

Abstract
With the advances in genomics research of the past decade, plant biology has seen numerous studies presenting large-scale quantitative analyses of gene expression. Microarray and next generation sequencing approaches are being used to investigate developmental, physiological and stress response processes, dissect epigenetic and small RNA pathways, and build large gene regulatory networks1-3. While these techniques facilitate the simultaneous analysis of large gene sets, they typically provide a very limited spatiotemporal resolution of gene expression changes. This limitation can be partially overcome by using either profiling method in conjunction with lasermicrodissection or fluorescence-activated cell sorting4-7. However, to fully understand the biological role of a gene, knowledge of its spatiotemporal pattern of expression at a cellular resolution is essential. Particularly, when studying development or the effects of environmental stimuli and mutants can the detailed analysis of a gene's expression pattern become essential. For instance, subtle quantitative differences in the expression levels of key regulatory genes can lead to dramatic phenotypes when associated with the loss or gain of expression in specific cell types.
Several methods are routinely used for the detailed examination of gene expression patterns. One is through analysis of transgenic reporter lines. Such analysis can, however, become time-consuming when analyzing multiple genes or working in plants recalcitrant to transformation. Moreover, an independent validation to ensure that the transgene expression pattern mimics that of the endogenous gene is typically required. Immunohistochemical protein localization or mRNA in situ hybridization present relatively fast alternatives for the direct visualization of gene expression within cells and tissues. The latter has the distinct advantage that it can be readily used on any gene of interest. In situ hybridization allows detection of target mRNAs in cells by hybridization with a labeled anti-sense RNA probe obtained by in vitro transcription of the gene of interest.
Here we outline a protocol for the in situ localization of gene expression in plants that is highly sensitivity and specific. It is optimized for use with paraformaldehyde fixed, paraffin-embedded sections, which give excellent preservation of histology, and DIG-labeled probes that are visualized by immuno-detection and alkaline-phosphatase colorimetric reaction. This protocol has been successfully applied to a number of tissues from a wide range of plant species, and can be used to analyze expression of mRNAs as well as small RNAs8-14.
Keywords: Plant Biology, Issue 57, In Situ hybridization, RNA localization, expression analysis, plant, DIG-labeled probe
Protocol
1. Sample preparation
Note: All steps up to and including the hybridization step are sensitive to RNAse activity. It is therefore essential to work in clean conditions. It is also quite common to make all solutions RNase-free by using DEPC-treated water and glassware baked at 180 °C for at least 3 hrs. Plastic containers are often cleaned through treatment with 0.2 N NaOH for 30 min. However, none of these precautions are essential and we typically use regular clean milliQ H2O for all solutions. We do keep separate reagents, boxes and glassware for in situ hybridizations and store these in a clean cabinet.
- Sample fixation
- Day 1: Prepare fresh 4% paraformaldehyde (PFA) fixative. For 500 ml, warm 400 ml 1x PBS (130 mM NaCl, 7 mM Na2HPO4, 3 mM NaH2PO4 pH7.0) to 60°C and dissolve two pellets of NaOH. In a fume hood, add 20 g of paraformaldehyde and mix thoroughly until dissolved. Place the solution on ice and when cooled adjust the pH to 7.2 with H2SO4 (1-2 drops for 100ml). Then adjust the volume to 500 mL with 1x PBS. Note: Do not use HCl to adjust the pH as this will release highly toxic fumes. Paraformaldehyde is toxic; this solution should therefore be prepared in a fume hood and disposed of properly. Also, Paraformaldehyde is stored at 4°C and should be bought new every 6 - 9 months.
- Harvest tissue samples and place them immediately in 15 mL fresh PFA fixative on ice in glass scintillation vials. If tissue dissection is required, this is best done on ice in cold fixative. Note: It is important to use a large excess of fixative. The general recommended ratio is to use 10 volumes of fixative to 1 volume of tissue.
- Apply a vacuum (˜500 mm Hg) to the samples while on ice. Hold a vacuum for 15-20 minutes; small bubbles should release from the samples but the fixative should not come to a boil. Release the vacuum slowly, and renew the PFA fixative to ensure the fixative remains at the right concentration. Repeat this step until the tissues sink after release of vacuum. Note: Fixation is required to fix and cross-link RNA molecules within the tissue. This step is critical as poorly fixed materials yield a low signal. Some tissues do not sink immediately but most will eventually sink overnight. To improve the infiltration of the fixative, the following detergents can be added: Triton up to 0.1% and/or Tween up to 0.1 - 0.3%. Alternatively, ethanol-based fixatives, such as formaldehyde-alcohol-acidic acid (FAA), can be used for tissues that are otherwise not easily infiltrated14,15.
- Replace the PFA fixative once more and keep the vials at 4°C overnight.
- Tissue embedding
- Day 2: Pre-cool 1x PBS and the ethanol series at 4°C.
- Rinse the samples twice for 30 min each in 1x PBS, on ice. Note: Again it is recommended to use a large excess of each solution.
- Dehydrate the samples by taking them through an ethanol series, on ice, as follow:
- 10% ethanol 30 min,
- 30% ethanol 30 min,
- 50% ethanol 1 hour,
- 70% ethanol 1 hour,
- 85% ethanol 1 hour,
- 95% ethanol 1hour,
- 3 changes of 100% ethanol for 1hour each
- At the end of the day, replace the ethanol by 0.1% Eosine in 100% ethanol overnight at 4°C. Eosine stains the cell wall making the samples more visible during embedding and sectioning. Notes: The times indicated here have been optimized for blocks of 3x3x3 mm but longer incubations may be required for bigger samples. Samples can be stored in 70% ethanol at 4°C for several months.
