terminal ligand binding domains of members of the mGlu receptor family have been solved, including that of the mGlu1 subtype100 and those of the mGlu3 and mGlu7 receptors.101 These structures have both aided the design of mGlu receptor ligands and permitted a retrospective analysis of the selectivity profile of some ligands which may lead to improved compounds in the future. Five cocrystal structures of the mGlu3 N-terminal domain were solved, including solutions with both l-glutamate and the more selective group II agonist DCG-IV (Figure 8). Interestingly, the hydrogen bonding network between l-glutamate and the binding site of mGlu3 was almost perfectly replicated in the DCG-IV costructure, with the exception of hydrogen bonds mediated through two water molecules to Ser278 and Arg64. Two of the three water molecules found in the l-glutamate costructure are absent in the DCG-IV structure because of the presence of the carboxylate side chain on the cyclized core of the ligand. Importantly, this moiety makes van der Waals interactions with the side chain of Tyr150 which are absent in the l-glutamate structure (Figure 8).101 While l-glutamate can be easily accommodated in the binding sites of both the mGlu1 and mGlu3 receptor, the binding of DCG-IV is impaired by the presence of Trp110 in mGlu1 in place of Tyr150 in mGlu3. At first site, this change appears relatively conservative, but the crystal structures reveal that the indole ring of Trp110 in mGlu1 is almost perpendicular to the phenol ring of Tyr150 of mGlu3; such an orientation sterically blocks the binding of DCG-IV (Figure 8).101 Similar observations have been made regarding the subtype selectivity of the bicyclic agonist LY404040.102 The binding of DCG-IV to mGlu7 also appears unfavorable because the van der Waals interaction with the Tyr150 of mGlu3 is absent in group III mGlu receptors.101 These observations almost certainly underlie the selectivity profile of DCG-IV for the mGlu2 and mGlu3 receptor subtypes. Homology models and the crystal structures are now being used to interpret data sets for some of the newer constrained bicyclic analogues of DCG-IV. LY379268, LY389795, LY404039, and LY404040 are all heterocyclic variants of the prototypical agonist LY354740.96−98 Structural insights have yielded very specific understanding of the pharmacology of these compounds: LY404040 and LY404039 are diastereomeric sulfoxide analogues of the sulfide-containing agonist LY389795. However, LY404040 has approximately 30-fold and 550-fold higher affinity at mGlu2 and mGlu3, respectively, than its diastereoisomer LY404039. This is due to the fact that the sulfoxide oxygen of LY404040 is oriented within hydrogen bonding distance of the phenol of Tyr326, an interaction that is not feasible for the diastereomer LY404039.98
Figure 8.

(A) ECD of the mGlu3 receptor crystal structure complex bound to glutamate (2E4V). (B) DCG-IV complex indicating differences between mGlu1 (yellow) and mGlu3 (red) that give rise to selectivity.
7. SBDD against GPCR Targets
It is beyond the scope of this Perspective to review the full scope of the many SBDD programs reported for GPCR targets in recent years. There has been extensive use of homology modeling and virtual screening, and a huge body of site directed mutagenesis data have been generated to support drug discovery efforts across a wide range of targets.103−116 However, in the following section we will briefly highlight some examples to illustrate strategies that have been employed and in particular where the new GPCR structural information described above has started to be used directly for SBDD.
7.1. β Adrenergic Receptors
The recent publication of the crystal structures of various antagonist and agonist ligands bound to the β1AR and β2AR has stimulated much interest in the academic community, leading to a spate of publications. Wacker et al.117 have described X-ray cocomplexes of three ligands 1–3 binding to β2AR-T4L (following up from their earlier crystal structures of carazolol 4 and timolol 5) and then carried out cross-docking of all five ligands (Scheme 1). As expected, the best docking scores were given from self-docking (ligand into its own structure), but good results were also achieved for other solutions. The ligand binding site was generally found to be quite rigid with ligands in similar positions relative to each other in each case. Kolb et al. have used the X-ray complex of β2AR (2RH1) for virtual screening of one million compounds with leadlike properties to examine the applicability of the system for hit discovery using docking protocols.118 Twenty-five virtual hits were selected and tested in a radioligand binding assay. Six hits (Ki of 9 nM to 3.2 μM) were confirmed (24% hit rate) falling into two main chemical classes, including 2 which has been crystallized by Wacker et al. above (Scheme 1) and 6 (Scheme 2). Similarly, Topiol et al. carried out docking and virtual screening using β2AR of both an in-house database and external database.40,56 Both databases gave good results; 36% and 12% hit rates for the in-house and commercial libraries, respectively (Ki of 0.1 nM to 21 μM and Ki of 14 nM to 4.3 μM), compared with 0.3% hit rate for screening of a set of randomly selected molecules. As well as rediscovering the well-known hydroxylamine chemotype for β2AR, such as the potent inhibitor 7 in Scheme 2, new chemical classes of hits were also discovered. Costanzi has examined how well homology models of β2AR-T4L based on bovine rhodopsin compare with the crystal structure solution.119 A recurring theme in modeling of GPCRs is the difficulty in correctly predicting the conformation and position of the extracellular loops. The best results were achieved building ECL2 de novo rather than basing the conformation on bovine rhodopsin where this loop partially occludes the binding site. The binding mode of the crystallographic ligand carazolol could be successfully recapitulated by docking into the model, particularly when SDM data were taken into account to manually adjust the model in the binding site. This work also serves as an example to illustrate that even quite small errors in the positions of residues within the ligand binding pocket will perturb ligand docking. To overcome this problem, generation of multiple models and consideration of SDM and SAR data are often important to generate useful results. de Graaf and Rognan have used the β2AR-T4L structure to develop a “customized” model that binds partial and full agonists.120 The rotameric states of Ser212 (5.43) and Ser215 (5.46) within the binding site were adjusted to facilitate H-bonding of the receptor to the catechol hydroxyl groups (or equivalent functionality) of agonist ligands. The modified receptor binding site then performed better than the X-ray structure in distinguishing partial/full agonists from decoy ligands in docking runs. The authors suggest that antagonist structures can be used as templates for agonist homology models and subsequent agonist ligand identification if appropriately modified, i.e., from knowledge of the effect of SDM on agonists vs antagonists. These modified models were then representative of “early activated” conformations for virtual screening. An example of drug design has been reported by Soriano-Ursúa and colleagues in which boron-containing analogues of β2AR agonists were proposed from docking against the β2 receptor binding site.121 One new agonist BR-AEA 8 (Scheme 2) was shown to be more potent than the corresponding diol agonists from which they were derived in a functional assay (relaxation of isolated guinea pig tracheal rings). The analogues were also shown to be competitively antagonized by β2AR antagonists. In another recent report, Hattori et al. have studied dockings of a number of β3AR agonists to rationalize observed selectivity using β2AR (2RH1) as the template for generation of a β3AR homology model.122 The potent and selective ligands, such as 9 (Scheme 2), included extensions designed to access a region of the binding site where the receptors were predicted to significantly differ at the entrance to the binding pocket.
Scheme 1. Antagonists of β2AR Used in Both Cocrystal Complexes and Comparative Docking Studies.

Scheme 2. Antagonists of β2AR (6 and 7) Identified by Virtual Screening and Agonists of β2AR (8) or β3AR (9) Designed Using the Available Crystal Structure Data.

7.2. Adenosine Receptors
The adenosine receptor family has been extensively studied over the years in search of both agonists to treat asthma and antagonists to treat Parkinson’s disease and cognitive disorders.67,123−130 Similar to the case for the β adrenergic receptors, the advent of a published crystal structure of the adenosine A2A receptor bound to the ligand ZM241385 has stirred considerable interest in the field. Michino et al. evaluated the value of GPCR structure prediction by initiating a community wide, blind prediction assessment of the ligand–receptor X-ray complex of ZM241385 to A2A-T4L.131 Twenty-nine groups participated, submitting 206 structural models before the release of the experimental coordinates for the crystal structure. The closest model had a ligand rmsd of 2.8 Å and a binding site rmsd of 3.4 Å. The results indicated that predictions, particularly of ligand–receptor binding mode and ECL conformations, remain challenging and additional insight from experimental data (such as SDM) on a receptor may be required to give good results. Carlsson et al.132 have used the adenosine A2A-T4L X-ray structure (3EML) to carry out a virtual screen of 1.4 million compounds and selected 20 for testing. Of these seven (35%) were found to be hits with affinities in the range 200 nM to 8.8 μM in a radioligand binding assay, of which 10 (Scheme 3) is an example. All of the hits were shown to be antagonists in a functional assay and most were selective versus the closely related adenosine A1 and A3 receptors. Analogues of the most potent hits were selected and tested, and a number of additional submicromolar hits were discovered. The binding modes of the hits were proposed, and all compounds appeared to form H-bonds with Asn253 (6.55) and Glu169 (ECL2) in a way related to the X-ray ligand ZM241385. Katritch et al. performed a virtual screen of adenosine A2A-T4L with 4.3 million compounds.67 Twenty-three of 56 experimentally tested molecules were active (41%), and affinities were in the range 32 nM to 10 μM, e.g., compound 11 (Scheme 3). All hits were again shown to be antagonists, but selectivity was relatively low against the adenosine A1 subtype in this case. Binding modes for representative hits were given and were generally consistent with those suggested by Carlsson et al. above. Serendipitously, one hit molecule was shown to be a very potent adenosine A1 receptor antagonist (Ki = 6 nM) with 20-fold selectivity versus the adenosine A2A receptor. Both of these studies of virtual screening versus the adenosine A2A receptor identified hits that were small polar molecules with respectable ligand efficiency values (LE).133 Ivanov et al. docked various known ligands into homology models of adenosine A2AR based on both bovine rhodopsin and β2AR-T4L.134 The model using β2AR-T4L as the template gave the best results. Problems with the bovine rhodopsin derived homology model were mainly related to the loop structure in the region of the binding site. Docking of the ligand ZM241385 could be further improved by adding crystallographically observed water molecules or by using constraints derived from SDM experiments. Docking of agonists (such as NECA) were also proposed and indicated that the ribose ring of the agonist interacted with Ser277 (7.42) and Thr88 (3.36), which have previously been identified by SDM studies as important for agonist but not antagonist affinity.68 An early example of the use of the A2AR-T4L protein–ligand structure being leveraged for drug design has been reported by Pastorin and co-workers.135 A new panel of triazolotriazine derivatives was designed from docking experiments with the program GOLD using the crystal structure coordinates and also homology models including the A3 receptor. The results allowed rationalization of the structure–activity relationships within the series as it developed and could account to some extent for selectivity between A2A and A3. A representative example is compound 12 (Scheme 3), which is a potent and selective adenosine A2A antagonist. A key observation was that the presence of a less bulky amino acid (Val169) in the ECL2 of the adenosine A3 receptor seemed to have a key influence over molecules containing larger substituents on the amino group of the heterocyclic core, modulating potency and selectivity for the A2A receptor versus the A3 subtype.
Scheme 3. Antagonists of the Adenosine A2A Receptor Identified by Virtual Screening or Designed Using the Recent Crystal Structure Data.

