

Abstract
This video will guide you through the process of culturing rat cortical neurons in the presence of a glial feeder layer, a system known as a bilaminar or co-culture model. This system is suitable for a variety of experimental needs requiring either a glass or plastic growth substrate and can also be used for culture of other types of neurons.
Rat cortical neurons obtained from the late embryonic stage (E17) are plated on glass coverslips or tissue culture dishes facing a feeder layer of glia grown on dishes or plastic coverslips (known as Thermanox), respectively. The choice between the two configurations depends on the specific experimental technique used, which may require, or not, that neurons are grown on glass (e.g. calcium imaging versus Western blot). The glial feeder layer, an astroglia-enriched secondary culture of mixed glia, is separately prepared from the cortices of newborn rat pups (P2-4) prior to the neuronal dissection.
A major advantage of this culture system as compared to a culture of neurons only is the support of neuronal growth, survival, and differentiation provided by trophic factors secreted from the glial feeder layer, which more accurately resembles the brain environment in vivo. Furthermore, the co-culture can be used to study neuronal-glial interactions1.
At the same time, glia contamination in the neuronal layer is prevented by different means (low density culture, addition of mitotic inhibitors, lack of serum and use of optimized culture medium) leading to a virtually pure neuronal layer, comparable to other established methods1-3. Neurons can be easily separated from the glial layer at any time during culture and used for different experimental applications ranging from electrophysiology4, cellular and molecular biology5-8, biochemistry5, imaging and microscopy4,6,7,9,10. The primary neurons extend axons and dendrites to form functional synapses11, a process which is not observed in neuronal cell lines, although some cell lines do extend processes.
A detailed protocol of culturing rat hippocampal neurons using this co-culture system has been described previously4,12,13. Here we detail a modified protocol suited for cortical neurons. As approximately 20x106 cells are recovered from each rat embryo, this method is particularly useful for experiments requiring large numbers of neurons (but not concerned about a highly homogenous neuronal population). The preparation of neurons and glia needs to be planned in a time-specific manner. We will provide the step-by-step protocol for culturing rat cortical neurons as well as culturing glial cells to support the neurons.
Protocol
1. Glia Dissection (~2 weeks before plating neurons)
To prepare for the dissection place sterile tools in 70% ethanol, add 4 mL cold dissection medium to 60 mm dishes (one dish per brain), and place 2.5% trypsin and DNase on ice to thaw (see details about surgical tools in table VI).
Use large scissors to decapitate 2-4 day old pups and place heads in a 100 mm sterile dish.
Use medium scissors to make a midline cut through the skin and pull back the skin to expose the skull. Use curved scissors to make a midline cut and two lateral cuts through the skull, without damaging the brain tissue underneath. Remove the upper skull to expose the brain.
Extract the brain and place it in its own 60 mm dish containing 4 mL cold dissection medium (see table I for composition). Place the dish under a stereomicroscope (Leica ZOOM 2000, zoom control: 7-30) and use No.5 forceps to separate the hemispheres, remove the midbrain, and peel off the meninges.
Collect all dissected cerebral cortices in a fresh 60 mm dish with cold dissection medium, and mince the tissue with curved scissors.
Transfer the minced tissue to a sterile 15 mL tube. Bring the volume to 4.5 mL with cold dissection medium and add 500 μL of 2.5% trypsin. Wrap the tube in Parafilm and float it in a 37°C water bath for 15 minutes, mixing by inversion every 5 minutes. (Note: here we describe the protocol we generally follow when using up to 5 brains; if more pups are used, volumes may need to be adjusted appropriately)
Transfer the tissue to a new 15 mL tube, minimizing the volume of trypsin-containing dissection medium carried over. Bring the volume to 4.8mL with fresh dissection medium and add 200 μL DNase to reach a final concentration of 60 ug/mL (stock solution: 3 mg of DNase and 5 mg MgSO4 in 2 mL of dissection medium). Triturate gently ~20 times with a sterile 5 mL pipette, then add 10 mL of glia plating medium (see table II for composition).
Allow large pieces of tissue to settle for 4-5 minutes, then transfer the top 80% of the cell suspension to a 50mL tube. Repeat the tissue trituration with an additional 2-3 mL of glia plating medium (and using a smaller pipette if needed); again, allow the tissue to settle, and add the top 80% to the previous collection. Bring the total volume to 20 mL with glia plating medium and centrifuge for 15 minutes at 280 g (or 1300 rpm in a IEC Centra CL2 centrifuge).
Aspirate the supernatant and resuspend the cell pellet in glia plating medium (roughly using 5-7 mL depending on the number of pups used); then add additional plating medium in order to have 10 mL for each brain dissected. Mix well and plate 10 mL of cell suspension in each 75 cm2 flask (i.e. one brain per flask).