- Day 3: The following morning, warm the samples to room temperature for one hour. Then replace the ethanol gradually with histoclear as follows:

- At the end of the day, add 1/4 volume of Paraplast Plus chips and place the vial in a 60°C oven. Also fill a beaker with Paraplast chips and replenish this frequently in order to always have freshly molten wax at hand. Note: The temperature of the oven is important and prolonged heating of Paraplast above 60°C should be avoided.
- Day 4: Gradually replace the histoclear with Paraplast by regularly adding more paraffin chips (every hour). At the end of the day, carefully pour off the histoclear/wax mixture and immediately replace it with pure molten wax. Leave the samples at 60°C overnight.
- Days 5 and 6: Change the Paraplast 3 times daily, about every 4 hours, with fresh molten wax keeping the samples at 60°C. Note: It is possible to change the wax only once a day, but the tissue preparation would take a few extra days to complete, as the wax needs to be changed at least 5 or 6 times. Vacuum infiltration of the tissue samples when these are in 100% EtOH and/or in wax can further improve the infiltration and embedding of tissues. Vacuum infiltration of samples in wax should be done at 60°C.
- Day 7: Change the wax once more. The smell of histoclear should now be gone completely and the samples can be embedded later that day.
- Set up a large warming plate at 60°C protected by aluminum foil.
- The method of embedding will vary depending on the type of tissue to be analyzed. The most important point at this step is to ensure that the samples are oriented correctly for sectioning later. One way is to use plastic molds and rings. Warm up the molds on the warming plate and pour molten wax into the bottom part of the mold. Transfer a tissue sample into the mold with warmed forceps and orient it into the desired position. Place the white ring onto the mold and add additional molten wax such that the tissues are covered completely. Carefully move the mold from the plate to the bench. Let the wax cool down gradually.
Another possibility is to distribute all the samples in a petri dish containing molten wax (the samples should be completely covered by wax) while working on the 60°C warmed plate. Orientate the samples with warm forceps. At the end, switch off the plate. When the wax is solidified, the petri dish can be moved to the bench and then store at 4°C. When ready for sectioning, small blocks containing a tissue sample can be cut out of the petri dish with a warmed blade and mounted onto a microtome specimen holder with an extra drop of molten wax. Let the sample harden for a few minutes before sectioning.
Note: Blocks can be stored at 4°C for 1 year or longer. For additional helpful tips on tissue fixation and embedding, see ref. 16
2. Probe preparation
Cloning Probes are usually generated to one or more specific regions of the gene of interest. Highly repetitive sequences should be excluded from the probe, but sequences corresponding to conserved protein domains, such as DNA-binding motifs in transcription factors, typically will yield gene specific expression patterns8,9,12,15 (Fig. 1). This because the RNAse A treatment in the post-hybridization steps (see below) reduces the level of probe cross-hybridization between gene family members. RNAse A cleaves at mismatches in RNA duplexes, fragmenting probes hybridized to transcripts with imperfect complementarity, which will be washed off in subsequent steps. In general, several probes may need to be tested for each gene of interest. Probes can be as short as 150 bases in length. Although there is no limit to the length of the probe, fragments shorter then 1.5 kb typically transcribe better. DNA fragments to be transcribed are cloned into a vector containing T3, T7, or SP6 promoters (e.g. pCRII-TOPO, Invitrogen). Alternatively, T3, T7 or SP6 promoter sequences can be included as a 5’-tail on the reverse primer used for amplifying the probe region. In every in situ hybridization experiment, we include a positive control probe for which the hybridization pattern is known and which works consistently, as well as a negative control (Figure 1D and H). The latter could be a random RNA probe that is known not to hybridize to any transcripts in the tissue of interest. Although it is common to use the sense probe of the gene under analysis, this is not necessary and any non-hybridizing RNA will suit the purpose. If available, tissues from a transcriptional null allele of the gene of interest would serve as an excellent negative control.
- In vitro transcription
- Linearize the plasmid by digesting approximately 5 μg DNA with a restriction enzyme that digests at the end of the insert opposite the site of the promoter. Do not use restriction enzymes that leave a 3'-overhang. This can lead to transcription artifacts because the polymerase can utilize the 3' overhang as a substrate to continue transcription. Alternatively, the probe region including the T3, T7 or SP6 promoter can be amplified by PCR to generate the DNA template for the in vitro transcription reaction.
- Check an aliquot on a gel to verify that the reaction has gone to completion.
- Extract the DNA with phenol/chloroform, precipitate with ethanol and resuspend the DNA pellet in milliQ H2O to a concentration of 1μg/μl. Alternatively, the DNA can be cleaned using standard DNA purification columns.
- Set up the in vitro transcription reaction by mixing:
- 1μl purified DNA (1μg/μl)
- 2μl 10x transcription buffer
- 2 μL 10x DIG RNA labeling mix
- 1μl RNAse OUT
- 2μl T3, T7 or SP6 RNA polymerase (20 U/μl)
- H2O up to 20μl.
- Remove the DNA template by adding 75 μL MilliQ H2O and 5 μL RQ1 DNAse (1 U/μl) to the transcription reaction and incubating the reaction at 37°C for 10 min.
