

Abstract
Standard slice electrophysiology has allowed researchers to probe individual components of neural circuitry by recording electrical responses of single cells in response to electrical or pharmacological manipulations1,2. With the invention of methods to optically control genetically targeted neurons (optogenetics), researchers now have an unprecedented level of control over specific groups of neurons in the standard slice preparation. In particular, photosensitive channelrhodopsin-2 (ChR2) allows researchers to activate neurons with light3,4. By combining careful calibration of LED-based photostimulation of ChR2 with standard slice electrophysiology, we are able to probe with greater detail the role of adult-born interneurons in the olfactory bulb, the first central relay of the olfactory system. Using viral expression of ChR2-YFP specifically in adult-born neurons, we can selectively control young adult-born neurons in a milieu of older and mature neurons. Our optical control uses a simple and inexpensive LED system, and we show how this system can be calibrated to understand how much light is needed to evoke spiking activity in single neurons. Hence, brief flashes of blue light can remotely control the firing pattern of ChR2-transduced newborn cells.
Protocol
1. Optical Calibration: Measuring LED Power
Attach a LED array to a heatsink actively cooled by a fan and affix this LED/heatsink aparatus to a collimating lens.
Replace lamp used in brightfield illumination with LED/heatsink/fan/lens apparatus. This apparatus must be carefully positioned so that the collimated LED beam travels along a straight optical path towards the condenser lens. Make sure the heatsink/fan is properly grounded to the system's common ground.
Drive the LED array with a power supply that can give fast and square pulses of current. This power supply may be controlled by a 5V TTL pulse originating from a pulse generator.
Center the collimated beam along the light path defined between the field diaphragm and the condensor lens. Ideally, the LED beam will slightly overfill the fully opened field diaphragm. Typically, a more tightly collimated LED beam will fill this aperture less, and will produce more power at the expense of uniformity. In our setup we increased light uniformity by choosing a collimating lens that projected a slightly expanded image of the LED array at its conjugate plane at the consenser diaphragm.
Achieve Kohler illumination by focusing the condenser so that the image of the field diaphragm (the diaphragm closest to the light source) is focused on the slice chamber (fig. 1). A thin tissue of lens paper can act as a projection screen to visualize the focused image of the field diaphragm at other depths.
Drill a series of pinholes of known diameter in an opaque material. Place one of these small pinholes over the sensor of an optical power-meter. Place the power meter on the specimen stage and center the power meter over the focused image of the field diaphragm by simply moving the power meter until it gives a maximum reading. Affix the power meter in this position.
Fully open all apertures (aperature diaphragm and field diaphragm). Systematically move the attached slice chamber/power meter relative to the optical lightpath and calculate the uniformity of optical power within the illuminated area. Build the uniformity plot for your system. If the microscope is properly setup with Kohler illumination centered at the objective's focus, maximum power should be directly underneath the objective, and regions outside of this focus should now receive a known amount of power according to the uniformity plot.
For each size of pinhole, build a standard curve of optical power vs. pinhole surface area. By adjusting the input current to the LED array, produce this curve at multiple power levels and from each curve calculate the power per mm2. If the LED array is to be used for patching optics, make sure to introduce optical elements necessary for patching (condensers, pinholes and filters) to know how much light is transmitted under patching illumination.
Flipping the power meter to face the objective, calculate the intensity of illumination at 470nm when the mercury lamp is turned on.
Add a live slice to the chamber, and rebuild standard curves to determine the light power transmitted through the scattering brain tissue.
2. Slicing Procedure and Electrophysiology
Part A: Slice Preparation
Anesthetize (60 mg/kg Ketamine and 2mg/kg Xylazine) and decapitate the mouse. Dissect the brain in artifical cerebral spinal fluid (ACSF, in mM: 124 NaCl, 3 KCl, 1.3 MgSO4, 26 NaHCO3, 1.25 NaHPO4, 20 glucose, 2 CaCl2; ~310mOsm, pH 7.4 when bubbled with a mixture of 95% O2 and 5% CO25,1), taking care not to damage the olfactory bulbs. Separate the two hemispheres and place on agar with the ventral surface even with one edge (for horizontal sections).