7.3. Dopamine Receptors
A comparison of the dopamine D3 receptor with dopamine D2 indicates that 17 of 18 primary contact residues are conserved in the dopamine D2 receptor subtype. This rationalizes the historical difficulty in designing subtype-selective ligands for the dopamine receptor.62,136,137 Homology modeling of the dopamine D2 receptor and docking of the dopamine D3-selective antagonist ligand R22 (13, Scheme 4) into the structure (D3) and the model (D2) have recently been performed.36 The results suggested that while the amine group of R22 binds to Asp110 (3.32) in the primary binding pocket as expected, the ligand might adopt an extended conformation that allows the indole-2-carboxamide to access an extended binding site formed by ECL1, ECL3, and residues at the top of TM1, TM2, and TM7, which are largely nonconserved between the two receptor subtypes.36 The interactions with this second binding site were proposed to be the drivers of selectivity for the dopamine D3 receptor. This bitopic mode of binding has been previously suggested for the muscarinic M2 receptor partial agonist McN-A-34335 and indeed may represent a general mechanism of action of many ligands that display high selectivity between closely related receptor subtypes (discussed earlier). An analysis of the homology model published by Ehrlich et al. based on the β2AR structure relative to the dopamine D3 receptor X-ray structure is discussed later in the section Discussion and Perspectives of this review (Figure 9).137
Figure 9.

(A) Comparison of a dopamine D3 receptor homology model based on the β2AR. (B) Homology model structure (green protein and green mesh carbon accessible surface from GRID calculated using the CH3 probe at 1 kcal/mol contour; piperazine ligand in green) and the X-ray structure of D3R (gray protein, X-ray ligand eticlopride and carbon accessible surface in transparent solid rendering). Two of the residues in the homology model that restrict the binding site are highlighted by being displayed in green colored stick.
7.4. CXCR1/2 Receptors
Allegretti et al. have developed a model of chemokine CXCR1 bound to (R)-ketoprofen 14 (Scheme 5), a potent noncompetitive inhibitor of CXCL8-induced human neutrophil chemotaxis.138 By use of these data, the binding site was proposed to be located in the TM bundle with ligands forming key interactions with Tyr46 (1.39), Lys99 (2.64), and Glu291 (7.35). The model was supported further by SDM and also ligand photoactivation studies. Compounds were then designed using the model, leading eventually to an agent that was selected as a clinical candidate for prevention of post ischemia–reperfusion injury. The derivative, called repertaxin 15, is an analogue of ibuprofen and is a rare example of where structure-based design methods have been reported to have aided in the identification of a GPCR drug candidate.
Scheme 5. Design of a Clinical Candidate for the CXCR1/2 Chemokine Receptor Driven by Homology Modeling and SDM Data.

7.5. Muscarinic M1 Receptor
The development of selective M1 muscarinic acetylcholine receptor (mAChR) agonists for cognitive disorders has long been a goal of the drug discovery community, but most attempts have resulted in compounds that display insufficient selectivity. The identification of M1 subtype selective agonists, AC-42 16 and 77-LH-28-117 (Scheme 6), that utilized an allosteric binding site on the receptor139,140 reignited interest in the field. Interestingly, the agonist activity of AC-42 was insensitive to mutation of Tyr381 (6.51) and Asn382 (6.52), which dramatically reduced the potency of carbachol.139 Recent efforts have utilized SDM and homology modeling of the M1 mAChR based on bovine rhodopsin and β2-AR to define the nature of this allosteric binding site.141 These studies suggested that the selective agonists still interact with the conserved Asp105 (3.32) to activate the receptor but make further interactions with residues at the extracellular end of TM2 and TM3, areas of the receptor that are less well-conserved across subtypes;141 again, it is this bitopic binding mode that likely confers the selectivity profile.
Scheme 6. Identification by Virtual Screening and Lead Optimization of a Selective Muscarinic M1 Receptor Agonist.

Using this information in combination with the crystal structure of rhodopsin allowed creation of a homology model of the M1 mAChR in which the important regions of receptor around the orthosteric binding site and the site at the extracellular end of TM domains 2, 3, and 7 were well-defined. This model of the receptor was then used for virtual screening to identify putatively selective M1 mAChR agonists; this approach yielded approximately 1000 hit compounds.142 Most notable was the benzimidazalone (18) (Scheme 6), which was weakly active at the M1 mAChR (EC50 = 400 nM) but was shown to be inactive at the M2–M5 mAChR subtypes. Optimization of the N-capping group and substitutions on the phenyl ring improved metabolic stability, ultimately leading to compound 19 (Scheme 6) which displayed increased M1 mAChR potency (EC50 = 8 nM), maintained subtype selectivity, and had low to moderate clearance in rat in vivo. Compound 19 was subsequently shown to be active in a rodent model of cognitive impairment and also by in vivo electrophysiology, suggesting that it would be suitable for further development.142 Another interesting development in this field has been the identification of BQCA 20 (and analogues), a subtype selective positive allosteric modulator that is thought to be able to engage with residues at the top of TM7 and in ECL2, binding concomitantly with acetylcholine to potentiate its agonist activity (Scheme 6).143
7.6. GPR109 Receptor
Boatman, Richman, and Semple recently reviewed the area of nicotinic acid receptor GPR109a and GPR109b research.108 They discussed the available mutagenesis and chimera studies that help define the binding site of nicotinic acid. Arg111 (3.36) in TM3 is considered the key polar interaction with the carboxylic acid of the ligand, and the pyridyl ring is suggested to sit in a box of aromatic residues on ECL1, ECL2, and TM7. Deng et al. have used a homology model to successfully optimize a series of anthranilic acid amides to derive potent GPR109a agonists.144 The model used bovine rhodopsin (IL9H) as the starting point, and the initial hit molecule 21 (Scheme 7) was proposed to bind to Arg111 (3.36) and Arg251 (6.55), again forming a salt bridge with the carboxylate of the ligand. Ser178 (ECL2) was proximal to the amide carbonyl of the hit and might also be involved in H-bonding. SDM studies were used to support the binding proposal and suggested that the amide of the ligand interacted with Ile254 (6.58), Phe255 (6.59), and Phe276 (7.35). This region was first probed with compound design, and a naphthyl group introduced into the ligand significantly improved agonist affinity. Next, targeting several polar groups at the mouth of the binding pocket with substituents on the naphthyl led to compound 22 (Scheme 7), which was a potent full agonist of GPR109a. This analogue also had a good ADME profile and in vivo efficacy.
Scheme 7. Identification of a GPR109a Agonist Optimized Using Homology Modeling of the Receptor.

7.7. Cannabinoid CB1 Receptors
The cannabinoid receptors are another important class of receptors that have received much attention in recent years.145−149 Tuccinardi et al. have constructed homology models of cannabinoid receptors CB1 and CB2 in the agonist conformation starting from bovine rhodopsin and using extensive SDM data to refine and improve confidence in the results.150 Docking of agonists was carried out, and selectivity for CB2 over CB1 was rationalized as being derived from Ser112 (3.31) and Phe197 (5.46) in CB2, which correspond to Gly195 (3.31) and Val282 (5.46) in CB1. Docking of a number of classes of known ligands seemed to support the hypothesis and to build on earlier models of these receptors.151 The binding modes and ligand receptor interactions of inverse agonists taranabant 23 and rimonabant 24 (Scheme 8) with the CB1 receptor have been studied by Lin et al.152 Ligand conformations were established by X-ray crystallography and NMR in solution. A homology model derived from bovine rhodopsin was then constructed, and new and historical SDM data were used to refine the ligand binding models. The key H-bonding interaction for taranabant was proposed as Ser383 (7.39), and the binding site for the two ligands appeared to largely overlap. However, the key polar interaction for rimonabant (as indicated from the SDM data) was Lys192 (3.28), so the ligands were proposed to differ in their key interactions in fine detail.
Scheme 8. Cannabinoid CB1 Receptor Antagonists Whose Binding Modes Have Been Rationalized by Homology Modeling and SDM Data.

7.8. Bradykinin B1 Receptor
Kuduk et al. have used a homology model of the bradykinin B1 receptor to design more potent analogues of a 2,3-diaminopyridine hit series.153,154 The homology model was derived from bovine rhodopsin and had previously been used in modeling of a series of dihydroquinoxalinones.155 The model was used to help design and rationalize the SAR of the 4-pyridylpiperazine analogue 26, derived from the unsubstituted compound 25 (Scheme 9); movement of the pyridyl nitrogen atom to the 2- or 3-position significantly reduced affinity. Mutagenesis of the site also supported the credibility of the model for SAR generation within this series of potent antagonists.
Scheme 9. Bradykinin B1 Receptor Antagonist Optimized Using a Homology Model of the Receptor.

7.9. 5HT Receptors
Sela and colleagues have outlined the drug design approach at EPIX Pharmaceuticals and described in silico screening of over 20 GPCRs.156 The focus on structure-based methods led to a good success rate in both screening and lead optimization for a range of targets. Four clinical candidates were identified in less than 4 years (PRX-08066, PRX-03140, PRX-07034, and PRX-00023) targeting the 5-HT receptor family.157 The philosophy of using rational approaches for hit and lead optimization throughout the drug discovery process suggests an advantage in using SBDD over empirical methods.
8. Discussion and Perspectives
SBDD using protein–ligand crystallography is now the established paradigm for enzyme targets, such as kinase, proteases, and phosphodiesterases, and is having a significant impact on the pipelines of pharmaceutical companies. Until very recently GPCR research has been limited to homology modeling supported by SDM studies and to empirical SAR used to develop ligand-based pharmacophores. The new era of protein–ligand X-ray crystallography for GPCR targets is quickly starting to open up the huge potential of SBDD to medicinal chemists for GPCR targets. Given the investment within the GPCR drug discovery community on SDM studies, there is also intriguing potential to couple X-ray crystallography results with extensive mutagenesis data for some receptors. For example, the adenosine receptor family has been exhaustively interrogated by SDM for many years and we are now in a position to rationalize these data with the crystal structures of both antagonist and agonist ligands. This level of understanding is probably unique in drug discovery, as in the enzyme drug discovery community SDM methods are rarely utilized, being seen as unnecessary where crystal structure data are available. However, a protein–ligand complex does not give any predictions of binding affinity in its own right, the value of individual interactions formed being a judgment of the viewer based on compound SAR and intuition. In our own laboratories we have developed a method called Biophysical Mapping in which a stabilized receptor (StaR) construct is studied by SDM in a biophysical screen using surface plasmon resonance (SPR).158 This direct binding approach does not require a radioligand or other labeling of the compounds being studied. This then allows panels of compounds to be screened and the contribution to their binding of individual residues to be rapidly enumerated. This method has been coupled with X-ray crystallography data for the adenosine A2A receptor and rapidly allowed optimization of a series of antagonists to yield a preclinical candidate targeted at the treatment of Parkinson’s disease (unpublished results). The use of this and other SDM approaches, linked with X-ray data, promises to revolutionize GPCR research.