- Purify the in vitro transcription reaction to eliminate non-incorporated nucleotides and small transcripts. One possibility is to use a size-exclusion Sephadex® column, such as the mini Quick Spin RNA Columns (Roche). Alternatively, the transcription reaction can be purified by a conventional LiCl/ethanol precipitation (see ref. 14). Keep 1μl to check on a gel later.
- Probes over 250 bases in length are usually partially hydrolized to obtain RNA molecules of approximately 150 nt and therefore small enough to efficiently penetrate the tissue sections. Add an equal volume of 2x carbonate buffer (80 mM NaHCO3, 120 mM Na2CO3) to the purified transcription reaction and incubate at 60°C. The incubation time should be calculated by the following formula: time = (Li - Lf) / (K x Li x Lf) where Li = initial probe length (in kb); Lf = final probe length (0.150 kb); K = 0.11 kb/minute. Keep 2 μL to check on a gel.
- Neutralize the hydrolysis reaction by adding 1/20 volume of 10% acetic acid.
- The probe is then purified by adding 1μl of tRNA (100mg/ml), 1/10 volume 3M NaAc pH 5.2, plus 2.5 volumes cold 100% ethanol, and incubating at -80°C for 30 min. Precipitate the RNA by centrifugation at 13,500 rpm for 20 min at 4°C. Discard the supernatant and rinse the RNA pellet with 500μl 70% ethanol. Centrifuge again, let the pellet air dry (typically about 15 min), and resuspend the RNA in 50μl 50% deionized formamide. The probe should be stored at -80°C until use.
- Check on a regular 1% TAE-agarose gel the final product as well as intermediate products from sections 2.2.4, 2.2.6 and 2.2.7 (Figure 2A). This allows monitoring of the efficiency of the in vitro transcription reaction. Note: The in vitro transcription reaction should yield approximately 1 μg of RNA per 1 μg of template.
- Dot Blot Analysis The probe concentration and DIG-UTP incorporation can be assessed by dot blot analysis (Figure 2B). Serial RNA dilutions are spotted onto a standard hybridization membrane alongside a labeled control RNA of known concentration. For instance, we use the DIG-UTP labeled control RNA probe included in the in vitro transcription kit (Roche). It is best to test multiple dilutions for each probe to establish an accurate concentration.
- Prepare blocking buffer by dissolving 0.5% of Blocking Reagent in 1x TBS buffer (100 mM Tris-HCl pH7.5, 150 mM NaCl) at 70°C, then let the solution cool down to room temperature.
- Spot 1μl of serial dilutions (typically 10-1 to 10-5) of the in vitro transcribed and control probes on an N+ nylon membrane. Fix the RNA by UV cross-linking. Note: A minimal surface of the membrane should be used to reduce the volume of buffers needed later.
- Equilibrate the membrane in 1x TBS for 5 min at room temperature (RT) on a shaking platform.
- Block the membrane using the blocking buffer for 30 min at RT on a shaking platform.
- Rinse the membrane in 1x TBS for 5 min.
- Incubate the membrane with anti-DIG antibody, diluting 1μl of Anti-digoxigenin-AP in 10 ml of 1x TBS, for 45 min at RT.
- Wash the membrane twice in 1x TBS for 15 min each.
- Equilibrate the membrane in 1x TN buffer (100mM Tris-HCl pH 9.5, 100mM NaCl) for 5 min.
- Stain the membrane by incubating with NBT/BCIP 1/50 (v/v) in 1x TN buffer. Staining can take 5-10 min. Afterwards rinse the membrane with water; it can then be dried and kept as a record. Note: NBT/BCIP is a substrate for the alkaline phosphatase conjugated to the anti-DIG antibody that upon hydrolysis produces a dark blue dye.
3. Sectioning
Warm the blocks to room temperature. If the blocks are too cold, individual sections may roll over without forming a wax ‘ribbon’. Trim the block into a trapezoid shape leaving about 1mm of wax around the sample.
Warm up a large slide warmer at 35-37°C. Note: Many protocols do this step at 42°C however reducing the temperature to 35-37°C prevents the formation of bubbles under the sections.
Place the block onto the microtome such that the longer of the two parallel faces is at the bottom. Carefully bring the block forward to the blade and make sure that the surface of the block is parallel to the blade. Section at a thickness of 8 - 10μm. The wax ribbons can be aligned on a non-sticky surface, such as aluminum foil or filter paper, to select sections of interest. This can be assessed using a nearby dissecting microscope. Note: The eosin staining of the tissue provides an indication whether the tissues have been properly infiltrated during the embedding. If the center of the block is not stained with eosin this usually indicates that the tissue is poorly fixed.
Mark Probe-on Plus slides with a pencil (most pens or markers are erased during the in situ hybridization protocol) and distribute 1mL of clean milliQ H2O onto it. Carefully float sections of interest on the surface of the water at room temperature (it is easier to manipulate the sections on the water when working at room temperature). Ensure that the shiny side (the bottom side as the ribbon comes off of the microtome) faces the water. Then transfer the slide slowly onto the slide warmer. Heating helps the sections to spread on the surface of the water. After 5 min, pour off the water carefully but in one smooth movement, so the ribbon is lowered down onto the slide. Let the slides dry for at least several hours or overnight at 37°C, so the tissue adheres. Note: Sectioned tissue can be stored in a box with silica desiccant for a few days to weeks at 4°C, however, it is best to use the slides as soon as possible.