Glue the agar and dorsal surface of each cortical hemisphere to the vibratome chuck, and slowly fill the bath with ice-cold ACSF. Slice from ventral surface in 300 μm sections, transferring each section to warmed (34-36°C) and oxygenated ACSF, allowing them to recover for 30-45 minutes.
Part B: Loose Patch Measurement of the Threshold to Spike
After bringing the slices to room temperature for 30 minutes, gently place a slice in the recording chamber of the microscope chamber under constant perfusion of oxygenated ACSF. At risk of excessively stimulating ChR2 infected neurons, the presence of EYFP-ChR2 can be confirmed under epifluorescence (fig. 2a)
Pull glass electrodes on a pipette puller (Sutter P-97). Fill this electrode with ACSF. When placed in the ACSF bath the tip resistance should be between 7-10 MOhms.
Under fluorescent illumination, locate in the slice a healthy ChR2-EYFP neuron with mature morphology. Also locate this neuron's soma under patching optics.
With light positive pressure passed through the electrode tip, lower the patch electrode towards the identified fluorescent neuron. When membrane contact is made, quickly release positive pressure and apply a small and brief amount of suction through the tip. A giga-ohm seal should be made between the plasma membrane and the walls of the patch electrode.
Even if a giga-seal is not formed, if the electrode is sufficiently close to a fluorescent neuron spiking activity should produce a measurable local field potential. Activate ChR2 in this neuron by flashing different doses of light. Because light-dose is a function of both LED power and duration, calculate how much light is necessary to evoke an action potential at multiple powers and durations (fig. 2b). Also observe how much spiking occurs under mercury lamp illumination.
3. Representative Results:
On our microscope (Olympus BX51WI), our LED is in line with 2 apertures and a condenser lens, thus maintaining the original lightpath of the factory-installed arc lamp. By closing both the field diaphragm and the aperature diaphragm, we can achieve brightfield contrast sufficient for patch clamp recordings (fig. 1b). With all diaphragms fully open we expose the slice to maximum light power for channelrhodopsin activation (fig. 1a). On our microscope, this patching configuration produces light-density that is approximately three orders of magnitude lower than the maximum full field density (4.1 μW/mm2 versus 6.88 mW/mm2).
We see robust labeling of adult-born olfactory bulb granule and periglomerular neurons weeks after lentiviral infection of migrating neuroblasts in the rostral migratory stream (fig. 2a) A loose-patch recording from a single adult-born ChR2-EYFP expressing granule cell indicates that a 5 ms stimulation at maximum power (6.88 mW/mm2) is sufficient to evoke spiking (fig. 2b). Since expression level varies between cells, the amount of light that passes the threshold to spike will vary and should be described statistically for each cell type of interest.
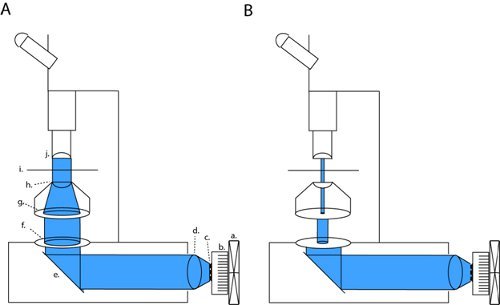 Figure 1. LED array setup for full-field photostimulation and patch-clamp slice electrophysiology. To activate channelrhodopsin (ChR2) we project a collimated beam through open back apertures and condenser optics (a). This configuration can be changed into high-contrast patching optics by fully closing the field diaphragm and modulating the width of the field diaphragm (b). Abbreviations: a. fan, b. heatsink, c. LED array, d. collimating lens, e. mirror, f. field diaphragm, g. aperture diaphragm, h. condenser lens, i. sample stage, j. objective.
Figure 1. LED array setup for full-field photostimulation and patch-clamp slice electrophysiology. To activate channelrhodopsin (ChR2) we project a collimated beam through open back apertures and condenser optics (a). This configuration can be changed into high-contrast patching optics by fully closing the field diaphragm and modulating the width of the field diaphragm (b). Abbreviations: a. fan, b. heatsink, c. LED array, d. collimating lens, e. mirror, f. field diaphragm, g. aperture diaphragm, h. condenser lens, i. sample stage, j. objective.