Move culture flasks to an incubator set at 37°C (5 to 10% CO2; proportional to the sodium bicarbonate content of the medium). Change culture medium after 24-48 hours, then every 3-4 days.
2. Secondary Glia (48 hours before plating neurons)
Prepare thermanox coverslips (if neurons will be plated on dishes) by adding 3 small drops of melted paraplast wax around the circumference of each washed and autoclaved thermanox. Cells will be plated on this side of the thermanox. Our thermanox are handmade reusable ~60 mm inserts cut from plastic (see table VI of reagents) with ~50 2 mm holes drilled through, although commercially available culture well inserts may be used instead.
Wash the primary glia with PBS two times, vigorously shaking each flask to remove any unattached cells. Each flask with confluent primary glia will provide roughly 10 million astroglia, therefore we regularly use 2-4 flasks per experiment.
Detach the cells with 0.05% tryspin-EDTA (1 mL10X 0.5% trypsin-EDTA stock solution diluted in 9 mL of PBS). Combine the cells from 2 flasks into one 50 mL conical tube and add an equal amount of glia plating medium (see table II of reagents). Centrifuge for 15 minutes at 280 g. Aspirate the supernatant and dissolve the glia pellet in a few mL of glia plating medium.
Count the cells using a hematocytometer and dilute the cell suspension as needed (~1x106 cells/mL); plate the cells either on 60mm dishes (if neurons will be plated on coverslips), or on thermanox coverslips (if neurons will be plated on 60 mm dishes). Cells should be plated at 500,000 cells per dish or thermanox using glia plating medium. In the case of thermanox coverslips, plate 600 μL of cells in a dropwise fashion, then after 2 hours flip thermanox and submerge cells in glia plating medium.
If preparing 60 mm dishes of secondary glia for neuronal coverslips, change medium to N2.1 the evening before the neuronal dissection.
3. Neuronal Dissection and Culture
Prepare glass coverslips (15 mm diameter) by adding 3 small drops of melted paraplast wax around the circumference of each sterile coverslip, in order to physically separate the cell layers and prevent cell scraping. Cells will be plated on this side of the coverslip.
Coat plates or coverslips with 0.5 mg/mL polylysine (either poly-L-lysine or poly-D-lysine dissolved in borate buffer; see table VI) for 2 hours or overnight. Wash with sterile water 2-3 times and allow to dry completely. Note: coverslips are placed in a hydrophobic petri dish to avoid spilling/spreading of the solutions during coating with poly-lysine or cell plating, which can easily occur if the coverslips are placed in tissue culture dishes.
Prepare for the neuronal dissection as for the glia dissection (step 1.1). Euthanize a 17-day pregnant rat, spray the fur with 70% ethanol, and cut medially through the skin to expose the uterus. Remove all fetuses and place them in a sterile 100 mm dish.
Rapidly dissect free and decapitate each fetus, placing each head in its own 60 mm dish with cold dissection medium (4 mL). We normally dissect 4-5 fetuses per experiment as approximately 20 million neurons are derived from each E17 fetus. If more embryos are needed, make sure that the whole procedure (from step 3.4 to 3.12) does not last more than 90-100 minutes.
Using a stereomicroscope and No.1/No.5 forceps isolate the cerebral cortices by pulling open the skin and skull, extracting the brain, separating the hemispheres, and removing the midbrain and meninges.
Collect all dissected cortices in a fresh 60 mm dish with cold dissection medium, and mince the tissue with curved scissors into ~1 mm pieces.
Transfer the minced tissue to a sterile 15 mL tube. Bring the volume to 4.5 mL with cold dissection medium and add 500 μL of 2.5% trypsin. Wrap the tube in Parafilm and float it in a 36°C water bath for 15 minutes, mixing by inversion every 5 minutes.
Transfer the tissue to a new 15 mL tube, minimizing the volume of trypsin-containing dissection medium carried over. Bring to 4.8 mL with fresh dissection medium and add 200 μL DNase to reach a final concentration of 60 ug/mL. Triturate roughly 10 times with a sterile 5 or 2 mL pipette; repeat this step as necessary using a fire-polished (cold) Pasteur pipette to dissociate most of the tissue. Then allow remaining pieces of tissue (generally very few, if any) to settle for 4-5 minutes.
Remove the upper single-cell suspension while leaving behind settled pieces of tissue, and place this solution in a new tube with an equal volume of neuron plating medium (see table III). Mix well but gently, then count the cells; plate on 15 mm coverslips (35,000 cells each in 200 μL) or 60 mm dishes (1x106 cells each in 3 mL) as needed. Transfer cells to the incubator (36.5°C, 5-10% CO2).