4. In situ Hybridization
- Section pre-treatment The volume required for each solution will obviously depend on the number of slides to be treated and the container size used. The sections should always be completely submerged. For a 25-slide rack and a neatly fitting container, 200-250 ml is sufficient. Because many of the incubation steps are very short, we usually prepare all possible solutions first and then start the slide pre-treatments. However, the acetic anhydride solution will need to be prepared immediately before use and protease is added at the time of use. All steps are performed at room temperature unless noted otherwise. Also, all buffers can be prepared at 10x concentration as a stock solution.
- Warm the protease buffer (100 mM Tris pH 8.0, 50 mM EDTA) in the container to 37°C.
- Deparaffinize and rehydrate the tissue sections as follows:
- histoclear - 10 min (use a glass dish)
- histoclear - 10 min (use a glass dish)
- 100% ethanol - 1 min
- 100% ethanol - 30 sec
- 95% ethanol - 30 sec
- 85% ethanol - 30 sec
- 70% ethanol - 30 sec
- 50% ethanol - 30 sec
- 30% ethanol - 30 sec
- 10% ethanol - 30 sec
- milliQ H2O - 1 min
- Incubate in 1x PBS for 2 min.
- Mix 625μl of protease 50mg/ml in 250 ml protease buffer pre-warmed to 37°C and incubate the slides for 20 min at 37°C. Note: The protease is required to digest fixed proteins and increase probe access to cellular RNAs. It is critical to control the time of incubation to maximize the hybridization signal without breakdown of the tissue, thus some optimization may be required for each tissue sample.
- Neutralize the protease activity in 0.2% glycine in 1x PBS for 2 min.
- Rinse the slides once in 1x PBS for 2 min.
- Treat the slides in 4% PFA solution for 10 min to re-fix the RNA which could be damaged by the protease treatment.
- Rinse the slides twice in 1x PBS for 2 min each.
- While the slides are in the PBS washes, make up 250 ml of 0.1 M triethanolamine solution pH 8.0 by mixing 12.5 ml of 2M triethanolamine (29.8g in 100ml milliQ H2O, pH8.0 with HCl) into 236.25 ml milliQ H2O in a glass dish. Add 1.25 ml acetic anhydride into the triethanolamine buffer immediately before putting the slides in, and stir well. After adding the slides, continue to stir slowly for 10 minutes. This requires that the slide rack is elevated in the container of triethanolamine / acetic anhydride. We use a clamping system with support stand. Note: In this step, positively charged amino groups that may lead to non-specific binding of the probe are acetylated. Also, acetic anhydride is toxic; this solution should therefore be prepared in a fume hood and disposed of properly.
- Rinse the slides twice in 1x PBS for 2 min.
- Dehydrate the tissue sections again:
- H2O - 1 min
- 10% ethanol - 30 sec
- 30% ethanol - 30 sec
- 50% ethanol - 30 sec
- 70% ethanol - 30 sec
- 85% ethanol - 30 sec
- 95% ethanol - 30 sec
- 100% ethanol - 30 sec
- 100% ethanol - 30 sec
- Hybridization
- Warm a heating block to 85°C.
- Prepare the hybridization buffer. For 10 ml, mix in a Falcon tube 1.25 ml in situ hybridization salts (3M NaCl, 100mM Tris-HCl pH 8.0, 100mM Na Phosphate pH6.8, 50mM EDTA), 5 ml deionized formamide, 2.5 ml 50 % Dextran sulfate, 250 μl 50x Denhardt, 125 μl 100mg/ml tRNA, and 875 μl H2O. Note: Dextran sulfate is very viscous, so it is best to preheat the solution to 60°C to make pipetting easier. Hybridization buffer can be store at -20°C.
- Prepare the different probes. For each slide, mix 1 μl probe with 9 μl milliQ H2O and 10 μl deionized formamide. Heat the probe at 85°C for 2-3 min, immediately chill on ice, centrifuge briefly, and add the probe to 80μl hybridization buffer per slide. Mix well by pipetting, but avoid making bubbles. Note: The amount of probe to be added per slide needs to be tested empirically. Generally, probes are used at a final concentration of 0.5 ng/kb/μl probe complexity. Since hybridizations are performed with 100 μl per slide, approximately 25 ng of probe is needed per slide for probes that are 0.5-kb in length and 50 ng of probe per slide for probes that are 1-kb long. For simplicity, we often try 0.5, 1, and 4 μl probe per slide.
- Place the slides on a clean surface and let them air dry completely for 5-10 min. Pipet the probe/hybridization buffer mix onto the right edge of the slide. Gradually lower a coverslip onto the slide; making sure that the hybridization solution covers all sections without any bubbles. Tap the coverslip lightly with your finger where bubbles form to displace them from all tissue sections. Do not pull the cover slip off, as this will damage the sections. Note: An alternate method commonly used for this step is to prepare "sandwiches" of two slides that are incubated with the same probe. For more details on this method, see ref. 14.
- Prepare a humidity chamber in a perfectly flat plastic box. Cover the bottom of the box with whatman paper moistened with milliQ water, covered the paper with parafilm leaving the corners uncovered; place the slides in the plastic box and seal the box tightly.
- Incubate the slides at the desired hybridization temperature, typically 50 - 55 °C, for 16-20 hours.
- For use the next morning, prepare 1 liter 0.2x SSC and store this at 55°C. In addition, prepare 1 liter NTE buffer (0.5 M NaCl, 10 mM Tris-HCl pH7.5, 1 mM EDTA pH8.0) and store at 37°C.