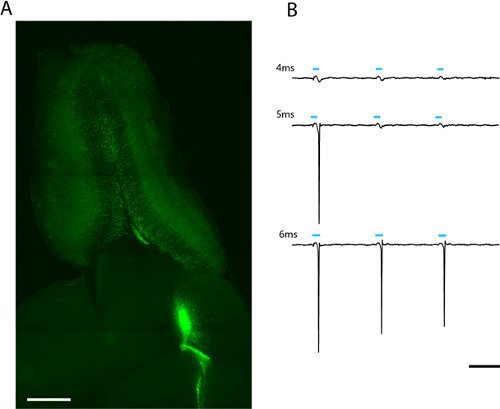 Figure 2. Image of a 300μm horizontal slice of olfactory bulb for patch clamp and whole-field photostimulation (a). Lentivirally infected adult-born granule cells expressing ChR2-EYFP can be seen radiating from the core of the olfactory bulb. The light-dose required to evoke spiking can be found by increasing the duration of the LED flash (b). The threshold for this granule cell was 5ms at full LED intensity (2.43mW/mm2). Scale in (a) = 500μm, scale in (b) = 50ms.
Figure 2. Image of a 300μm horizontal slice of olfactory bulb for patch clamp and whole-field photostimulation (a). Lentivirally infected adult-born granule cells expressing ChR2-EYFP can be seen radiating from the core of the olfactory bulb. The light-dose required to evoke spiking can be found by increasing the duration of the LED flash (b). The threshold for this granule cell was 5ms at full LED intensity (2.43mW/mm2). Scale in (a) = 500μm, scale in (b) = 50ms.
Discussion
Recent years have seen an explosion in the popularity of optogenetic tools for neuroscience research6. As a result, it is increasingly important to lower the barrier of entry for labs wishing to begin using these new tools. Here we describe how to conduct a simple and low cost retrofitting and calibration of a conventional patch-clamp rig so that it can do full-field optical stimulation of channelrhodopsin-expressing neurons. In particular, we apply this technique to the study of olfactory bulb adult neurogenesis to specifically control newly born neurons with light.
We demonstrate how to determine if a LED light source produces sufficient power for ChR2 activation, and demonstrate how the intrinsically rapid on/off speed of LEDs can easily regulate the dosage of light given to neurons. While our lab has successfully used this technique, there are alternative ways of exposing neurons to controlled doses of light each with its own benefits and drawbacks.
If more optical power is needed, many mercury lamps will produce considerably more light than an LED array, and a microscope's objective and filter sets are already present to focus the desired wavelengths onto the sample. However, with this solution neither the power nor the duration of exposure can be easily controlled. Length of exposure would need to be controlled by an external shutter, and these are often more expensive than a LED array, and the power would need to be attenuated by adding neutral density filters to the light path – at risk of causing vibrations that would reduce the success rate of patch-clamp experiments.
Our implementation also makes it difficult to control the spatial extent of illumination. If spot illumination is desired to target specific groups of ChR2 expressing neurons, one solution is to scan a laser spot with a galvometer scanning mirror to different locations on the slice. At this point, however, the microscope begins to resemble a conventional confocal microscope in both complexity and cost. A recently developed alternative is to focus the image of a micro-led array onto the sample7, or to shape a uniform field of excitation with a digital micromirror device8.
LED arrays have distinct advantages that make them uniquely suited to channelrhodopsin stimulation. Their on/off speed occurs on nanosecond timescales and so this speed can be used to finely tune the duration of light exposure to biological tissue. In addition, the color of LEDs can be chosen to fit specific excitation bands without the need for filtering. With the advent of red-shifted channelrhodopsin mutants, it would be simple to use two LEDs of different colors to individually excite two different populations of neurons. Alternatively, white LED arrays - like the one used in this report that combines blue (450nm peak) and green (550nm peak) gallium nitride diodes - are broad-spectrum which gives the investigator the freedom to target a wide range of excitation bands.