The detailed understanding we now have of the β-adrenergic and adenosine A2A receptors, as well as structures of rhodopsin and opsin, greatly improves our ability to produce quality homology models of GPCRs for use in virtual screening and SBDD. Models created between 2000 and 2008 used bovine rhodopsin as the starting point for model generation and were limited by the lack of data on a system with a noncovalently bound ligand.159−161 Homology modeling can now be carried out with much more confidence to derive protein–ligand binding site information for GPCRs of interest, but a careful choice of which template to use as the basis of any new homology model must be made.57,162 There is a very large body of literature on the modeling of GPCRs, their binding sites, and protein–ligand interactions, and the area has been extensively reviewed.102,121,130,157,163 Virtual screening applications, leading to the identification of hit series, have been a focus of interest in the past decade, and with the advent of the new GPCR structures even more effort can be expected in this area.56,67,112,113,117,118,132,164
However, while homology models of GPCRs have been useful to generate hypotheses for SAR and selectivity and with mixed success for virtual screening, modeling of extracellular loops remains a critical issue. Often the model will also have an induced fit to the ligand(s) used during its generation. The recent availability of X-ray structures with noncovalently bound ligands such as the β1AR and β2AR structures in addition to rhodopsin structures improves our ability to generate good models, but we believe additional experimental information from SDM is key to giving higher confidence binding modes for ligands. This allows much more confidence in models for compound SAR and allows virtual screening constraints to be applied to enable the discovery of new ligands (e.g, for our own adenosine A2A project mentioned above). As an example of a model generated from the new βAR structures, in Figure 9 we compared a homology model of a ligand bound to the dopamine D3 receptor generated by Ehrlich et al. from the β2AR structure with the dopamine D3 receptor X-ray structure.137 The overall model is very close to the X-ray solution, as can be seen from the transmembrane backbone helices (rmsd of 1.4 Å using Maestro from Schrödinger). However, at a detailed level, there are some constrictions to the modeled binding site from particular residues that affect the docking of other ligands such as the X-ray ligand eticlopride. This is a general issue when using any homology model in which the backbone atoms may be slightly shifted and the amino acid side chain conformations are derived from a library or related structure with an optional induced fit to a particular ligand. Indeed, even in experimental structures a different ligand can induce changes within a given binding site. In Figure 9 the homology model is compared to the X-ray structure using GRID carbon accessible surfaces.50,51 It can be seen that part of the region occupied by the X-ray ligand eticlopride is occluded in the model (highlighted by displaying in stick are two of the side chain residues in the homology model causing this). An attempt to dock eticlopride into the homology model using GLIDE XP (Schrödinger) produced poses where the ligand had to shift away from the occluded region. This example shows how challenging it is to produce a general model for ligand docking, even when very similar to the X-ray structure, and how small differences between the model and the X-ray structure can perturb the results. This example reinforces the need to regularly challenge homology models using SDM and compound SAR to iteratively improve the working model being used and also highlights the value of actual experimental crystal structures over modeling.
Water molecules play a key role in the ligand binding, either as part of the binding process (interstitial) or most frequently by providing free energy gains upon their displacement. A thorough consideration of waters can be very important to improve docking and design predictions. Often the limited resolution of protein X-ray structures means that only some, or none, of the waters in the binding site are resolved, and even then their experimental basis (electron density) can be very weak. This is a problem for most current GPCR structures. A new approach called WaterMap from Schrödinger that uses molecular dynamics simulations165 provides a computational method to quantify water energies for apo or liganded structures in the binding site relative to bulk solvent. In Figure 10 the adenosine A2A receptor binding site (PDB code 3EML) with the ZM241385 ligand is shown with water positions calculated (with no ligand present), color-coded to show the most “unhappy” waters vs bulk solvent waters as red and then yellow (based on total free energy, summing the entropic and enthalpic components calculated). There are a large number (∼75%) of “unhappy” waters deep in the pocket, many unexploited by the ZM241385 ligand. This might then explain why hits with relatively high potency and ligand efficiency can be identified for this target. The GRID maps in the figure also highlight the liphophilic and water probe hotspots; some “unhappy” waters are in lipophilic hotspots, but predictions based only on this perform poorly because the energies do not take into account the whole network (a better correspondence has been noted in some enzyme binding sites, where more waters occur on the protein surface, but false positives and negatives still occur). The various GRID probes (lipophilic, H-bond acceptor and donor, etc.) also provide a key indication of what chemical groups would be complementary to the protein, working well in partnership with WaterMap in guiding design. WaterMap calculations can also be done with the ligand present and suggest regions to explore and any ligand destabilization of the remaining water network. These new computational tools, used to assist in optimization of homology models and throughout SBDD campaigns, offer great potential to maximize the utility of the newly available GPCR X-ray structural information.
Figure 10.

Adenosine A2A binding site from the 3EML structure with the ZM241385 ligand with waters calculated (with no ligand present) using the WaterMap program from Schrödinger shown.165 These are color coded to show the most “unhappy” vs bulk solvent as red (>3.5 kcal/mol), then yellow (2.2–3.5 kcal/mol), with gray intermediate (−1 to 2.2 kcal), and blue “happy” (<1 kcal/mol). GRID maps highlight the shape (Csp3 (C3) at 1 kcal/mol in light gray) of the lipophilic hotspots (aromatic C–H probe (C1=) in yellow at −2.5 kcal/mol) and the water probe hotspots (in green at −6.6 kcal/mol). Note the large number of “unhappy” waters deep in the binding site that are not exploited by ZM241385.
Figure 11 describes a cartoon model of our current understanding of the antagonist to agonist transition for family A GPCRs with small molecule natural ligands. Although preliminary, the recent progress with X-ray structures of systems with both agonists and antagonists bound, detailed earlier, allows some proposals to be made on a general mechanism of this crucial process. The antagonist GPCR conformation is relatively open and available for the diffusion of ligands into the TMD binding site. There is a large body of evidence that GPCR ligands bind to a common site on the extracellular face in the center of the seven helices, and a general feature is the presence of conserved hydrogen bonding or charged residues to interact with the natural ligand.166 For example, in aminergic GPCRs there is an acidic residue at position 3.32 always present to bind to the basic amine in the ligands, and in many receptors there is a hydrogen bonding residue at position 6.55; these important clusters of residues have recently been termed “chemoprints”.103,163 This region in the orthosteric site is labeled as the H-bonding subsite in Figure 11. Deeper in the pocket, the TMD site is often largely hydrophobic or aromatic in character, and this region has previously been recognized and called the “conserved” site.167 In Figure 11 this area is labeled as the lipophilic subsite. The two regions (H-bonding and lipophilic) of the orthosteric site of the CB1 receptor have previously been called aromatic and polar microdomains.145 At the entrance to the pocket, adjacent to ECL2 we propose that there will generally be an allosteric binding site where negative allosteric modulators or the extended part of bitopic ligands bind but small molecule orthosteric ligands do not interact. This has been previously called the “variable” region, as it was recognized as a potential site for larger ligands to derive selectivity for a specific receptor subtype.167 Also encompassed by this proposed allosteric site is a region recently called the “minor groove” suggested as a selectivity pocket and also to have a role in signaling and receptor activation, next to TM2, TM7, and ECL2.168 In family A peptide receptors the allosteric site in Figure 11 would actually be part of the “address” region for the peptide ligand while the area labeled the orthosteric site in Figure 11 would be the “message” region (discussed earlier).73 After initial binding of the natural agonist to this form of the receptor, conformational changes probably driven by formation of new polar interactions with the ligand and a closing-in around the binding site will occur to form the receptor–agonist complex. As described earlier, although the changes within the binding site on complexation of agonists are quite subtle, there seem to be key polar interactions formed to agonists that are not involved in antagonist binding. Indeed, antagonists also tend to be larger, holding open the receptor to prevent the conformational changes involved in agonist signaling. In Figure 11 we therefore propose the formation of a “signaling subsite” on transition to the receptor–agonist complex, which may itself be a precursor to the proposed rotation of the toggle switch and further changes to the so-called microswitches upon G protein binding on the intracellular surface.45 The changes in the allosteric site (now the site of engagement of PAMs) are uncertain, but this site must have an overall different shape such that PAMs stabilize the agonist conformation of the receptor. Rather than considering the conformation of the receptor binding to NAMs or PAMs as discrete forms in solution, our suggestion is that these modulators bind to the same conformations as antagonists or agonists, respectively, but are noncompetitive with respect to the orthosteric binding site, i.e., that there is a simple two-state model operating. This is consistent with our observations upon conformational thermostabilization of multiple receptors to form StaRs, where either the agonist or antagonist state appears to be trapped by a wide range of ligands, with no evidence for trapping of any other conformations during these studies. Finally, agonists appear to engage in the H-bonding subsite in a manner similar to that of antagonists, and this suggests that agonists can bind to both conformations of the receptor and act to pull the equilibrium over to the active state. Antagonists and inverse agonists, however, in the model proposed here, will only be capable of binding to the antagonist state, as they tend to be larger than agonists and sterically blocked from binding to the agonist state.
Figure 11.

Simplified description of family A GPCR binding sites in the antagonist and agonist conformational states. Antagonists bind to both H-bonding and lipophilic subsites in a more open form of the orthosteric pocket of the receptor. NAMs are proposed to occupy an allosteric pocket at the entrance to the receptor adjacent to ECL2. Agonists trigger a change in receptor conformation on binding to the antagonist state in which the volume of the binding site decreases and new polar interactions are formed deep inside the pocket close to the toggle switch W6.48. PAMs are proposed to stabilize the agonist state by binding to the allosteric pocket in an alternative conformation.
The analysis above suggests that subtype selectivity might best be derived by accessing an allosteric site of either antagonist or agonist receptor conformations and that targeting the orthosteric site may not be profitable. However, if we compare this situation to the kinase enzyme family, we can draw some parallels. In kinases close to the hinge that binds the adenine of ATP, many kinases are similar and within kinase subtypes this part of the active site is very similar indeed. However, it has been shown many times that small differences in this region between close family members can be used to derive selectivity without necessarily requiring large extended ligands that access more variable regions outside this region of the binding site.169 Figure 12 shows the binding sites of β1AR and β2AR overlaid.58,60 Very close to the ligands there are two residue changes between β1 and β2: Phe325/Tyr308 (7.35) and Val172/Thr164 (4.56). The Tyr308 (7.35) in β2AR forms a hydrogen bond to Asn293 (6.55), which is missing in β1AR where the Phe325 (7.35) cannot make this interaction. This has the effect of changing the positions of the aromatic rings of Phe325 (7.35) and Tyr308 (7.35) relative to each other and also of the nearby Asn310/293 (6.55) between the two systems. In β1AR the Asn310 (6.55) is capable of forming a weak hydrogen bond to the ligand cyanopindolol, while in β2AR this residue is not available to bind to the ligand in the same way (Figure 12). Overall, the surface of the ligand-binding pocket made by these residue pairs has a subtly different shape and polarity, which might be expected to allow for the design of selective ligands. In our own research on the structure based optimization of a series of adenosine A2A antagonists, selectivity was achieved over other adenosine receptor subtypes “inside” the orthosteric site, taking advantage of similar small differences between the subtypes and without accessing the allosteric region at the entrance to the binding site (unpublished results).
Figure 12.

Potential for selectivity in β-adrenergic receptors: superimposition of crystal structures of cyanopindolol in β1AR (yellow) and carazolol in β2AR (blue) adrenoceptors. The surface of the pocket in β1AR slightly differs from the shape of the pocket in β2AR because of the lack of the phenol hydroxyl group in Phe325 (7.35) (β1) vs Tyr308 (7.35) (β2). This single atom change also affects the H-bonding of the adjacent Asn residue (6.55) in the binding site.