- Post-hybridization treatment
- Remove the coverslips carefully to not damage the sections. Place the slides in a rack and wash them twice in 0.2x SSC at 55°C for one hour. Note: Coverslips often fall off easily when the slides are placed upright. If the coverslip remains attached, immerse the slide in warm 0.2x SSC for 1 or 2 min to wash the coverslip off. Avoid pulling the coverslip of the slide, as this can cause damage to the sections.
- In the meantime, prepare 500 ml washing buffer (1% BSA, 0.3% triton in TBS buffer) and 100 ml blocking solution (0.5% blocking reagent in TBS buffer). Stir the blocking powder into TBS that is slowly heated to 70°C and let it cool down to room temperature once dissolved. Note: The volumes of washing buffer and blocking solution needed will vary depending on the size of the flat plastic box used in steps 4.3.8 -11. It will take some time to completely dissolve Triton, so prepare enough solution in advance.
- Equilibrate the slides in 1x NTE for 2 min.
- Proceed to the RNAse A treatment by transferring the slides into 1x NTE buffer to which 20μg/ml RNAse A is added just prior use, incubate at 37°C for 30 min. Note: RNAse A cleaves free single-stranded RNA as well as at mismatches in RNA duplexes. This step helps reduce the level of non-specific binding of the probe, as cleaved probed fragments are washed off in the last 0.2x SSC wash (see 4.3.6).
- Rinse the slides twice in 1x NTE for 2 min each.
- Wash the slides in fresh 0.2x SSC at 55°C for one hour.
- Equilibrate the slides in 1x PBS for 5 min at room temperature.
- Transfer the slides from the rack onto the bottom of a flat plastic box and cover them with a minimal volume of blocking solution. Block the slides for 45 min on a slowly shaking platform.
- Transfer the slides to a second clean flat plastic box and wash them for 45 min with washing buffer on a shaking platform.
- Proceed to the antibody incubation. Dilute the anti-digoxigenin antibody in washing buffer (1:5 v/v) and either apply a minimal volume directly onto each slide or place the slide into a flat plastic box and cover them with a minimal volume of antibody solution. Incubate the slides in the dark for 2-3 hours at room temperature or overnight at 4°C.
- Wash the slides 4 times for 15 min each with washing buffer on shaking platform. Use a minimal volume of washing buffer and a flat plastic box. Clean the plastic box between each of the washes. Note: Cleaning the box between washes will reduced the general background on the slide. To simplify this, we use two identical flat boxes and transfer the slides into a clean box after each wash.
- Transfer the slides into 1x TBS for 2 min.
- Equilibrate the slides in TN buffer twice for 2 min each.
- Prepare staining solution immediately before use by adding 20 μl NBT/BCIP per 1 ml TN buffer. Pour the staining solution in a small plastic weighing dish. Sandwich two slides together with the sections facing each other. Dip one long side of the sandwich in the solution, allowing capillary action to pull up the solution. Drain the slides on a Kimwipe and fill the slides with antibody solution again. You may need to tap the slides as the solution flows in to avoid bubbles. Place the slides in a plastic box and store at room temperature in the dark.
- Day 10 to Day 15: Refresh the staining solution twice daily for 1-5 days by rinsing the slides in TN buffer and preparing new sandwiches containing fresh staining solution. Note: Replacing the staining solution at regular intervals will improve the signal to background ratio. Development of the signal can be monitored under a compound or stereo-microscope. It is possible to photograph the sections while they are in TN buffer in between rounds of staining or once they are transferred into water just before permanent mounting.
- Mounting
- Once the signal is obvious, rinse the slides in milliQ H2O and dehydrate them quickly through an ethanol series:
- 10% ethanol - 10 s
- 30% ethanol - 10 s
- 50% ethanol - 10 s
- 70% ethanol - 5 s
- 80% ethanol - 5 s
- 95% ethanol - 5 s
- 100% ethanol - 5 s
- histoclear - 2 min
- histoclear - 2 min
- Mount the slides by placing one or two drops of Toluene-based mounting medium on each slide and slowly lowering the coverslip onto the slide avoiding the formation of bubbles. Let the mounting medium harden overnight before analyzing the result under a compound microscope. Once mounted, slides can be stored for years at room temperature.
5. Representative Results:
After 1-5 days, a red-purple signal will develop in those cells where the probe has hybridized to complementary transcripts (Figure 1). The color signal thus provides a direct visualization at cellular resolution of the expression pattern of the gene of interest. A weaker background staining may also develop, resulting from the non-specific binding of the probe or staining of the plant tissues (Figure 3). Such background signal might be relatively more pronounced in the small, less determined cells of the tissue, but background staining would also be observed in sections hybridized to the negative and positive control probes and would not be reproducible between experiments.