After optical calibration, each cell type that expresses channelrhodopsin must be characterized to determine how much light is required to evoke an action potential. We describe a simple electrophysiological characterization using the loose-patch method, which has the advantage of avoiding changing the cell’s internal dynamics with the pipette solution. However, in a full study whole-cell recordings are necessary to determine how much light produces a given amount of depolarization, and the stability of these evoked potentials over time. Unfortunately the sensitivity of spiking activity to light dose is depends on a number of parameters outside of experimental control. These include the level of ChR2 expression (a function of construct copy number and developmental regulation of the viral promoter), and the intrinsic biophysical properties of the neuron9.
The fluorescent intensity of the ChR2-EYFP fusion may be an indicator of how much light is needed to bring the neuron to threshold. However, there are nonlinearities in this sort of estimate caused by internalized aggregates of channel, and by shunting conductances caused by activating channels distal to the spike initiation zone. Ultimately, for every type of neuron a statistical characterization of the population will have to be made. This suffers for the caveat that dimly fluorescent cells are difficult to patch, and so viral constructs may benefit from an additional fluorescent label that is somatically restricted10.
It is clearly redundant to patch a ChR2 infected neuron if light can be used to control its spiking activity. A better use of optogenetics in-vitro is to investigate the synaptic connectivity of an optogenetically targeted population with other members of the circuit. In our hands, we have coupled full field stimulation of ChR2 adult-born granule cells with patch clamp recordings from mitral cells to measure the properties of the newly formed synapse between these two cell types 4. Because the probability of connectivity between a single granule and mitral cell is very low, our experiment was only possible by simultaneous activation of many hundreds ChR2 expressing granule cells. Future discovery of new genetic markers and new viral delivery methods to label specific neuronal populations will increase the usefulness of in-vitro optogenetics in different systems. In particular, this technique is ideally suited to discover sparse but strong connections between unexpected neuronal subsets11,12. As the variety of uses for this technique grows, so will the need for simple and low-cost hardware to control the light delivered onto in-vitro slices.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by the life insurance company "AG2R-La-Mondiale", Ecole des Neurosciences de Paris (ENP), the Agence Nationale de la Recherche "ANR-09-NEUR-004" in the frame of "ERA-NET NEURON" of FP7 program by the European Commission, and the Pasteur Foundation. Sebastien Wagner was supported by the Letten Foundation.
References
- Nissant A. Adult neurogenesis promotes synaptic plasticity in the olfactory bulb. Nature Neuroscience. 2009;12:728–730. doi: 10.1038/nn.2298. [DOI] [PubMed] [Google Scholar]
- Apicella A. Pyramidal Cells in Piriform Cortex Receive Convergent Input from Distinct Olfactory Bulb Glomeruli. Journal of Neuroscience. 2010;30:14255–14260. doi: 10.1523/JNEUROSCI.2747-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES. genetically targeted optical control of neural activity. Nature. 2005;8:1263–1263. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Bardy C. where new inhibitory neurons release neurotransmitters in the adult olfactory bulb. The Journal of Neuroscience. 2010;30:17023–17034. doi: 10.1523/JNEUROSCI.4543-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS. Functional maturation of the first synapse in olfaction: development and adult neurogenesis. The Journal of neuroscience. 2008;28:2919–2932. doi: 10.1523/JNEUROSCI.5550-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. Circuit-breakers: optical technologies for probing neural signals and systems. Nature reviews. Neuroscience. 2007;8:577–581. doi: 10.1038/nrn2192. [DOI] [PubMed] [Google Scholar]
- Grossman N. Multi-site optical excitation using ChR2 and micro-LED array. Journal of neural engineering. 2010;7:16004–16004. doi: 10.1088/1741-2560/7/1/016004. [DOI] [PubMed] [Google Scholar]
- Dhawale AK. Non-redundant odor coding by sister mitral cells revealed by light addressable glomeruli in the mouse. Nature neuroscience. 2010;13:1404–1412. doi: 10.1038/nn.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick JP. Functional control of transplantable human ESC-derived neurons via optogenetic targeting. Stem cells. 2010;28:2008–2016. doi: 10.1002/stem.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N. Neurons born in the adult dentate gyrus form functional synapses with target cells. Nature Neuroscience. 2008;11:901–907. doi: 10.1038/nn.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Channelrhodopsin-2 Localised to the Axon Initial Segment. PLoS ONE. 5:e13761–e13761. doi: 10.1371/journal.pone.0013761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]