With the advent of SBDD for GPCRs, fragment based drug discovery (FBDD) becomes an attractive opportunity for the GPCR drug discovery community.170−172 Fragments are simply low molecular weight organic molecules, usually defined as 100–250 Da in size, and as such are much smaller than hits typically identified by other methods such as HTS. As we have seen, the GPCR binding sites for family A receptors are large and generally hydrophobic but contain key H-bonding polar residues for both binding and signaling. GPCR systems should be highly suitable for FBDD approaches, and fragments with high ligand efficiency (LE) may be detectable.133 Often, however, fragments bind promiscuously and might bind to multiple receptors, potentially making triaging of hits (often done by evaluating selectivity) more challenging. Albert et al. have described their experiences of FBDD and gave high concentration screening (HCS) results for some GPCR targets, indicating that respectable hit rates can be achieved.173 Unfortunately, false positives could not be readily identified, as there were no orthogonal biophysical assays in which to evaluate the hits and no access to X-ray cocomplexes available at the time. In one example, the melanocortin 4 receptor, 60 confirmed hits were identified from a library of 660 fragments screened at 1 mM. Near-neighbor screening of commercial analogues produced hits with more conventional potency (10–50 μM) but at the expense of a significant reduction in LE, suggesting that the larger and more active analogues were not binding optimally in the receptor. In another more recent example, a ligand-based NMR method has been successfully applied to GPR40 to identify hits, and this method has the potential for use in FBDD.174 In the authors’ own research, we have used StaRs to several GPCRs in both families A and B as reagents for fragment based screening using high-concentration screens, surface plasmon resonance (Biacore), and target-immobilized NMR (TINS NMR) based screening.175 SPR screening of an A2A-StaR and TINS NMR screening of a β1AR StaR both identified hits that could be validated by showing activity in orthogonal assays. Both of these biophysical methods depend on immobilizing the stabilized receptor on a surface and then flowing a solution of fragments in buffer through the instrument. Fragment binding is then detected by either an increase in mass bound to the chip (response units) or a change in the NMR signal of the fragment. For example, simple xanthines were readily identified as binders to the A2A-StaR and a medium resolution crystal structure for caffeine was subsequently solved in the A2A-StaR X-ray system.66 These findings using StaR proteins for FBDD methodologies are promising, suggesting that leveraging of the range of biophysical tools currently available for soluble proteins can now be contemplated for GPCR targets.
In conclusion, the field of GPCR drug discovery is set to have a new lease of life, invigorated by a quantum leap of progress in structural biology over the past 3 years. The new era of SBDD for GPCRs should now allow even some of the most challenging targets to yield to the efforts of medicinal chemists. This very well established drug family can expect to see many more first in class agents and better molecules with improved physicochemical properties and selectivity profiles, derived from the careful, rational optimization of hits using structure based and fragment based methods. In particular, a disciplined emphasis on maintaining high ligand efficiency and ligand lipophilicity efficiency should allow the next generation of research on this target family to profit from improved clinical success rates and lead to safe and efficacious medicines over the next decade.5,133 These are exciting times.
Acknowledgments
The authors thank Andrea Bortolato and Ben Tehan for help producing the figures and Frank Blaney and Malcolm Weir for helpful discussions.
Abbreviations Used
- GPCR
G-protein-coupled receptor
- SBDD
structure-based drug design
- FBDD
fragment-based drug discovery
- LE
ligand efficiency
- MWT
molecular weight
- PDB
Protein Data Bank
- TMD
seven-transmembrane spanning domain
- ECL
extracellular loop
- ECD
extracellular domain
- ICL
intracellular loop
- PAM
positive allosteric modulator
- NAM
negative allosteric modulator
- NBE
new biological entity
- NCE
new chemical entity
- GABA
γ-aminobutyric acid
- GLP 1
glucagon like peptide 1
- CRF 1
corticotrophin releasing factor 1
- LCP
lipidic cubic phase
- StaR
stabilized receptor
- βAR
β adrenergic receptor
- T4L
T4-fusion lysozyme
- SDM
site-directed mutagenesis
- HTS
high-throughput screening
- CGRP
calcitonin gene-related peptide
- CLR
calcitonin-receptor-like receptor
- mGlu
metabotropic glutamate
- TINS-NMR
target-immobilized NMR
- NECA
N-ethylcarboxyadenosine
- RAMP1
receptor activity modifying protein 1
- SDF-1
stromal-derived factor 1.
Biographies
Miles Congreve is Head of Chemistry at Heptares Therapeutics Ltd. in Welwyn Garden City, U.K., where he is responsible for the company’s medicinal chemistry projects targeting GPCRs using SBDD approaches. He was previously Director of Chemistry at Astex Therapeutics in Cambridge, U.K., working in the field of fragment based drug discovery (2001–2008). Before joining Astex, he held various positions at GlaxoSmithKline (1993–2001), working on a broad range of medicinal chemistry and hits to leads projects at Ware and then Stevenage before moving to Cambridge in 1999 to run the GlaxoWellcome chemistry research facility embedded in the Department of Chemistry at University of Cambridge, U.K. His Ph.D. studies were also carried out at the University of Cambridge (1990–1993), supervised by Professor Andrew Holmes.
Christopher J. Langmead is Head of Pharmacology at Heptares Therapeutics Ltd. He is responsible for directing the in vitro and in vivo pharmacology and ADMET studies for Heptares’ structure-based GPCR drug discovery projects. He previously led teams involved with biochemical pharmacology in psychiatry research at GlaxoSmithKline (1998–2009), working at all stages of preclinical drug discovery. He received his B.A. in Pharmacology from the University of Cambridge, U.K., and a Ph.D. from University College London in molecular pharmacology of G-protein-coupled receptors.
Jonathan S. Mason is Head of Computational Chemistry at Heptares Therapeutics Ltd. and a consultant Chief Scientist (Predictive Technologies & Drug Design, Discovery Chemistry & DMPK) at Lundbeck Research, Copenhagen, Denmark, with responsibilities that include SBDD projects and fragment-based drug discovery approaches. He has previously led teams involved with computational chemistry, structural biology, medicinal informatics, and knowledge discovery at Pfizer in the U.K. (Executive Director MISD) and Bristol-Myers Squibb in the U.S. (Director SB&M), following many years at Rhône-Poulenc Rorer (now Sanofi-Aventis) in the U.K., France, and U.S. building and leading computer-assisted drug design teams following 5 years as a medicinal chemist. His Ph.D. studies were carried out at Queen Mary, University of London (1976–1979), supervised by Dr. Martin F. Ansell.
Fiona H. Marshall is a founder and Chief Scientific Officer at Heptares Therapeutics Ltd. Fiona has over 20 years’ experience working on G-protein-coupled receptor drug discovery. She spent 12 years at GlaxoSmithKline where she was Head of Molecular Pharmacology. Her group was responsible for the discovery of the GABAB receptor heterodimer, the identification of RAMPs, the cloning of the CGRP and adrenomedullin receptors, the identification of the nicotinic acid receptor, and the deorphanization of GPR41 and GPR43. She was Director of Discovery Pharmacology at Europe for Millennium Pharmaceuticals and then spent several years as an independent consultant. Fiona has a B.Sc. in Biochemistry from the University of Bath, U.K., and a Ph.D. in Neuroscience from University of Cambridge, U.K.
References
- Bikker J. A.; Trumpp-Kallmeyer S.; Humblet C. G-Protein coupled receptors: models, mutagenesis, and drug design. J. Med. Chem. 1998, 41 (16), 2911–2927. [DOI] [PubMed] [Google Scholar]
- Henderson R.; Schertler G. F. The structure of bacteriorhodopsin and its relevance to the visual opsins and other seven-helix G-protein coupled receptors. Philos. Trans. R. Soc. London, Ser. B 1990, 326 (1236), 379–389. [DOI] [PubMed] [Google Scholar]
- Schertler G. F.; Villa C.; Henderson R. Projection structure of rhodopsin. Nature 1993, 362 (6422), 770–772. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46 (1–3), 3–26. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6 (11), 881–890. [DOI] [PubMed] [Google Scholar]
- Empfield J. R.; Leeson P. D. Lessons learned from candidate drug attrition. IDrugs 2010, 13 (12), 869–873. [PubMed] [Google Scholar]
- Lagerstrom M. C.; Schioth H. B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discovery 2008, 7 (4), 339–357. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. The druggable genome. Nat. Rev. Drug Discovery 2002, 1 (9), 727–730. [DOI] [PubMed] [Google Scholar]
- Briones M.; Bajaj M. Exenatide: a GLP-1 receptor agonist as novel therapy for type 2 diabetes mellitus. Expert Opin. Pharmacother. 2006, 7 (8), 1055–1064. [DOI] [PubMed] [Google Scholar]
- Lewiecki E. M. Emerging drugs for postmenopausal osteoporosis. Expert Opin. Emerging Drugs 2009, 14 (1), 129–144. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Subramanian G.; Hall S.; Lin J.; Laoui A.; Vaidehi N. Allosteric antagonist binding sites in class B GPCRs: corticotropin receptor 1. J. Comput.-Aided Mol. Des. 2010, 24 (8), 659–674. [DOI] [PubMed] [Google Scholar]
- Tellew J. E.; Lanier M.; Moorjani M.; Lin E.; Luo Z.; Slee D. H.; Zhang X.; Hoare S. R.; Grigoriadis D. E. St; Denis Y.; Di Fabio R.; Di Modugno E.; Saunders J.; Williams J. P. Discovery of NBI-77860/GSK561679, a potent corticotropin-releasing factor (CRF1) receptor antagonist with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2010, 20 (24), 7259–7264. [DOI] [PubMed] [Google Scholar]
- Yona S.; Lin H. H.; Siu W. O.; Gordon S.; Stacey M. Adhesion-GPCRs: emerging roles for novel receptors. Trends Biochem. Sci. 2008, 33 (10), 491–500. [DOI] [PubMed] [Google Scholar]
- Pin J. P.; Galvez T.; Prezeau L. Evolution, structure, and activation mechanism of family 3/C G-protein-coupled receptors. Pharmacol. Ther. 2003, 98 (3), 325–354. [DOI] [PubMed] [Google Scholar]
- McLean B. N. Intrathecal baclofen in severe spasticity. Br. J. Hosp. Med. 1993, 49 (4), 262–267. [PubMed] [Google Scholar]