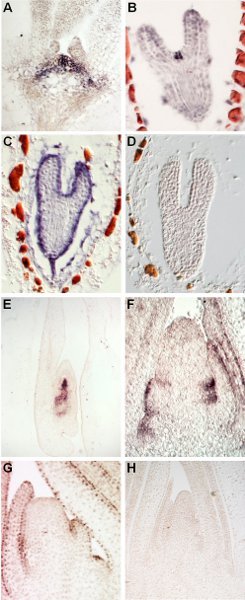 Figure 1. Representative results obtained with different probes on Arabidopsis and maize tissues. In situ hybridizations of Arabidopsis (A-D) and maize (E-H) tissues with distinct gene-specific probes illustrating the type of results that can be expected upon the successful completion of the outlined protocol. (A) Localization of SHOOTMERISTEMLESS1 (STM) in the Arabidopsis vegetative shoot apex; note the presence of purple blue signal in the indeterminate cells of the meristem and the lack of signal in the surrounding leaves. (B-D) Sections of Arabidopsis torpedo stage embryos showing CLAVATA3 (B) expression in the few embryonic stem cells, AtML1 (C) expression in the epidermal layer, and the lack of signal in sections probed with a random negative control RNA (D). (E) Localization of the STM homolog knotted1 in the developing maize embryo; note the strong signal specifically in the embryonic meristem and root. (F-H) Longitudinal sections through the vegetative shoot apex from maize showing distinct in situ hybridization patterns for arf3a (F) and ocl4 (G) and no hybridization signal for the negative control probe (H). arf3a is expressed on the abaxial/bottom leaf surface, whereas the AtML1 homolog ocl4 is expressed specifically in the epidermis.
Figure 1. Representative results obtained with different probes on Arabidopsis and maize tissues. In situ hybridizations of Arabidopsis (A-D) and maize (E-H) tissues with distinct gene-specific probes illustrating the type of results that can be expected upon the successful completion of the outlined protocol. (A) Localization of SHOOTMERISTEMLESS1 (STM) in the Arabidopsis vegetative shoot apex; note the presence of purple blue signal in the indeterminate cells of the meristem and the lack of signal in the surrounding leaves. (B-D) Sections of Arabidopsis torpedo stage embryos showing CLAVATA3 (B) expression in the few embryonic stem cells, AtML1 (C) expression in the epidermal layer, and the lack of signal in sections probed with a random negative control RNA (D). (E) Localization of the STM homolog knotted1 in the developing maize embryo; note the strong signal specifically in the embryonic meristem and root. (F-H) Longitudinal sections through the vegetative shoot apex from maize showing distinct in situ hybridization patterns for arf3a (F) and ocl4 (G) and no hybridization signal for the negative control probe (H). arf3a is expressed on the abaxial/bottom leaf surface, whereas the AtML1 homolog ocl4 is expressed specifically in the epidermis.
The following gene fragments were used as probe: STM: the cDNA region encompassing amino acids 81 - 382, which includes the conserved homeodomain; CLV3: the full length cDNA; AtML1: the third exon; kn1: the entire open reading frame; arf3a: nucleotides 9 - 225 of cDNA clone HM004539; ocl4: the 3' 1.7 kb of the full length cDNA, which includes several conserved protein domains.
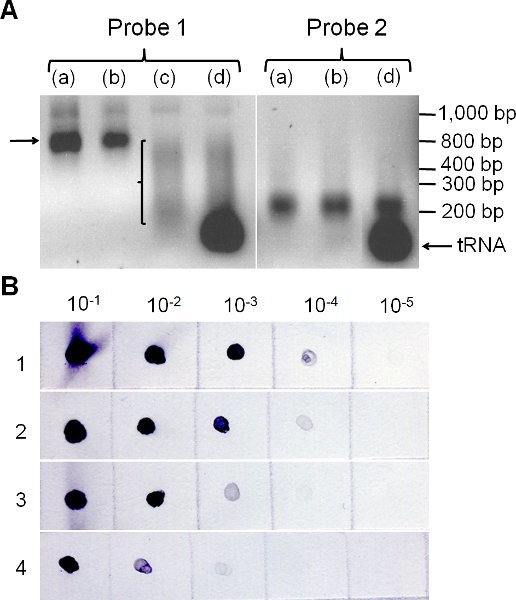 Figure 2. Checking points while making in vitro transcription probes. (A) In situ hybridization probes are checked on a standard agarose gel after in vitro transcription (a), after DNase treatment and subsequent purification of the in vitro transcription reaction (b), after carbonate hydrolysis –if required- (c) and when ready for use (d). For probes over 250 bp in length, such as probe 1, the carbonate hydrolysis will yield a range of smaller probe fragments (bracket). (B) Colorimetric quantification of probes by dot blot analysis. Dilutions ranging from 10-1 to 10-5 of a 100 ng/μl control probe (1) and of three newly DIG-labeled probes (2-4) are spotted on a transfer membrane, incubated with anti-DIG antibody, and assayed using the colorimetric assay outlined in section 2.3 of the protocol. The analysis suggests the following estimated concentrations: probe 2, ˜100 ng/μl; probe 3, ˜10 ng/μl probe 4, ˜1 ng/μl. Probe 4 is unlikely to yield a good in situ hybridization signal.
Figure 2. Checking points while making in vitro transcription probes. (A) In situ hybridization probes are checked on a standard agarose gel after in vitro transcription (a), after DNase treatment and subsequent purification of the in vitro transcription reaction (b), after carbonate hydrolysis –if required- (c) and when ready for use (d). For probes over 250 bp in length, such as probe 1, the carbonate hydrolysis will yield a range of smaller probe fragments (bracket). (B) Colorimetric quantification of probes by dot blot analysis. Dilutions ranging from 10-1 to 10-5 of a 100 ng/μl control probe (1) and of three newly DIG-labeled probes (2-4) are spotted on a transfer membrane, incubated with anti-DIG antibody, and assayed using the colorimetric assay outlined in section 2.3 of the protocol. The analysis suggests the following estimated concentrations: probe 2, ˜100 ng/μl; probe 3, ˜10 ng/μl probe 4, ˜1 ng/μl. Probe 4 is unlikely to yield a good in situ hybridization signal.