After 3-4 hours combine the neuron layer and the secondary glia as follows. For neurons on 60 mm dishes, wash the cells with warm DMEM, change the medium to 4mL of serum free medium (N2, see table IV), and add one thermanox with glia to each plate (glial cells should be facing the neurons). For neuronal coverslips, transfer 3 to 5 coverslips to each 60 mm dish of secondary glia, with the neurons facing down.
24 hours after plating the neurons, add 10 μM of cytosine-β-D-arabinofuranoside to each dish, in order to prevent glia proliferation. We prepare 4 mM stock solutions, dilute 1:10 with N2 medium, and add 25 uL/mL of diluted solution to each dish.
Cells are generally used for experiments after 7-10 days in culture, although exact time depends on desired differentiation stage; they have been grown for up to 4 weeks without a significant decrease in survival. After the first week, it is recommended that one-half volume of media be replaced every week.
4. Representative Results:
A few hours after neurons have attached to the substrate, they begin to extend lamelliopodia. Processes continue to elongate (Fig 2A) and polarize over several days in culture, and extended axons and dendrites are clearly visible (Fig 2B; MAP2 staining). As neurons mature the dendritic arbors become more elaborate and they develop synaptic contacts; about 2-3 weeks after plating many dendritic spines are visible (Fig 2C)11.
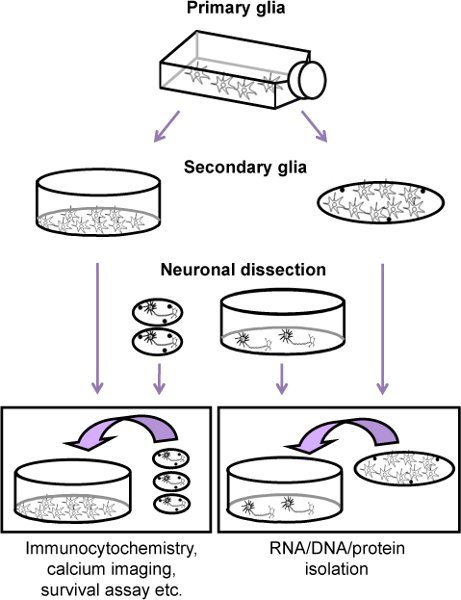 Figure 1. Flow chart of the procedure for culturing rat cortical neurons along with a glial feeder layer (secondary glia). First, 11 days before the neuronal dissection, a primary glia culture is prepared from neonatal rats (P2-4). When the cells are confluent (roughly nine days after plating the primary glia and two days before the neuronal dissection), astrocytes are mechanically separated from oligodendrocyte precursor cells and microglia and plated either on dishes or thermanox (secondary glia). Neurons are then obtained from the cortices of E17 embryos and plated either on coverslips or on dishes (coated with poly-lysine). Four hours after plating, the neuronal layer is added to the secondary glia.
Figure 1. Flow chart of the procedure for culturing rat cortical neurons along with a glial feeder layer (secondary glia). First, 11 days before the neuronal dissection, a primary glia culture is prepared from neonatal rats (P2-4). When the cells are confluent (roughly nine days after plating the primary glia and two days before the neuronal dissection), astrocytes are mechanically separated from oligodendrocyte precursor cells and microglia and plated either on dishes or thermanox (secondary glia). Neurons are then obtained from the cortices of E17 embryos and plated either on coverslips or on dishes (coated with poly-lysine). Four hours after plating, the neuronal layer is added to the secondary glia.
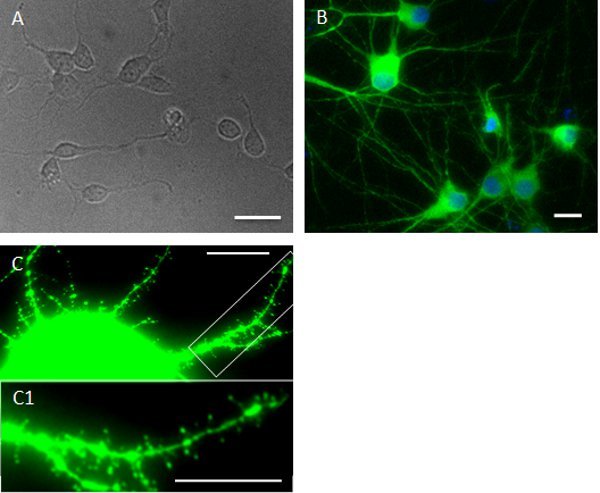 Figure 2. Examples of cultures at various stages (e.g. 5 hours; 12 DIV; 21 DIV). Figures are representative images of cortical neurons on coverslips at different time points. After neurons attach to the substrate neurons begin to extend lamelliopodia. Phase contrast image shows a few extended neurites five hours after plating (A). The image in the middle (B, from Cook et al. 2010, with permission) shows 12 DIV cortical neurons fixed and immunostained with the neuronal dendritic marker MAP2. Hoechst staining was used as a counterstaining. As neurons mature dendritic spines also become visible (C). Scale bars: A, B = 25 μm; C = 10 μm.