- Iqbal J.; Zaidi M.; Schneider A. E. Cinacalcet hydrochloride (Amgen). IDrugs 2003, 6 (6), 587–592. [PubMed] [Google Scholar]
- Kenakin T. Principles: receptor theory in pharmacology. Trends Pharmacol. Sci. 2004, 25 (4), 186–192. [DOI] [PubMed] [Google Scholar]
- Park P. S.; Lodowski D. T.; Palczewski K. Activation of G protein-coupled receptors: beyond two-state models and tertiary conformational changes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 107–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhalfioui F.; Magnin T.; Wagner R. From purified GPCRs to drug discovery: the promise of protein-based methodologies. Curr. Opin. Pharmacol. 2009, 9 (5), 629–635. [DOI] [PubMed] [Google Scholar]
- Keov P.; Sexton P. M.; Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 2011, 60 (1), 24–35. [DOI] [PubMed] [Google Scholar]
- La Motta C.; Sartini S.; Morelli M.; Taliani S.; Da Settimo F. Allosteric modulators for adenosine receptors: an alternative to the orthosteric ligands. Curr. Top. Med. Chem. 2010, 10 (10), 976–992. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Jones C. K.; Lindsley C. W. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci. 2009, 30 (3), 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetz J. A. Allosteric modulation of dopamine receptors. Mini-Rev. Med. Chem. 2005, 5 (6), 555–561. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Christopoulos A.; Lindsley C. W. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery 2009, 8 (1), 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E. F.; Heaton W. H.; Miller M.; Fox J.; Balandrin M. F.; Van Wagenen B. C.; Colloton M.; Karbon W.; Scherrer J.; Shatzen E.; Rishton G.; Scully S.; Qi M.; Harris R.; Lacey D.; Martin D. Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J. Pharmacol. Exp. Ther. 2004, 308 (2), 627–635. [DOI] [PubMed] [Google Scholar]
- Urwyler S. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol. Rev. 2011, 63 (1), 59–126. [DOI] [PubMed] [Google Scholar]
- Langmead C. J.; Christopoulos A. Allosteric agonists of 7TM receptors: expanding the pharmacological toolbox. Trends Pharmacol. Sci. 2006, 27 (9), 475–481. [DOI] [PubMed] [Google Scholar]
- May L. T.; Leach K.; Sexton P. M.; Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 1–51. [DOI] [PubMed] [Google Scholar]
- Lazareno S.; Popham A.; Birdsall N. J. Allosteric interactions of staurosporine and other indolocarbazoles with N-[methyl-(3)H]scopolamine and acetylcholine at muscarinic receptor subtypes: identification of a second allosteric site. Mol. Pharmacol. 2000, 58 (1), 194–207. [DOI] [PubMed] [Google Scholar]
- Leach K.; Loiacono R. E.; Felder C. C.; McKinzie D. L.; Mogg A.; Shaw D. B.; Sexton P. M.; Christopoulos A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 2010, 35 (4), 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A.; Ruegg D.; Litschig S.; Stoehr N.; Stierlin C.; Heinrich M.; Floersheim P.; Prezeau L.; Carroll F.; Pin J. P.; Cambria A.; Vranesic I.; Flor P. J.; Gasparini F.; Kuhn R. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J. Biol. Chem. 2000, 275 (43), 33750–33758. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H.; Rowe B. A.; Morales S.; Chavez-Noriega L. E.; Yin R.; Jachec C.; Rao S. P.; Bain G.; Pinkerton A. B.; Vernier J. M.; Bristow L. J.; Varney M. A.; Daggett L. P. Pharmacological characterization and identification of amino acids involved in the positive modulation of metabotropic glutamate receptor subtype 2. Mol. Pharmacol. 2003, 64 (4), 798–810. [DOI] [PubMed] [Google Scholar]
- Binet V.; Brajon C.; Le Corre L.; Acher F.; Pin J. P.; Prezeau L. The heptahelical domain of GABA(B2) is activated directly by CGP7930, a positive allosteric modulator of the GABA(B) receptor. J. Biol. Chem. 2004, 279 (28), 29085–29091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. J.; Tomkinson N. P.; Wiley K. E.; Brammall A.; Bowers L.; Grahames C.; Gaw A.; Meghani P.; Shelton P.; Wright T. J.; Mallinder P. R. Identification of a putative intracellular allosteric antagonist binding-site in the CXC chemokine receptors 1 and 2. Mol. Pharmacol. 2008, 74 (5), 1193–1202. [DOI] [PubMed] [Google Scholar]
- Valant C.; Gregory K. J.; Hall N. E.; Scammells P. J.; Lew M. J.; Sexton P. M.; Christopoulos A. A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J. Biol. Chem. 2008, 283 (43), 29312–29321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. Structure of the human dopamine d3 receptor in complex with a d2/d3 selective antagonist. Science 2010, 330 (6007), 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. A.; Stevens R. C. Discovery of new GPCR biology: one receptor structure at a time. Structure 2009, 17 (1), 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Rasmussen S. G.; Kobilka B. K. The structure and function of G-protein-coupled receptors. Nature 2009, 459 (7245), 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B.; Schertler G. F. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol. Sci. 2008, 29 (2), 79–83. [DOI] [PubMed] [Google Scholar]
- Sabio M.; Topiol S. W.. X-ray Structure Developments for GPCR Drug Targets. In GPCR Molecular Pharmacology and Drug Targeting: Shifting Paradigms and New Directions; Gilchrist A., Ed.; John Wiley and Sons: Hoboken, NJ, 2010; pp 434–459. [Google Scholar]
- Ballesteros J. A.; Weinstein H.; Stuart C. S.. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure–Function Relations in G Protein-Coupled Receptors. In Methods in Neurosciences; Academic Press: San Diego, CA, 1995; Vol. 25, pp 366–428. [Google Scholar]
- Bridges T. M.; Lindsley C. W. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem. Biol. 2008, 3 (9), 530–541. [DOI] [PubMed] [Google Scholar]
- Valant C.; Sexton P. M.; Christopoulos A. Orthosteric/allosteric bitopic ligands: going hybrid at GPCRs. Mol. Interventions 2009, 9 (3), 125–135. [DOI] [PubMed] [Google Scholar]
- Holst B.; Nygaard R.; Valentin-Hansen L.; Bach A.; Engelstoft M. S.; Petersen P. S.; Frimurer T. M.; Schwartz T. W. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 2010, 285 (6), 3973–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R.; Frimurer T. M.; Holst B.; Rosenkilde M. M.; Schwartz T. W. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 2009, 30 (5), 249–259. [DOI] [PubMed] [Google Scholar]
- Standfuss J.; Edwards P. C.; D’Antona A.; Fransen M.; Xie G.; Oprion D.; Schertler G. F. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 2011, 471 (7340), 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R.; Mahalingam M.; Ludeke S.; Huber T.; Siebert F.; Sakmar T. P. Functional role of the “ionic lock”—an interhelical hydrogen-bond network in family A heptahelical receptors. J. Mol. Biol. 2008, 380 (4), 648–655. [DOI] [PubMed] [Google Scholar]
- Hofmann K. P.; Scheerer P.; Hildebrand P. W.; Choe H. W.; Park J. H.; Heck M.; Ernst O. P. A G protein-coupled receptor at work: the rhodopsin model. Trends Biochem. Sci. 2009, 34 (11), 540–552. [DOI] [PubMed] [Google Scholar]
- Scheerer P.; Park J. H.; Hildebrand P. W.; Kim Y. J.; Krauss N.; Choe H. W.; Hofmann K. P.; Ernst O. P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455 (7212), 497–502. [DOI] [PubMed] [Google Scholar]
- Goodford P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28 (7), 849–857. [DOI] [PubMed] [Google Scholar]
- Sciabola S.; Stanton R. V.; Mills J. E.; Flocco M. M.; Baroni M.; Cruciani G.; Perruccio F.; Mason J. S. High-throughput virtual screening of proteins using GRID molecular interaction fields. J. Chem. Inf. Model. 2010, 50 (1), 155–169. [DOI] [PubMed] [Google Scholar]
- Palczewski K.; Kumasaka T.; Hori T.; Behnke C. A.; Motoshima H.; Fox B. A.; Le Trong I.; Teller D. C.; Okada T.; Stenkamp R. E.; Yamamoto M.; Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science 2000, 289 (5480), 739–745. [DOI] [PubMed] [Google Scholar]
- Patny A.; Desai P. V.; Avery M. A. Homology modeling of G-protein-coupled receptors and implications in drug design. Curr. Med. Chem. 2006, 13 (14), 1667–1691. [DOI] [PubMed] [Google Scholar]
- Barton N.; Blaney F. E.; Garland S.; Tehan B.; Wall I. Seven transmembrane G protein-coupled receptors: insights for drug design from structure and modeling. Compr. Med. Chem. II 2007, 4, 669–701. [Google Scholar]
- Garland S.; Blaney F.. GPCR Homology Model Development and Application. In Burger’s Medicinal Chemistry, Drug Discovery and Development, 7th ed.; Abraham D. J., Rotella D. P.; John Wiley and Sons: Hoboken, NJ, 2010; pp 279–300. [Google Scholar]
- Sabio M.; Jones K.; Topiol S. Use of the X-ray structure of the beta2-adrenergic receptor for drug discovery. Part 2: Identification of active compounds. Bioorg. Med. Chem. Lett. 2008, 18 (20), 5391–5395. [DOI] [PubMed] [Google Scholar]
- Mobarec J. C.; Sanchez R.; Filizola M. Modern homology modeling of G-protein coupled receptors: which structural template to use?. J. Med. Chem. 2009, 52 (16), 5207–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T.; Serrano-Vega M. J.; Baker J. G.; Moukhametzianov R.; Edwards P. C.; Henderson R.; Leslie A. G.; Tate C. G.; Schertler G. F. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 2008, 454 (7203), 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; Choi H. J.; Rosenbaum D. M.; Kobilka T. S.; Thian F. S.; Edwards P. C.; Burghammer M.; Ratnala V. R.; Sanishvili R.; Fischetti R. F.; Schertler G. F.; Weis W. I.; Kobilka B. K. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 2007, 450 (7168), 383–387. [DOI] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G.; Thian F. S.; Kobilka T. S.; Choi H. J.; Kuhn P.; Weis W. I.; Kobilka B. K.; Stevens R. C. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318 (5854), 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson N.; Jazayeri A.; Errey J.; Baig A.; Hurrell E.; Zhukov A.; Langmead C. J.; Weir M.; Marshall F. H. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology 2011, 60 (1), 36–44. [DOI] [PubMed] [Google Scholar]
- Lopez L.; Selent J.; Ortega R.; Masaguer C. F.; Dominguez E.; Areias F.; Brea J.; Loza M. I.; Sanz F.; Pastor M. Synthesis, 3D-QSAR, and structural modeling of benzolactam derivatives with binding affinity for the D(2) and D(3) receptors. ChemMedChem 2010, 5 (8), 1300–1317. [DOI] [PubMed] [Google Scholar]