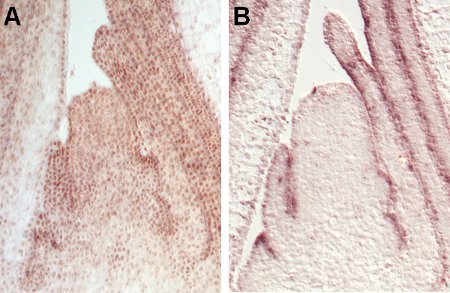 Figure 3. Comparison of a non-specific probe to a well-working probe. Longitudinal sections through the vegetative shoot apex from maize showing a non-specific background signal (A) and a specific in situ hybridization signal for the OCL5 probe in the outer cell layer (B).
Figure 3. Comparison of a non-specific probe to a well-working probe. Longitudinal sections through the vegetative shoot apex from maize showing a non-specific background signal (A) and a specific in situ hybridization signal for the OCL5 probe in the outer cell layer (B).
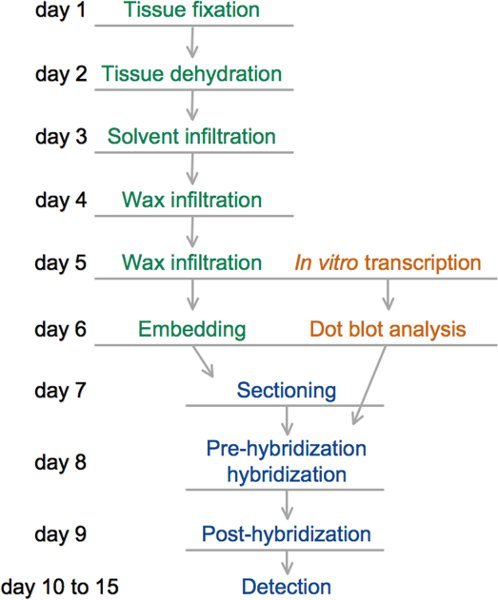 Figure 4. Diagram showing the timeline of steps in the in situ hybridization protocol. Tissue embedding steps are indicated in green, probe-preparation steps in orange, and the in situ hybridization steps in blue. This timeline considers that the DNA template for the in vitro transcription of the probe is available. Parafin-embedded tissue blocks and DIG-labeled probes can be prepared beforehand and stored at 4°C and -80°C, respectively, until the time of use. The in situ hybridization protocol can then be completed in as little as four days.
Figure 4. Diagram showing the timeline of steps in the in situ hybridization protocol. Tissue embedding steps are indicated in green, probe-preparation steps in orange, and the in situ hybridization steps in blue. This timeline considers that the DNA template for the in vitro transcription of the probe is available. Parafin-embedded tissue blocks and DIG-labeled probes can be prepared beforehand and stored at 4°C and -80°C, respectively, until the time of use. The in situ hybridization protocol can then be completed in as little as four days.
Discussion
The in situ hybridization method outlined here allows the direct visualization of the spatiotemporal expression pattern of any gene of interest with great cellular resolution, high sensitivity and specificity. The protocol does not allow a quantitative comparison of expression levels between genes. However, the method's high sensitivity and resolution can reveal gradients of gene expression within a tissue8, 18, which is not possible with most other methods of analyzing gene expression.
The protocol includes many steps, which can make trouble-shooting problematic. The most critical steps of the protocol are the fixation of the tissue and the selection and labeling of the probe. Poorly infiltrated tissues and probes with low DIG incorporation will yield very weak signals that might be difficult to detect above background. For each new gene of interest, it is advisable to try a few distinct probes derived from different regions of the transcript. Occasionally, two or three probes targeting different smaller regions of the gene may need to be combined to obtain a strong and specific hybridization signal. In addition, the hybridization conditions, such as the temperature and composition of the hybridization buffer, can be optimized for each gene or tissue to lower or increase the hybridization stringency. Also the protease treatment step is a variable and can be changed to adapt the protocol for different plant species or for the detection of transcripts with very low abundance. The inclusion of both positive and negative control probes will be helpful in identifying problematic points of the protocol.
The procedure is optimized for paraffin-embedded plant tissue sections but with minor modifications can be used also for whole-mount in situ hybridization. Although widely used in animal research19, whole mount in situ hybridization will be limited to plant tissues that can be easily infiltrated, such as embryos, roots or young meristematic tissues20,21. The protocol can also be easily modified to simultaneously detect two distinct mRNAs or localize transcripts alongside proteins15,22,23. Finally, it is possible to extend the application of this method to the visualization of small RNA expression patterns in plants. miRNA expression patterns have been determined using this protocol in both maize and Arabidopsis8,14. More recently, the in vitro transcribed probes have been replaced by commercially available DIG-labeled locked nucleic acid (LNA) oligo probes that increase signal sensitivity18, 24.
Disclosures
No conflicts of interest declared.
Acknowledgments
Research in the laboratory of M.T. is supported by grants from the National Science Foundation (DBI-0820610 and IOS-1022102) and the NY Department of Health (NYSTEM-C024308). C.M. received postdoctoral fellowships from the Spanish Ministry of Education and Science (2007-0937) and the Foundation Rafael del Pino, Spain.