Figure 2. Examples of cultures at various stages (e.g. 5 hours; 12 DIV; 21 DIV). Figures are representative images of cortical neurons on coverslips at different time points. After neurons attach to the substrate neurons begin to extend lamelliopodia. Phase contrast image shows a few extended neurites five hours after plating (A). The image in the middle (B, from Cook et al. 2010, with permission) shows 12 DIV cortical neurons fixed and immunostained with the neuronal dendritic marker MAP2. Hoechst staining was used as a counterstaining. As neurons mature dendritic spines also become visible (C). Scale bars: A, B = 25 μm; C = 10 μm.
Discussion
This protocol provides a method for culturing rat primary cortical neurons in the presence of glia cells, while allowing the neurons to be easily isolated for experimental analysis. The glia support development of a healthy neuronal phenotype, while also modulating neuronal responses to experimental treatments in a physiologically relevant manner. Additionally, by passaging the primary glia before culturing with neurons this cell population becomes selectively enriched in astrocytes, thereby preventing inflammatory microglial stimulation. For certain types of experiments, however, different proportions of glial cells may be desired, thus additional glia cultures may be performed to optimize this cellular composition. This protocol may also be adapted for culturing neurons derived from other brain regions, such as the hippocampus, to fit particular experimental demands
Major concerns of this technique include preventing bacterial contamination and preventing glial proliferation into the neuronal layer. This system does not include antibiotic agents, thus sterile techniques are especially critical at all stages. Glial proliferation is prevented by the addition of the anti-mitotic agent cytosine arabinofuranoside, the low density plating and the serum-free culture medium; following this protocol astrocytes should compose no more than 3-5% of cells in the neuronal layer1,4 and less than 1% microglia should be detected. Immunopanning or other techniques can be used to remove even this low glia contamination4.
This technique can also be modified for co-culturing any variety of adherent cell types when isolating a single type for analysis is desirable. For example, this system was successfully adapted for analyzing the effects of osteoblasts on Ca2+ signaling in bone-metastatic cancer cells14.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors thank previous laboratory members who contributed to refinement of this protocol and the NIH for support over the years (DA19808 and DA15014 to OM). Anna Abt1 is a fellow of the "Interdisciplinary and Translational Research Training in neuroAIDS" (T32-MH078795); thus, this work was supported in part by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award 5T32MH079785.
References
- Cook A. Interactions between chemokines: regulation of fractalkine/CX3CL1 homeostasis by SDF/CXCL12 in cortical neurons. J. Biol. Chem. 2010;285:10563–10563. doi: 10.1074/jbc.M109.035477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai J, Burbassi S, Rubin J, Meucci O. CXCL12 inhibits expression of the NMDA receptor's NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell. Death. Dis. 2010;1:e33–e33. doi: 10.1038/cddis.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta R. Morphine increases brain levels of ferritin heavy chain leading to inhibition of CXCR4-mediated survival signaling in neurons. J. Neurosci. 2009;29:2534–2544. doi: 10.1523/JNEUROSCI.5865-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 1998;95:14500–14500. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ. Regulation of neuronal P53 activity by CXCR 4. Mol. Cell. Neurosci. 2005;30:58–66. doi: 10.1016/j.mcn.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP. Modulation of neuronal CXCR4 by the micro-opioid agonist DAMGO. J. Neurovirol. 2006;12:492–500. doi: 10.1080/13550280601064798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S. Role of the transcription factor E2F1 in CXCR4-mediated neurotoxicity and HIV neuropathology. Neurobiol. Dis. 2007;25:17–26. doi: 10.1016/j.nbd.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ. The chemokine receptor CXCR4 regulates cell-cycle proteins in neurons. J. Neurovirol. 2003;9:300–314. doi: 10.1080/13550280390201010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ. The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell. Death. Differ. 2008;15:1663–1672. doi: 10.1038/cdd.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Vaidya A, Meucci O. CXCL12-mediated regulation of ANP32A/Lanp, a component of the inhibitor of histone acetyl transferase (INHAT) complex, in cortical neurons. J. Neuroimmune. Pharmacol. 2011;6:163–170. doi: 10.1007/s11481-010-9228-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat. Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Goslin K, Banker G. In: Culturing Nerve Cells. Second Edition. Goslin K, Banker G, editors. The MIT Press; 1998. pp. 339–370. [Google Scholar]
- D'Ambrosio J, Fatatis A. Osteoblasts modulate Ca2+ signaling in bone-metastatic prostate and breast cancer cells. Clin. Exp. Metastasis. 2009;26:955–964. doi: 10.1007/s10585-009-9286-3. [DOI] [PubMed] [Google Scholar]