- Pooput C.; Rosemond E.; Karpiak J.; Deflorian F.; Vilar S.; Costanzi S.; Wess J.; Kirk K. L. Structural basis of the selectivity of the beta(2)-adrenergic receptor for fluorinated catecholamines. Bioorg. Med. Chem. 2009, 17 (23), 7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa H.; Isogaya M.; Nagao T.; Kurose H. The role of the seventh transmembrane region in high affinity binding of a beta 2-selective agonist TA-2005. Mol. Pharmacol. 1998, 53 (1), 128–134. [DOI] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y.; Lane J. R.; Ijzerman A. P.; Stevens R. C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322 (5905), 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré A. S.; Robertson N.; Errey J. C.; Ng I.; Hollenstein K.; Tehan B.; Hurrell E.; Bennet K.; Congreve M.; Magnani F.; Tate C. G.; Weir M.; Marshall F. H.. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Rueda M.; Lam P. C.; Yeager M.; Abagyan R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins 2010, 78 (1), 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. K.; Gao Z. G.; Van Rompaey P.; Gross A. S.; Chen A.; Van Calenbergh S.; Jacobson K. A. Modeling the adenosine receptors: comparison of the binding domains of A2A agonists and antagonists. J. Med. Chem. 2003, 46 (23), 4847–4859. [DOI] [PubMed] [Google Scholar]
- Dal Ben D.; Lambertucci C.; Marucci G.; Volpini R.; Cristalli G. Adenosine receptor modeling: What does the A2A crystal structure tell us?. Curr. Top. Med. Chem. 2010, 10, 993–1018. [DOI] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; Hamel D. J.; Kuhn P.; Handel T. M.; Cherezov V.; Stevens R. C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330 (6007), 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salchow K.; Bond M. E.; Evans S. C.; Press N. J.; Charlton S. J.; Hunt P. A.; Bradley M. E. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br. J. Pharmacol. 2010, 159 (7), 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews G.; Jones C.; Wreggett K. A. An intracellular allosteric site for a specific class of antagonists of the CC chemokine G protein-coupled receptors CCR4 and CCR5. Mol. Pharmacol. 2008, 73 (3), 855–867. [DOI] [PubMed] [Google Scholar]
- Portoghese P. S. Bivalent ligands and the message–address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci. 1989, 10 (6), 230–235. [DOI] [PubMed] [Google Scholar]
- Werge T. M. Identification of an epitope in the substance P receptor important for recognition of the common carboxyl-terminal tachykinin sequence. J. Biol. Chem. 1994, 269 (35), 22054–22058. [PubMed] [Google Scholar]
- Veldkamp C. T.; Seibert C.; Peterson F. C.; De la Cruz N. B.; Haugner J. C. 3rd; Basnet H.; Sakmar T. P.; Volkman B. F. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci. Signaling 2008, 1 (37), ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Lewis I.; Kim K. S.; Rajarathnam K.; Gong J. H.; Dewald B.; Moser B.; Baggiolini M.; Sykes B. D. Structure–activity relationships of chemokines. J. Leukocyte Biol. 1995, 57 (5), 703–711. [DOI] [PubMed] [Google Scholar]
- Smith S. O. Structure and activation of the visual pigment rhodopsin. Annu. Rev. Biophys. 2010, 39, 309–328. [DOI] [PubMed] [Google Scholar]
- Park J. H.; Scheerer P.; Hofmann K. P.; Choe H. W.; Ernst O. P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 2008, 454 (7201), 183–187. [DOI] [PubMed] [Google Scholar]
- Weis W. I.; Kobilka B. K. Structural insights into G-protein-coupled receptor activation. Curr. Opin. Struct. Biol. 2008, 18 (6), 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; Choi H. J.; Fung J. J.; Pardon E.; Casarosa P.; Chae P. S.; Devree B. T.; Rosenbaum D. M.; Thian F. S.; Kobilka T. S.; Schnapp A.; Konetzki I.; Sunahara R. K.; Gellman S. H.; Pautsch A.; Steyaert J.; Weis W. I.; Kobilka B. K. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469 (7329), 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Zhang C.; Lyons J. A.; Holl R.; Aragao D.; Arlow D. H.; Rasmussen S. G.; Choi H. J.; Devree B. T.; Sunahara R. K.; Chae P. S.; Gellman S. H.; Dror R. O.; Shaw D. E.; Weis W. I.; Caffrey M.; Gmeiner P.; Kobilka B. K. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 2011, 469 (7329), 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T.; Moukhametzianov R.; Baker J. G.; Nehme R.; Edwards P. C.; Leslie A. G.; Schertler G. F.; Tate C. G. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 2011, 469 (7329), 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Wu H.; Katritch V.; Han G. W.; Jacobson K. A.; Gao Z. G.; Cherezov V.; Stevens R. C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332 (6027), 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T. Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G.; Tate C.. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature [Online early access] DOI: 10.1038/nature10136. Published Online: May 18, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C.; Reedtz-Runge S.; Rudolph R.; Stubbs M. T. Passing the baton in class B GPCRs: peptide hormone activation via helix induction?. Trends Biochem. Sci. 2009, 34 (6), 303–310. [DOI] [PubMed] [Google Scholar]
- Hansen K. B.; Knop F. K.; Holst J. J.; Vilsboll T. Treatment of type 2 diabetes with glucagon-like peptide-1 receptor agonists. Int. J. Clin. Pract. 2009, 63 (8), 1154–1160. [DOI] [PubMed] [Google Scholar]
- Pioszak A. A.; Parker N. R.; Suino-Powell K.; Xu H. E. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J. Biol. Chem. 2008, 283 (47), 32900–32912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak A. A.; Xu H. E. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 2008, 105 (13), 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace C. R.; Perrin M. H.; DiGruccio M. R.; Miller C. L.; Rivier J. E.; Vale W. W.; Riek R. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 2004, 101 (35), 12836–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foord S. M.; Marshall F. H. RAMPs: accessory proteins for seven transmembrane domain receptors. Trends Pharmacol. Sci. 1999, 20 (5), 184–187. [DOI] [PubMed] [Google Scholar]
- Ho T. W.; Edvinsson L.; Goadsby P. J. CGRP and its receptors provide new insights into migraine pathophysiology. Nat. Rev. Neurol. 2010, 6 (10), 573–582. [DOI] [PubMed] [Google Scholar]
- ter Haar E.; Koth C. M.; Abdul-Manan N.; Swenson L.; Coll J. T.; Lippke J. A.; Lepre C. A.; Garcia-Guzman M.; Moore J. M. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure 2010, 18 (9), 1083–1093. [DOI] [PubMed] [Google Scholar]
- Lisman J. E.; Coyle J. T.; Green R. W.; Javitt D. C.; Benes F. M.; Heckers S.; Grace A. A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31 (5), 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabet I.; Parmentier M. L.; De Colle C.; Bockaert J.; Acher F.; Pin J. P. Comparative effect of L-CCG-I, DCG-IV and gamma-carboxy-l-glutamate on all cloned metabotropic glutamate receptor subtypes. Neuropharmacology 1998, 37 (8), 1043–1051. [DOI] [PubMed] [Google Scholar]
- Ishida M.; Saitoh T.; Shimamoto K.; Ohfune Y.; Shinozaki H. A novel metabotropic glutamate receptor agonist: marked depression of monosynaptic excitation in the newborn rat isolated spinal cord. Br. J. Pharmacol. 1993, 109 (4), 1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monn J. A.; Valli M. J.; Massey S. M.; Wright R. A.; Salhoff C. R.; Johnson B. G.; Howe T.; Alt C. A.; Rhodes G. A.; Robey R. L.; Griffey K. R.; Tizzano J. P.; Kallman M. J.; Helton D. R.; Schoepp D. D. Design, synthesis, and pharmacological characterization of (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740): a potent, selective, and orally active group 2 metabotropic glutamate receptor agonist possessing anticonvulsant and anxiolytic properties. J. Med. Chem. 1997, 40 (4), 528–537. [DOI] [PubMed] [Google Scholar]
- Monn J. A.; Valli M. J.; Massey S. M.; Hansen M. M.; Kress T. J.; Wepsiec J. P.; Harkness A. R.; Grutsch J. L. Jr.; Wright R. A.; Johnson B. G.; Andis S. L.; Kingston A.; Tomlinson R.; Lewis R.; Griffey K. R.; Tizzano J. P.; Schoepp D. D. Synthesis, pharmacological characterization, and molecular modeling of heterobicyclic amino acids related to (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740): identification of two new potent, selective, and systemically active agonists for group II metabotropic glutamate receptors. J. Med. Chem. 1999, 42 (6), 1027–1040. [DOI] [PubMed] [Google Scholar]
- Monn J. A.; Massey S. M.; Valli M. J.; Henry S. S.; Stephenson G. A.; Bures M.; Herin M.; Catlow J.; Giera D.; Wright R. A.; Johnson B. G.; Andis S. L.; Kingston A.; Schoepp D. D. Synthesis and metabotropic glutamate receptor activity of S-oxidized variants of (−)-4-amino-2-thiabicyclo-[3.1.0]hexane-4,6-dicarboxylate: identification of potent, selective, and orally bioavailable agonists for mGlu2/3 receptors. J. Med. Chem. 2007, 50 (2), 233–240. [DOI] [PubMed] [Google Scholar]
- Patil S. T.; Zhang L.; Martenyi F.; Lowe S. L.; Jackson K. A.; Andreev B. V.; Avedisova A. S.; Bardenstein L. M.; Gurovich I. Y.; Morozova M. A.; Mosolov S. N.; Neznanov N. G.; Reznik A. M.; Smulevich A. B.; Tochilov V. A.; Johnson B. G.; Monn J. A.; Schoepp D. D. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat. Med. 2007, 13 (9), 1102–1107. [DOI] [PubMed] [Google Scholar]
- Kunishima N.; Shimada Y.; Tsuji Y.; Sato T.; Yamamoto M.; Kumasaka T.; Nakanishi S.; Jingami H.; Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000, 407 (6807), 971–977. [DOI] [PubMed] [Google Scholar]
- Muto T.; Tsuchiya D.; Morikawa K.; Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 2007, 104 (10), 3759–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topiol S.; Sabio M.; Uberti M. Exploration of structure-based drug design opportunities for mGluRs. Neuropharmacology 2011, 60 (1), 93–101. [DOI] [PubMed] [Google Scholar]
- Costanzi S.; Mamedova L.; Gao Z. G.; Jacobson K. A. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J. Med. Chem. 2004, 47 (22), 5393–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Martinez M.; Marty A.; Jourdan M.; Escrieut C.; Archer E.; Gonzalez-Muniz R.; Garcia-Lopez M. T.; Maigret B.; Herranz R.; Fourmy D. Combination of molecular modeling, site-directed mutagenesis, and SAR studies to delineate the binding site of pyridopyrimidine antagonists on the human CCK1 receptor. J. Med. Chem. 2005, 48 (15), 4842–4850. [DOI] [PubMed] [Google Scholar]
- Betz S. F.; Lio F. M.; Gao Y.; Reinhart G. J.; Guo Z.; Mesleh M. F.; Zhu Y. F.; Struthers R. S. Determination of the binding mode of thienopyrimidinedione antagonists to the human gonadotropin releasing hormone receptor using structure–activity relationships, site-directed mutagenesis, and homology modeling. J. Med. Chem. 2006, 49 (21), 6170–6176. [DOI] [PubMed] [Google Scholar]
- Barry G. D.; Suen J. Y.; Low H. B.; Pfeiffer B.; Flanagan B.; Halili M.; Le G. T.; Fairlie D. P. A refined agonist pharmacophore for protease activated receptor 2. Bioorg. Med. Chem. Lett. 2007, 17 (20), 5552–5557. [DOI] [PubMed] [Google Scholar]