References
- Rensink WA, Buell CR. Microarray expression profiling resources for plant genomics. Trends. Plant. Sci. 2005;10:603–609. doi: 10.1016/j.tplants.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010;42:1060–1067. doi: 10.1038/ng.703. [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SM, Orlando DA, Lee JY, Wang JY, Koch J, Dinneny JR, Mace D, Ohler U, Benfey PN. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science. 2007;318:801–806. doi: 10.1126/science.1146265. [DOI] [PubMed] [Google Scholar]
- Brooks L, Strable J, Zhang X, Ohtsu K, Zhou R, Sarkar A, Hargreaves S, Elshire RJ, Eudy D, Pawlowska T, Ware D, Janick-Buckner D. Microdissection of shoot meristem functional domains. PLoS. Genet. 2009;5:e1000476–e1000476. doi: 10.1371/journal.pgen.1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith DW, Birnbaum K. Global studies of cell type-specific gene expression in plants. Annu. Rev. Plant. Biol. 2006;57:451–475. doi: 10.1146/annurev.arplant.57.032905.105302. [DOI] [PubMed] [Google Scholar]
- Ohtsu K, Smith M, Emrich S, Borsuk L, Zhou R, Chen T, Zhang X, Timmermans M, Beck J, Buckner B, Janick-Buckner D, Nettleton D, Scanlon M, Schnable P. Global gene expression analysis of the shoot apical meristem of maize (Zea mays L.) Plant. J. 2007;52:391–404. doi: 10.1111/j.1365-313X.2007.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez MT, Kui JS, Thomas J, Heller BA, Timmermans MCP. microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature. 2004;428:84–88. doi: 10.1038/nature02363. [DOI] [PubMed] [Google Scholar]
- Kramer EM, Irish VF. Evolution of genetic mechanisms controlling petal development. Nature. 1999;399:144–148. doi: 10.1038/20172. [DOI] [PubMed] [Google Scholar]
- Stuurman J, Jäggi F, Kuhlemeier C. Shoot meristem maintenance is controlled by a GRAS-gene mediated signal from differentiating cells. Genes Dev. 2002;16:2213–2218. doi: 10.1101/gad.230702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, McCormick S, Timmermans M, Sinha N. The expression domain of PHANTASTICA determines leaflet placement in compound leaves. Nature. 2003;424:438–443. doi: 10.1038/nature01820. [DOI] [PubMed] [Google Scholar]
- Kramer EM, Holappa L, Gould B, Jaramillo MA, Setnikov D, Santiago PM. Elaboration of B gene function to include the identity of novel floral organs in the lower eudicot Aquilegia. Plant Cell. 2007;19:750–766. doi: 10.1105/tpc.107.050385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman ZB, Cohen O, Alvarez JP, Abu-Abied M, Pekker I, Paran I, Eshed Y, Zamir D. The making of a compound inflorescence in tomato and related nightshades. PLoS Biol. 2008;6:e288–e288. doi: 10.1371/journal.pbio.0060288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidner C, Timmermans MCP. In situ hybridization as a tool to study the role of microRNAs in plant development. Methods. Mol. Biol. 2006;342:159–179. doi: 10.1385/1-59745-123-1:159. [DOI] [PubMed] [Google Scholar]
- Jackson D. Double labeling of KNOTTED1 mRNA and protein reveals multiple potential sites of protein trafficking in the shoot. 2002;129:1423–1429. doi: 10.1104/pp.006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen WA. Botanical Histochemistry. San Francisco: Freeman; 1962. pp. 408–408. [Google Scholar]
- Jackson D. In situ hybridization in plants. In: Bowles DJ, Gurr SJ, McPherson M, editors. Molecular Plant Pathology: A Practical Approach. Oxford: University Press; 1991. pp. 163–174. [Google Scholar]
- Chitwood DH, Nogueira FT, Howell Montgomery, Carrington TA, C J, Timmermans MCP. Pattern formation via small RNA mobility. Genes. Dev. 2009;23:549–554. doi: 10.1101/gad.1770009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piette D, Hendrickx M, Willems E, Kemp CR, Leyns L. An optimized procedure for whole-mount in situ hybridization on mouse embryos and embryoid bodies. Nat. Protoc. 2008;3:1194–1201. doi: 10.1038/nprot.2008.103. [DOI] [PubMed] [Google Scholar]
- Blilou I, Xu J, Wildwater M, Willemsen V, Paponov I, Friml J, Heidstra R, Aida M, Palme K, Scheres B. The PIN auxin efflux facilitator network controls growth and patterning in Arabidopsis roots. Nature. 2005;433:39–44. doi: 10.1038/nature03184. [DOI] [PubMed] [Google Scholar]
- Jackson D, Hake S. Control of phyllotaxy in maize by the abphyl1 gene. Development. 1999. pp. 126–315. [DOI] [PubMed]
- Chuck G, Whipple C, Jackson D, Hake S. The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development. 2010;137:1243–1250. doi: 10.1242/dev.048348. [DOI] [PubMed] [Google Scholar]
- Long JA, Barton MK. The development of apical embryonic pattern in Arabidopsis. Development. 1998;125:3027–3035. doi: 10.1242/dev.125.16.3027. [DOI] [PubMed] [Google Scholar]
- Nogueira FT, Chitwood DH, Madi S, Ohtsu K, Schnable PS, Scanlon MJ, Timmermans MCP. Regulation of small RNA accumulation in the maize shoot apex. PLoS Genet. 2009;5:e1000320–e1000320. doi: 10.1371/journal.pgen.1000320. [DOI] [PMC free article] [PubMed] [Google Scholar]