- Dukat M.; Mosier P. D.; Kolanos R.; Roth B. L.; Glennon R. A. Binding of serotonin and N1-benzenesulfonyltryptamine-related analogs at human 5-HT6 serotonin receptors: receptor modeling studies. J. Med. Chem. 2008, 51 (3), 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatman P. D.; Richman J. G.; Semple G. Nicotinic acid receptor agonists. J. Med. Chem. 2008, 51 (24), 7653–7662. [DOI] [PubMed] [Google Scholar]
- Kane B. E.; McCurdy C. R.; Ferguson D. M. Toward a structure-based model of salvinorin A recognition of the kappa-opioid receptor. J. Med. Chem. 2008, 51 (6), 1824–1830. [DOI] [PubMed] [Google Scholar]
- Harterich S.; Koschatzky S.; Einsiedel J.; Gmeiner P. Novel insights into GPCR-peptide interactions: mutations in extracellular loop 1, ligand backbone methylations and molecular modeling of neurotensin receptor 1. Bioorg. Med. Chem. 2008, 16 (20), 9359–9368. [DOI] [PubMed] [Google Scholar]
- Cavasotto C. N.; Orry A. J.; Murgolo N. J.; Czarniecki M. F.; Kocsi S. A.; Hawes B. E.; O’Neill K. A.; Hine H.; Burton M. S.; Voigt J. H.; Abagyan R. A.; Bayne M. L.; Monsma F. J. Jr. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 2008, 51 (3), 581–588. [DOI] [PubMed] [Google Scholar]
- Engel S.; Skoumbourdis A. P.; Childress J.; Neumann S.; Deschamps J. R.; Thomas C. J.; Colson A. O.; Costanzi S.; Gershengorn M. C. A virtual screen for diverse ligands: discovery of selective G protein-coupled receptor antagonists. J. Am. Chem. Soc. 2008, 130 (15), 5115–5123. [DOI] [PubMed] [Google Scholar]
- McRobb F. M.; Capuano B.; Crosby I. T.; Chalmers D. K.; Yuriev E. Homology modeling and docking evaluation of aminergic G protein-coupled receptors. J. Chem. Inf. Model. 2010, 50 (4), 626–637. [DOI] [PubMed] [Google Scholar]
- Kneissl B.; Leonhardt B.; Hildebrandt A.; Tautermann C. S. Revisiting automated G-protein coupled receptor modeling: the benefit of additional template structures for a neurokinin-1 receptor model. J. Med. Chem. 2009, 52 (10), 3166–3173. [DOI] [PubMed] [Google Scholar]
- Pecic S.; Makkar P.; Chaudhary S.; Reddy B. V.; Navarro H. A.; Harding W. W. Affinity of aporphines for the human 5-HT2A receptor: insights from homology modeling and molecular docking studies. Bioorg. Med. Chem. 2010, 18 (15), 5562–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente T.; Martin-Fontecha M.; Sallander J.; Benhamu B.; Campillo M.; Medina R. A.; Pellissier L. P.; Claeysen S.; Dumuis A.; Pardo L.; Lopez-Rodriguez M. L. Benzimidazole derivatives as new serotonin 5-HT6 receptor antagonists. Molecular mechanisms of receptor inactivation. J. Med. Chem. 2010, 53 (3), 1357–1369. [DOI] [PubMed] [Google Scholar]
- Wacker D.; Fenalti G.; Brown M. A.; Katritch V.; Abagyan R.; Cherezov V.; Stevens R. C. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc. 2010, 132 (33), 11443–11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P.; Rosenbaum D. M.; Irwin J. J.; Fung J. J.; Kobilka B. K.; Shoichet B. K. Structure-based discovery of beta2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 2009, 106 (16), 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S. On the applicability of GPCR homology models to computer-aided drug discovery: a comparison between in silico and crystal structures of the β2-adrenergic receptor. J. Med. Chem. 2008, 51 (10), 2907–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C.; Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J. Med. Chem. 2008, 51 (16), 4978–4985. [DOI] [PubMed] [Google Scholar]
- Soriano-Ursua M. A.; Trujillo-Ferrara J. G.; Correa-Basurto J. Homology modeling and flex-ligand docking studies on the guinea pig beta(2) adrenoceptor: structural and experimental similarities/ differences with the human beta(2). J. Mol. Model. 2009, 15 (10), 1203–1211. [DOI] [PubMed] [Google Scholar]
- Hattori K.; Orita M.; Toda S.; Imanishi M.; Itou S.; Nakajima Y.; Tanabe D.; Washizuka K.; Araki T.; Sakurai M.; Matsui S.; Imamura E.; Ueshima K.; Yamamoto T.; Yamamoto N.; Ishikawa H.; Nakano K.; Unami N.; Hamada K.; Matsumura Y.; Takamura F. Discovery of highly potent and selective biphenylacylsulfonamide-based beta3-adrenergic receptor agonists and molecular modeling based on the solved X-ray structure of the beta2-adrenergic receptor: part 6. Bioorg. Med. Chem. Lett. 2009, 19 (16), 4679–4683. [DOI] [PubMed] [Google Scholar]
- Gao Z. G.; Chen A.; Barak D.; Kim S. K.; Muller C. E.; Jacobson K. A. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J. Biol. Chem. 2002, 277 (21), 19056–19063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielan L.; Bolcato C.; Federico S.; Cacciari B.; Bacilieri M.; Klotz K. N.; Kachler S.; Pastorin G.; Cardin R.; Sperduti A.; Spalluto G.; Moro S. Combining selectivity and affinity predictions using an integrated support vector machine (SVM) approach: an alternative tool to discriminate between the human adenosine A(2A) and A(3) receptor pyrazolo-triazolo-pyrimidine antagonists binding sites. Bioorg. Med. Chem. 2009, 17 (14), 5259–5274. [DOI] [PubMed] [Google Scholar]
- Colotta V.; Lenzi O.; Catarzi D.; Varano F.; Filacchioni G.; Martini C.; Trincavelli L.; Ciampi O.; Pugliese A. M.; Traini C.; Pedata F.; Morizzo E.; Moro S. Pyrido[2,3-e]-1,2,4-triazolo[4,3-a]pyrazin-1-one as a new scaffold to develop potent and selective human A3 adenosine receptor antagonists. Synthesis, pharmacological evaluation, and ligand–receptor modeling studies. J. Med. Chem. 2009, 52 (8), 2407–2419. [DOI] [PubMed] [Google Scholar]
- Lenzi O.; Colotta V.; Catarzi D.; Varano F.; Poli D.; Filacchioni G.; Varani K.; Vincenzi F.; Borea P. A.; Paoletta S.; Morizzo E.; Moro S. 2-Phenylpyrazolo[4,3-d]pyrimidin-7-one as a new scaffold to obtain potent and selective human A3 adenosine receptor antagonists: new insights into the receptor-antagonist recognition. J. Med. Chem. 2009, 52 (23), 7640–7652. [DOI] [PubMed] [Google Scholar]
- Cheong S. L.; Dolzhenko A.; Kachler S.; Paoletta S.; Federico S.; Cacciari B.; Klotz K. N.; Moro S.; Spalluto G.; Pastorin G. The significance of 2-furyl ring substitution with a 2-(para-substituted) aryl group in a new series of pyrazolo-triazolo-pyrimidines as potent and highly selective hA(3) adenosine receptors antagonists: new insights into structure–affinity relationship and receptor–antagonist recognition. J. Med. Chem. 2010, 53 (8), 3361–3375. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Xu Z.; Liu G.; Tang Y. Insights into binding modes of adenosine A(2B) antagonists with ligand-based and receptor-based methods. Eur. J. Med. Chem. 2010, 45 (8), 3459–3471. [DOI] [PubMed] [Google Scholar]
- Jaakola V. P.; Ijzerman A. P. The crystallographic structure of the human adenosine A2A receptor in a high-affinity antagonist-bound state: implications for GPCR drug screening and design. Curr. Opin. Struct. Biol. 2010, 20 (4), 401–414. [DOI] [PubMed] [Google Scholar]
- Michino M.; Abola E.; Brooks C. L. 3rd; Dixon J. S.; Moult J.; Stevens R. C. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR dock 2008. Nat. Rev. Drug Discovery 2009, 8 (6), 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J.; Yoo L.; Gao Z. G.; Irwin J. J.; Shoichet B. K.; Jacobson K. A. Structure-based discovery of A(2A) adenosine receptor ligands. J. Med. Chem. 2010, 53, 3748–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9 (10), 430–431. [DOI] [PubMed] [Google Scholar]
- Ivanov A. A.; Barak D.; Jacobson K. A. Evaluation of homology modeling of G-protein-coupled receptors in light of the A(2A) adenosine receptor crystallographic structure. J. Med. Chem. 2009, 52 (10), 3284–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorin G.; Federico S.; Paoletta S.; Corradino M.; Cateni F.; Cacciari B.; Klotz K. N.; Gao Z. G.; Jacobson K. A.; Spalluto G.; Moro S. Synthesis and pharmacological characterization of a new series of 5,7-disubstituted-[1,2,4]triazolo[1,5-a][1,3,5]triazine derivatives as adenosine receptor antagonists: a preliminary inspection of ligand–receptor recognition process. Bioorg. Med. Chem. 2010, 18 (7), 2524–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobrath J. V.; Wang S. Computational elucidation of the structural basis of ligand binding to the dopamine 3 receptor through docking and homology modeling. J. Med. Chem. 2006, 49 (15), 4470–4476. [DOI] [PubMed] [Google Scholar]
- Ehrlich K.; Gotz A.; Bollinger S.; Tschammer N.; Bettinetti L.; Harterich S.; Hubner H.; Lanig H.; Gmeiner P. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J. Med. Chem. 2009, 52 (15), 4923–4935. [DOI] [PubMed] [Google Scholar]
- Allegretti M.; Bertini R.; Cesta M. C.; Bizzarri C.; Di Bitondo R.; Di Cioccio V.; Galliera E.; Berdini V.; Topai A.; Zampella G.; Russo V.; Di Bello N.; Nano G.; Nicolini L.; Locati M.; Fantucci P.; Florio S.; Colotta F. 2-Arylpropionic CXC chemokine receptor 1 (CXCR1) ligands as novel noncompetitive CXCL8 inhibitors. J. Med. Chem. 2005, 48 (13), 4312–4331. [DOI] [PubMed] [Google Scholar]
- Spalding T. A.; Trotter C.; Skjaerbaek N.; Messier T. L.; Currier E. A.; Burstein E. S.; Li D.; Hacksell U.; Brann M. R. Discovery of an ectopic activation site on the M(1) muscarinic receptor. Mol. Pharmacol. 2002, 61 (6), 1297–1302. [DOI] [PubMed] [Google Scholar]
- Langmead C. J.; Fry V. A.; Forbes I. T.; Branch C. L.; Christopoulos A.; Wood M. D.; Herdon H. J. Probing the molecular mechanism of interaction between 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine (AC-42) and the muscarinic M(1) receptor: direct pharmacological evidence that AC-42 is an allosteric agonist. Mol. Pharmacol. 2006, 69 (1), 236–246. [DOI] [PubMed] [Google Scholar]
- Lebon G.; Langmead C. J.; Tehan B. G.; Hulme E. C. Mutagenic mapping suggests a novel binding mode for selective agonists of M1 muscarinic acetylcholine receptors. Mol. Pharmacol. 2009, 75 (2), 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzik B.; Garzya V.; Shi D.; Walker G.; Lauchart Y.; Lucas A. J.; Rivero R. A.; Langmead C. J.; Watson J.; Wu Z.; Forbes I. T.; Jin J. 2′ Biaryl amides as novel and subtype selective M1 agonists. Part II: Further optimization and profiling. Bioorg. Med. Chem. Lett. 2010, 20 (12), 3545–3549. [DOI] [PubMed] [Google Scholar]
- Ma L.; Seager M. A.; Wittmann M.; Jacobson M.; Bickel D.; Burno M.; Jones K.; Graufelds V. K.; Xu G.; Pearson M.; McCampbell A.; Gaspar R.; Shughrue P.; Danziger A.; Regan C.; Flick R.; Pascarella D.; Garson S.; Doran S.; Kreatsoulas C.; Veng L.; Lindsley C. W.; Shipe W.; Kuduk S.; Sur C.; Kinney G.; Seabrook G. R.; Ray W. J. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106 (37), 15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q.; Frie J. L.; Marley D. M.; Beresis R. T.; Ren N.; Cai T. Q.; Taggart A. K.; Cheng K.; Carballo-Jane E.; Wang J.; Tong X.; Waters M. G.; Tata J. R.; Colletti S. L. Molecular modeling aided design of nicotinic acid receptor GPR109A agonists. Bioorg. Med. Chem. Lett. 2008, 18 (18), 4963–4967. [DOI] [PubMed] [Google Scholar]
- Gonzalez A.; Duran L. S.; Araya-Secchi R.; Garate J. A.; Pessoa-Mahana C. D.; Lagos C. F.; Perez-Acle T. Computational modeling study of functional microdomains in cannabinoid receptor type 1. Bioorg. Med. Chem. 2008, 16 (8), 4378–4389. [DOI] [PubMed] [Google Scholar]
- Diaz P.; Phatak S. S.; Xu J.; Astruc-Diaz F.; Cavasotto C. N.; Naguib M. 6-Methoxy-N-alkyl isatin acylhydrazone derivatives as a novel series of potent selective cannabinoid receptor 2 inverse agonists: design, synthesis, and binding mode prediction. J. Med. Chem. 2009, 52 (2), 433–444. [DOI] [PubMed] [Google Scholar]
- Szabo G.; Kiss R.; Payer-Lengyel D.; Vukics K.; Szikra J.; Baki A.; Molnar L.; Fischer J.; Keseru G. M. Hit-to-lead optimization of pyrrolo[1,2-a]quinoxalines as novel cannabinoid type 1 receptor antagonists. Bioorg. Med. Chem. Lett. 2009, 19 (13), 3471–3475. [DOI] [PubMed] [Google Scholar]
- Lange J. H.; Coolen H. K.; van der Neut M. A.; Borst A. J.; Stork B.; Verveer P. C.; Kruse C. G. Design, synthesis, biological properties, and molecular modeling investigations of novel tacrine derivatives with a combination of acetylcholinesterase inhibition and cannabinoid CB1 receptor antagonism. J. Med. Chem. 2010, 53 (3), 1338–1346. [DOI] [PubMed] [Google Scholar]
- El Bakali J.; Muccioli G. G.; Renault N.; Pradal D.; Body-Malapel M.; Djouina M.; Hamtiaux L.; Andrzejak V.; Desreumaux P.; Chavatte P.; Lambert D. M.; Millet R. 4-Oxo-1,4-dihydropyridines as selective CB2 cannabinoid receptor ligands: structural insights into the design of a novel inverse agonist series. J. Med. Chem. 2010, 53 (22), 7918–7931. [DOI] [PubMed] [Google Scholar]
- Tuccinardi T.; Ferrarini P. L.; Manera C.; Ortore G.; Saccomanni G.; Martinelli A. Cannabinoid CB2/CB1 selectivity. Receptor modeling and automated docking analysis. J. Med. Chem. 2006, 49 (3), 984–994. [DOI] [PubMed] [Google Scholar]
- Antel J.; Gregory P. C.; Nordheim U. CB1 cannabinoid receptor antagonists for treatment of obesity and prevention of comorbid metabolic disorders. J. Med. Chem. 2006, 49 (14), 4008–4016. [DOI] [PubMed] [Google Scholar]
- Lin L. S.; Ha S.; Ball R. G.; Tsou N. N.; Castonguay L. A.; Doss G. A.; Fong T. M.; Shen C. P.; Xiao J. C.; Goulet M. T.; Hagmann W. K. Conformational analysis and receptor docking of N-[(1S,2S)-3-(4-chlorophenyl)-2-(3-cyanophenyl)-1-methylpropyl]-2-methyl-2 -{[5-(trifluoromethyl)pyridin-2-yl]oxy}propanamide (taranabant, MK-0364), a novel, acyclic cannabinoid-1 receptor inverse agonist. J. Med. Chem. 2008, 51 (7), 2108–2114. [DOI] [PubMed] [Google Scholar]
- Kuduk S. D.; Di Marco C. N.; Chang R. K.; Wood M. R.; Kim J. J.; Schirripa K. M.; Murphy K. L.; Ransom R. W.; Tang C.; Torrent M.; Ha S.; Prueksaritanont T.; Pettibone D. J.; Bock M. G. 5-Piperazinyl pyridine carboxamide bradykinin B1 antagonists. Bioorg. Med. Chem. Lett. 2006, 16 (10), 2791–2795. [DOI] [PubMed] [Google Scholar]
- Huang H.; Player M. R. Bradykinin B1 receptor antagonists as potential therapeutic agents for pain. J. Med. Chem. 2010, 53 (15), 5383–5399. [DOI] [PubMed] [Google Scholar]
- Su D. S.; Markowitz M. K.; DiPardo R. M.; Murphy K. L.; Harrell C. M.; O’Malley S. S.; Ransom R. W.; Chang R. S.; Ha S.; Hess F. J.; Pettibone D. J.; Mason G. S.; Boyce S.; Freidinger R. M.; Bock M. G. Discovery of a potent, non-peptide bradykinin B1 receptor antagonist. J. Am. Chem. Soc. 2003, 125 (25), 7516–7517. [DOI] [PubMed] [Google Scholar]
- Sela I.; Golan G.; Strajbl M.; Rivenzon-Segal D.; Bar-Haim S.; Bloch I.; Inbal B.; Shitrit A.; Ben-Zeev E.; Fichman M.; Markus Y.; Marantz Y.; Senderowitz H.; Kalid O. G protein coupled receptors -in silico drug discovery and design. Curr. Top. Med. Chem. 2010, 10 (6), 638–656. [DOI] [PubMed] [Google Scholar]
- Senderowitz H.; Marantz Y. G protein-coupled receptors: target-based in silico screening. Curr. Pharm. Des. 2009, 15 (35), 4049–4068. [DOI] [PubMed] [Google Scholar]
- Zhukov A.; Andrews S. P.; Errey J. C.; Robertson N.; Tehan B.; Mason J. S.; Marshall F. H.; Weir M.; Congreve M.. J. Med. Chem. 2011, DOI: 10.1021/jm2003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupi X.; Dolker N.; Lopez-Rodriguez M. L.; Campillo M.; Ballesteros J. A.; Pardo L. Structural models of class a G protein-coupled receptors as a tool for drug design: insights on transmembrane bundle plasticity. Curr. Top. Med. Chem. 2007, 7 (10), 991–998. [DOI] [PubMed] [Google Scholar]
- Radestock S.; Weil T.; Renner S. Homology model-based virtual screening for GPCR ligands using docking and target-biased scoring. J. Chem. Inf. Model 2008, 48 (5), 1104–1117. [DOI] [PubMed] [Google Scholar]
- Li Y. Y.; Hou T. J.; Goddard W. A. 3rd. Computational modeling of structure–function of G protein-coupled receptors with applications for drug design. Curr. Med. Chem. 2010, 17 (12), 1167–1180. [DOI] [PubMed] [Google Scholar]
- Yarnitzky T.; Levit A.; Niv M. Y. Homology modeling of G-protein-coupled receptors with X-ray structures on the rise. Curr. Opin. Drug Discovery Dev. 2010, 13 (3), 317–325. [PubMed] [Google Scholar]
- Klabunde T.; Giegerich C.; Evers A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. J. Med. Chem. 2009, 52 (9), 2923–2932. [DOI] [PubMed] [Google Scholar]
- Topiol S.; Sabio M. Use of the X-ray structure of the beta2-adrenergic receptor for drug discovery. Bioorg. Med. Chem. Lett. 2008, 18 (5), 1598–1602. [DOI] [PubMed] [Google Scholar]
- Higgs C.; Beuming T.; Sherman W. Hydration site thermodynamics explain SARs for triazolylpurines analogues binding to the A2A receptor. ACS Med. Chem. Lett. 2010, 1, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater R. P. Location and nature of the residues important for ligand recognition in G-protein coupled receptors. J. Mol. Recognit. 2005, 18 (1), 60–72. [DOI] [PubMed] [Google Scholar]
- Bondensgaard K.; Ankersen M.; Thogersen H.; Hansen B. S.; Wulff B. S.; Bywater R. P. Recognition of privileged structures by G-protein coupled receptors. J. Med. Chem. 2004, 47 (4), 888–899. [DOI] [PubMed] [Google Scholar]
- Rosenkilde M. M.; Benned-Jensen T.; Frimurer T. M.; Schwartz T. W. The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol. Sci. 2010, 31 (12), 567–574. [DOI] [PubMed] [Google Scholar]
- Akritopoulou-Zanze I.; Hajduk P. J. Kinase-targeted libraries: the design and synthesis of novel, potent, and selective kinase inhibitors. Drug Discovery Today 2009, 14 (5–6), 291–297. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Chessari G.; Tisi D.; Woodhead A. J. Recent developments in fragment-based drug discovery. J. Med. Chem. 2008, 51 (13), 3661–3680. [DOI] [PubMed] [Google Scholar]
- Chessari G.; Woodhead A. J. From fragment to clinical candidate—a historical perspective. Drug Discovery Today 2009, 14 (13–14), 668–675. [DOI] [PubMed] [Google Scholar]
- Schulz M. N.; Hubbard R. E. Recent progress in fragment-based lead discovery. Curr. Opin. Pharmacol. 2009, 9 (5), 615–621. [DOI] [PubMed] [Google Scholar]
- Albert J. S.; Blomberg N.; Breeze A. L.; Brown A. J.; Burrows J. N.; Edwards P. D.; Folmer R. H.; Geschwindner S.; Griffen E. J.; Kenny P. W.; Nowak T.; Olsson L. L.; Sanganee H.; Shapiro A. B. An integrated approach to fragment-based lead generation: philosophy, strategy and case studies from AstraZeneca’s drug discovery programmes. Curr. Top. Med. Chem. 2007, 7 (16), 1600–1629. [DOI] [PubMed] [Google Scholar]
- Bartoschek S.; Klabunde T.; Defossa E.; Dietrich V.; Stengelin S.; Griesinger C.; Carlomagno T.; Focken I.; Wendt K. U. Drug design for G-protein-coupled receptors by a ligand-based NMR method. Angew. Chem., Int. Ed. 2010, 49 (8), 1426–1429. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Rich R. L.; Myszka D. G.; Figaroa F.; Siegal G.; Marshall F. H. Fragment screening of stabilized G-protein-coupled receptors using biophysical methods. Methods Enzymol. 2011, 493, 115–136. [DOI] [PubMed] [Google Scholar]
- Okada T.; Fujiyoshi Y.; Silow M.; Navarro J.; Landau E. M.; Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc. Natl. Acad. Sci. U.S.A. 2002, 99 (9), 5982–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Edwards P. C.; Burghammer M.; Villa C.; Schertler G. F. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004, 343 (5), 1409–1438. [DOI] [PubMed] [Google Scholar]
- Okada T.; Sugihara M.; Bondar A. N.; Elstner M.; Entel P.; Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J. Mol. Biol. 2004, 342 (2), 571–583. [DOI] [PubMed] [Google Scholar]
- Salom D.; Lodowski D. T.; Stenkamp R. E.; Le Trong I.; Golczak M.; Jastrzebska B.; Harris T.; Ballesteros J. A.; Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (44), 16123–16128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standfuss J.; Xie G.; Edwards P. C.; Burghammer M.; Oprian D. D.; Schertler G. F. Crystal structure of a thermally stable rhodopsin mutant. J. Mol. Biol. 2007, 372 (5), 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T.; Hiraki K.; Takahashi N.; Hori T.; Ago H.; Masuda K.; Takio K.; Ishiguro M.; Miyano M. Crystal structure of squid rhodopsin with intracellularly extended cytoplasmic region. J. Biol. Chem. 2008, 283 (26), 17753–17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M.; Kouyama T. Crystal structure of squid rhodopsin. Nature 2008, 453 (7193), 363–367. [DOI] [PubMed] [Google Scholar]
- Hanson M. A.; Cherezov V.; Griffith M. T.; Roth C. B.; Jaakola V. P.; Chien E. Y.; Velasquez J.; Kuhn P.; Stevens R. C. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 2008, 16 (6), 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch M. P.; Zou Y.; Rasmussen S. G.; Liu C. W.; Nygaard R.; Rosenbaum D. M.; Fung J. J.; Choi H. J.; Thian F. S.; Kobilka T. S.; Puglisi J. D.; Weis W. I.; Pardo L.; Prosser R. S.; Mueller L.; Kobilka B. K. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 2010, 463 (7277), 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino C. L.; Riley C. K.; Looney J.; Crouch R. K.; Okada T. Binding of more than one retinoid to visual opsins. Biophys. J. 2010, 99 (7), 2366–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H. W.; Kim Y. J.; Park J. H.; Morizumi T.; Pai E. F.; Krauss N.; Hofmann K. P.; Scheerer P.; Ernst O. P. Crystal structure of metarhodopsin II. Nature 2011, 471 (7340), 651–655. [DOI] [PubMed] [Google Scholar]